
Abstract
Plant protection products (PPPs) and the active substance(s) contained within them are rigorously and comprehensively tested prior to registration to ensure that human health is not impacted by their use. In recent years, there has been a widespread drive to have more relevant testing strategies (e.g., ILSI/HESI-ACSA and new EU Directives), which also take account of animal welfare, including the 3R (replacement, refinement, and reduction) principles. The toxicity potential of one such new active substance, sulfoxaflor, a sulfoximine insecticide (CAS #946578-00-3), was evaluated utilizing innovative testing strategies comprising: (1) an integrated testing scheme to optimize information obtained from as few animals as possible (i.e., 3R principles) through modifications of standard protocols, such as enhanced palatability study design, to include molecular endpoints, additional neurotoxicity and immunotoxicity parameters in a subchronic toxicity study, and combining multiple test guidelines into one study protocol; (2) generation of toxicokinetic data across dose levels, sexes, study durations, species, strains and life stages, without using satellite animals, which was a first for PPP development, and (3) addition of prospective mode of action (MoA) endpoints within repeat dose toxicity studies as well as proactive inclusion of specific MoA studies as an integral part of the development program. These novel approaches to generate key data early in the safety evaluation program facilitated informed decision-making on the need for additional studies and contributed to a more relevant human health risk assessment. This supplement also contains papers which describe in more detail the approach taken to establish the MoA and human relevance framework related to toxicities elicited by sulfoxaflor in the mammalian toxicology studies:
Introduction
Plant protection products (PPPs) and the active substance(s) contained within them are tested rigorously prior to registration to ensure that human health is not negatively impacted by their use. For decades, an extensive battery of mammalian toxicology tests has been required, aiming to generate data on potential adverse effects: for example, acute oral/dermal toxicity, skin and eye irritation, skin sensitization, genetic toxicity, systemic toxicity, developmental and reproductive toxicity, neurotoxicity, and carcinogenicity; in a range of species, durations, life stages and endpoints. However, as summarized by CitationCarmichael et al. (2006), “Not all studies conducted in a registration dossier are currently used in quantitative risk assessment for agricultural chemicals. The current regulatory schemes operate on the principle of covering all of the hazard endpoints that could be critical in several species and for multiple durations. From this list, a choice of studies is made to derive reference doses.” Although the data generated through this testing paradigm have been extensive, the relevance of data from high-dose animal studies to human health outcomes is not well characterized and further experiments are often required to determine the mechanism causing the observed adverse effects. As noted by the CitationCouncil of Canadian Academies (2012) “Even when the current testing approach is successful at defining the hazard, the information is usually of a specific nature, which may not be useful for extrapolation to other species, other life stages, or susceptible populations.”
In addition, PPPs are often developed for a global market and the testing requirements for registration often vary across geographies and include specific regional testing requirements (). The toxicity testing strategy for a PPP under development, through the discovery phase of a new active substance to regulatory approval of the active and its products, is usually designed to take these various testing requirements into account. As illustrates, it is often necessary to conduct certain toxicity studies to satisfy the requirements of a single regulatory authority, in addition to conducting the standard testing package that is needed by all regulatory authorities. For example, the 1-year chronic dog study is not routinely required by the US EPA, EU or China but is usually conducted for a global PPP registration because regions such as Brazil and Japan still require this study, despite questions surrounding the scientific value of this study type (CitationKobel et al. 2010).
Table 1. Summary of global regulatory authorities and PPP testing requirements.
Therefore, in recent years, there has been a widespread drive to have more relevant testing strategies (e.g., International Life Sciences Institute/Health and Environmental Sciences Institute—Agricultural Chemical Safety Assessment Technical Committee [ILSI/HESI-ACSA] and new EU Directives [1107/2009 EC]), which also take account of animal welfare, including the 3R (replacement, refinement, and reduction) principles. The ACSA Technical Committee of ILSI/HESI was established in 2000, charged with proposing an updated testing scheme for PPPs that incorporated the current understanding of toxicity and exposure science (CitationCarmichael et al. 2006). The Committee concluded that a tiered approach to the toxicity testing of PPPs would be appropriate and proposed a number of possible refinements to the current testing paradigm (CitationBarton et al. 2006, CitationCarmichael et al. 2006, CitationCooper et al. 2006, CitationDoe et al. 2006). In contrast to initiatives aimed at reducing unnecessary toxicity testing, it is also becoming much more common for active substances to be tested above the “standard” toxicity testing requirements and incorporated higher-tier investigative studies such as mode-of-action (MoA) work to enable registration of the new PPPs. This increase in higher-tier and customized study testing is driven by a general trend to obtain a mechanistic understanding underlying hazard characteristics of molecules to inform risk management decisions. These additional MoA data can be useful to support or refute the human relevance of a toxicity finding, which may lead to cessation of further development of a new active substance or performing further research to elucidate the MoA. The MoA/human relevance framework (HRF) was developed by the International Programme on Chemical Safety (IPCS) of the World Health Organization (WHO) (CitationBoobis et al. 2006, Citation2008, CitationSonich-Mullin et al. 2001) and ILSI (CitationMeek et al. 2003, CitationSeed et al. 2005) and can be used as a template upon which to elucidate the human relevance of effects observed in animals. A classic example of this is rodent liver effects driven by a constitutive androstane receptor (CAR)-mediated MoA, which would be considered not relevant to humans, and therefore, this finding is not used to derive the reference doses (RfDs) or to drive hazard classifications that are part of the risk assessment process to safeguard human health.
New testing paradigm
In recent years, our laboratory implemented innovations in regulatory toxicity testing of PPPs. The foundation of this strategy was grounded in the guiding principle of incorporating new techniques with robust scientific scrutiny, while concurrently striving to replace, refine, and reduce (3Rs) animal use whenever possible through integrated testing. The approaches fall under three main headings:
1) toxicity testing incorporating 3R principles,
2) integrated toxicokinetic (TK) investigations across studies, and
3) prospective MoA investigations
This introductory paper will describe examples of these approaches from the toxicology program of a new sulfoximine insecticide, sulfoxaflor (CAS #946578-00-3, XDE- 208, X11422208, XR-208, [1-(6-trifluormethylpyridin-3-yl)ethyl)](methyl)-oxido-l4-sulfanylidenecyanamide). Sulfoxaflor displays a high level of biological activity in sap-feeding insects (Zhu et al. 2010), as one of a new class of insecticidal molecules (sulfoximines) that are chemically distinct and appear to exert insecticidal actions through an interaction with nAChRs that is different from the neonicotinoids and also possibly from other sulfoximines (CitationZhu et al. 2011, CitationLonghurst et al. 2012, CitationBabcock et al. 2011, CitationWatson et al. 2011).
This special edition also contains papers that describe in more detail the approach taken to establish the MoA and HRF related to toxicities elicited by sulfoxaflor in the mammalian toxicology studies:
4) developmental toxicity in rats mediated via the fetal muscle nAChR (CitationEllis-Hutchings et al. 2014),
5) liver tumors in rodents mediated via CAR/PXR (CitationLeBaron et al. 2014), and
6) Leydig cell tumors in Fischer 344 rats (CitationRasoulpour et al. 2014)
Integrated toxicity testing incorporating 3R principles
The 3Rs are a widely accepted ethical framework for conducting scientific experiments using animals humanely (CitationRussell and Burch 1959). The following definitions (from the UK National Centre for the Replacement, Refinement and Reduction of Animals in Research Citation[NC3Rs], 2011) describes the concept as follows:
Replacement refers to methods that avoid or replace the use of animals in an area where they would otherwise have been used.
Reduction refers to methods which minimize animal use and enable researchers to obtain comparable levels of information from fewer animals or to obtain more information from the same number of animals, thereby reducing future use of animals.
Refinement refers to improvements to scientific procedures and husbandry which minimize actual or potential pain, suffering, distress or lasting harm and/or improve animal welfare in situations where the use of animals is unavoidable.
Approaches to the 3Rs described in the current paper include modifying standard palatability studies to provide additional information, integrating additional testing parameters into standard toxicity studies, conducting dietary developmental toxicity studies, and obtaining MoA data from regulatory studies.
Modified approach to determine palatability
Rodents. Palatability studies are dose range-finding studies traditionally designed to identify the ‘Maximum Tolerated Dose’ (MTD) in the chosen species/strain of test animal in preparation for conducting repeat-dose studies. These studies are usually the first dietary administration of an active substance, which traditionally involves administering high doses to determine the MTD that can be used to set dose levels in subsequent studies. Due to the nature of these studies, they have the potential to use significant numbers of animals, with previous study designs often using both sexes with 5 animals/group. Therefore, palatability studies within our new testing strategy have been conducted in a staged manner, with an ‘Up-and-Down’-like method utilized in rodents to decide on the next dose level as key data became available. For example, the process was used until an ‘MTD’ was determined in the design of the sulfoxaflor-administered mouse palatability study ().
Figure 1. The mouse palatability study was conducted in a stepwise manner using a minimum number of animals and also minimizing excessively high-dose levels.
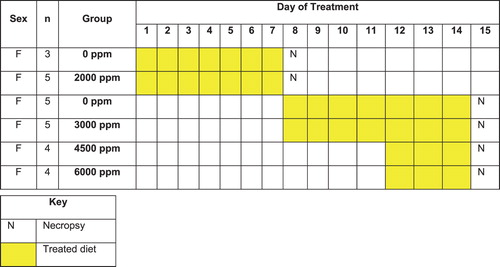
Initially, groups of five female mice were given a diet containing 0 or 2000 ppm sulfoxaflor (previously determined as the maximum palatable dietary concentration in rats) daily for 7 days. The mice were necropsied on study Day 8 (along with a group of three control mice, which were initially excluded from the study following randomization). The original five control mice remained for comparison on study with subsequent treatment groups. As no significant effects were observed in mice given 2000 ppm, an additional group of five female mice received diets containing 3000 ppm of sulfoxaflor daily for up to 7 days. Again, no significant effects were noted; therefore, two additional groups of four female mice received diets containing 4500 or 6000 ppm sulfoxaflor and were terminated after 3 days on study because of unacceptable decreases in feed consumption and body weights. The five control mice given untreated feed throughout the study were necropsied the same time. Animals were evaluated by daily body weights, feed consumption and cage-side examinations. A complete necropsy was conducted on all animals, and liver weights were recorded at necropsy. Samples of liver tissue from animals in the control, 4500 ppm and 6000 ppm groups were collected after 3 days of treatment for possible future enzyme analyses. Blood samples were collected from all animals (treated and controls) at necropsy and stored at − 80°C for possible TK analysis. Histological evaluation of the liver from all animals was conducted.
This method allowed a reduction in the number of animals used and reduced potential unnecessary overt toxicity by ‘ramping up’ the dose levels rather than initiating higher-dose groups with no prior knowledge of the potential toxicity from repeated doses. Only females were used in this study, as they are often considered more sensitive than males to palatability effects (CitationLaviano et al. 1996), which further reduced the number of animals used. In this manner, 3000 ppm was identified as the appropriate high dietary concentration to use in the sulfoxaflor-administered 28-day mouse study, which is a key aim of the palatability studies. This method was the first step in a default, ‘triggered’ palatability approach which is now standard at Dow. In the default study design, a group of three control and a single treatment group (e.g., 500 mg/kg/day) of female rats are administered test material for 14 days. Two additional groups of three female rats are added to this study on study Day 8 and all animals undergo necropsy at the same time on Day 15. This staggered start for the two additional dose groups allows more precise dose–response information as body weight and feed consumption effects in the initial treatment group drive dose levels for the staggered start groups. For example, if the initial group is administered 500 mg/kg/day and has a slight feed consumption response, compared to controls, the additional groups can be administered 250 and 750 mg/kg/day, which start on study Day 8. If 500 mg/kg/day induces pronounced toxicity, the additional groups can be 100 and 250 mg/kg/day. Overall, the study design not only reduces animal use but also potentially creates less distress in animals on study by using a triggered design.
Dogs. In the sulfoxaflor dog palatability study, the aim was also to use as few animals as possible to identify appropriate dose levels for the subsequent 28-day study, in alignment with the aim of reducing the use of this species in toxicity testing (CitationHasiwa et al. 2011). The design of this study was also driven by the finding that sulfoxaflor was unpalatable and difficult to administer to dogs. The study consisted of six male and three female beagle dogs in total, aiming at re-using the same dogs with a period of recovery between treatments where possible, and was conducted over six phases, with recovery periods (7 days) between each phase:
Phase A (6 males, 2 females) evaluated palatability of the test material in ground diet offered for 3 days at dose levels of 300, 750, or 1500 ppm.
Phase B (6 males) evaluated palatability of the test material in ground diet offered for 3 days at dose levels of 300 or 750 ppm with bacon flavoring added.
Phase C (4 males, 2 females) evaluated palatability of the test material in pre-formulated pelletized diet offered for 3 days at dose levels of 300 or 750 ppm with bacon flavoring added.
Phase D (4 males) evaluated tolerability of the test material administered in a gelatin capsule twice a day for 3 days at dose levels of 10 or 25 mg/kg/day (5 or 12.5 mg/kg/dose).
Phase E (2 males, 1 females) evaluated palatability of the test material in ground diet offered for 2 hours per day for 2, 4, or 5 days, as dry diet or with water added to form a slurry, at respective dose levels of 50, 100, or 250 ppm.
Phase F (6 males, 3 females) evaluated tolerability of the test material administered in a gelatin capsule after offering canned diet once per day for 11 days at dose levels of 10 or 15 mg/kg/day.
For this palatability and tolerability study, all animals survived each exposure phase, but none of the exposure procedures produced food consumption results that were considered acceptable to sustain repeated exposure (28 or 90 days). Therefore, the 28-day study was conducted with further probe studies at the beginning of this study, both parts of the study being conducted with the same animals. Six female animals were assigned to one of two treatment courses; females only were considered adequate for this experiment as no significant sex differences had been observed in the previous study. Group assignments are identified in : three animals were given ad libitum dietary administration for 6 days and three animals were given oral (capsule) administration at 15 mg/kg body weight twice daily for 6 days. Following the initial part of the study, these animals were not dosed for 2 weeks, and then, these same two groups of three female Beagle dogs were administered the test material as presented in . For this second tolerability/palatability phase of the study, exposure at 500 ppm via diet or 15 mg/kg/day via capsule was not well tolerated as determined by insufficient food consumption. Exposure at 100 ppm (Group 4 of the probe study) via the diet was well tolerated as determined by adequate food consumption, but the amount of test material consumed was not sufficient to justify it as a potential highest dose for subsequent studies. Oral gavage of 15 mg/kg/day technical-grade sulfoxaflor was tolerated by the dogs based on the adequate food consumption and tolerable in-life clinical signs. Based on this information, oral gavage exposure was determined to be the most appropriate route of test material exposure in Beagle dogs and a 90-day and 1-year dog study was successfully completed. In totality, the probe work to identify an acceptable way to administer sulfoxaflor to dogs to conduct the 90-day and 1-year studies was identified using a total of 15 dogs.
Figure 2. The dog palatability study was conducted in a stepwise manner using a minimum number of animals, with no separate control group but the individual animals acting as their own controls by using pre-exposure food consumption values. Severe tolerability issues were encountered with administration of the test material to dogs, hence the need to assess dietary (ad libitum, restricted, flavored, and tinned feed), capsule and gavage dosing as the route of administration.
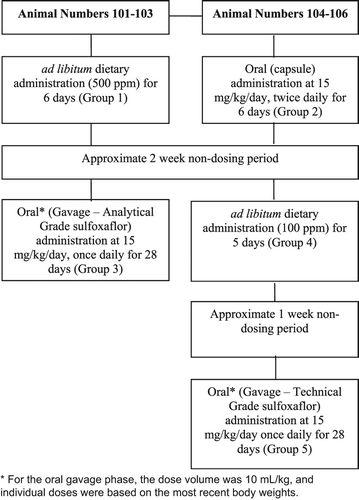
Table 2. Animal assignment for the probe part of the 28-day dog study.
The key advantage of this approach was flexibility, when dealing with a compound exhibiting palatability and tolerability issues in dogs. Previously palatability studies in dogs had used animals for one treatment period with termination afterwards. Given the success of the sulfoxaflor testing strategy, this 3Rs-centric approach to obtain palatability and short-term toxicity data in dogs could have applicability to future active substances with differing palatability and toxicity properties.
As palatability studies are the first to employ repeat-dose dietary administration of an active substance, these studies were also utilized to obtain additional information early in the testing program. For example, blood samples were collected from animals (rat, mouse, dog, and rabbit) during the course of the studies to determine the steady-state systemic exposure for improved interpretation of any observed toxicity. In mice, target organ data were generated and tissue samples were taken for future MoA work if necessary (this approach is discussed further in Integrated MoA studies).
Integrated toxicity testing
A typical PPP toxicity program will use a significant number of animals. Consistent with the commitment to 3Rs, one of the ways to reduce animal use is integration and merging of multiple smaller studies into a fewer large ones. While there are benefits to this approach from an animal use standpoint, there are also significant benefits from a scientific standpoint to help interpret findings, that is, a finding from a smaller study could be placed into a better context within a larger study. A number of mandatory toxicity studies are amenable to additions that make separate studies unnecessary.
A key example for integrated testing is the 90-day rat repeat-dose oral toxicity study. This is the key toxicology study providing information relating to toxic effects and potential health hazards likely to arise from repeated exposures over a subchronic time period (CitationOECD, 2002). It is required by all regulatory bodies (e.g., European Union, United States, Canada, Australia, China and Japan) and has relatively harmonized study guidelines (OPPTS 870.3100 [rodent]; OECD 408, EEC, Part B.26, JMAFF [Subchronic Oral Toxicity Study]).
The revision of the OECD Test Guideline for 90-day oral toxicity studies in rodents (Test Guideline 408, adopted on September 21, 1998) placed additional emphasis on neurological, immunological, and reproductive endpoints. It is stated that the “study should allow for the identification of chemicals with the potential to cause neurotoxic, immunological or reproductive organ effects, which may warrant further in-depth investigation.” In addition to the required 90-day rodent study for general toxicity, the US EPA OPPTS (2007 revision to 40 CFR part 158, pesticide data requirements) requires an active substance effects on repeat-dose neurotoxicity and immunotoxicity endpoints to be assessed (OPPTS 870.6200 Neurotoxicity Screening Battery and OPPTS 870.7800 Immunotoxicity, respectively).
In this toxicologic program, it was considered appropriate to take this integration aim further and cover additional endpoint requirements within one study. Thus, the 90-day rat study, based upon the OECD 408 test guideline, conducted for sulfoxaflor included many integrated parts ():
neurotoxicity—similar to OECD 424 and OPPTS 870.6200 guidelines;
immunotoxicity—based on OPPTS guideline 870.7800; and
TK/metabolism (blood and urine at weeks 4 and 13);
Figure 3. The 90-day rat study design for sulfoxaflor was enhanced with a number of ‘add-ons’ including endpoints for toxicokinetics (indicated in red text), neurotoxicity (pink text), and immunotoxicity (blue-colored text). toxicokinetics, TK; sheep red blood cell, SRBC; functional observational battery, FOB; plaque-forming cell, PFC.
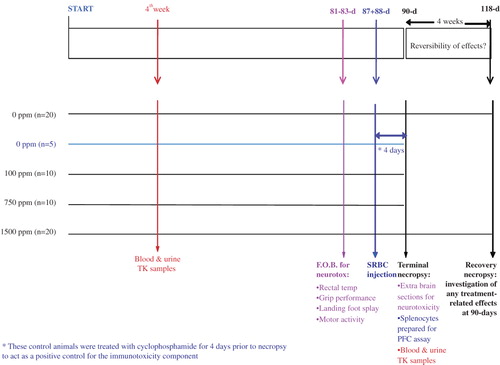
This integrated approach used 130 rats in total, compared to the 250 that would have been used if separate studies were conducted (i.e., standard 90-day dietary toxicity study: 120 rats; Immunotoxicity 870.7800: 50 rats; and Neurotoxicity OPPTS 870.6200: 80 rats).
Neurotoxicity. For the neurotoxicity component, parameters evaluated included a functional observational battery (FOB; cage-side, hand-held, and open-field observations, rectal temperature, fore- and hindlimb grip performance, and landing foot splay), motor activity, and histopathologic examinations, which included a detailed review of the nervous system. For sulfoxaflor, the FOB and motor activity were conducted pre-exposure and prior to necropsy (Days 81–83). It is possible to include additional FOB and motor activity assessments at weeks 2, 6, and 12 post-exposure to be even closer to the EPA/OECD neurotoxicity test guidelines (OECD and EPA guidelines for subchronic neurotoxicity studies require FOB and motor activity assessments during the pre-exposure period, as well as weeks 1–2 [OECD only], 4, 8, and 12–13 post-exposure); although this was not done for sulfoxaflor, additional FOB timepoints are now evaluated routinely in our laboratory.
Due to the need to collect data on multiple tissues (e.g., organ weights), perfusion fixation could not be included in these subchronic studies. Furthermore, both central and peripheral nervous system tissues were embedded in paraffin prior to sectioning, instead of plastic embedding, which is often used for peripheral nerves. However, the standard subchronic histopathology requirements were specifically expanded for sulfoxaflor to enhance the opportunity to detect potential neurotoxicity by including additional sections of the brain and spinal cord, additional peripheral nerves and cranial nerves and ganglia. Nine cross-sections of the brain were prepared to include the following structures: olfactory bulb, cerebrum (frontal, parietal, temporal, and occipital lobes), thalamus/hypothalamus, midbrain, pons, cerebellum, and medulla oblongata. Sections from peripheral and cranial nerves were included as follows: sciatic, tibial (proximal and distal—at the knee and calf muscle branches, respectively), sural and optic; the trigeminal nerves with ganglia and the spinal ganglia (cervical and lumbar). Paraffin-embedded tissues were sectioned approximately 6 μm thick, stained with hematoxylin and eosin, and examined by a veterinary pathologist using a light microscope.
Tissues were examined and peer-reviewed by Board Certified (American College of Veterinary Pathologists) veterinary pathologists with experience evaluating numerous perfusion and immersion fixed CNS and PNS tissues from studies over the last 20 years. Despite the identified methodological differences from standard subchronic neurotoxicity studies, these pathologists felt confident in the sensitivity of the neuropathological assessments. In addition, perfusion fixation was used in the developmental neurotoxicity study, in which no neuropathology was detected. For these reasons and also for reasons of animal welfare, an additional stand-alone neurotoxicity study based on the US EPA/OECD guidelines (OPPTS 870.6200, OECD 424) was not conducted.
Immunotoxicity. The integrated approach to immunotoxicity had been described previously by CitationLadics et al. (1995), and this approach was followed in the current study to allow a compliant OPPTS 870.7800 to be conducted within the 90-day rat study (10 animals per group [5/sex/dose]) compared to the guideline recommended 8 total animals per group). In order to obtain data on the functional responsiveness of the immune system, all rats designated for immunotoxicity assessments (including five animals per sex to serve as a immunotoxicity positive control [IPC] group) were immunized 4 days prior to necropsy (males—Day 87 and females—Day 88) with a single, 0.5-ml intravenous (i.v.) injection of 2 × 108 sheep red blood cells (SRBC), via the lateral tail vein (). CitationLadics et al. (1995) showed that injection of SRBC in 90-day study rats did not alter hematological or clinical chemistry parameters or the weights or morphology of routine protocol tissues (with the expected exception of the spleen) compared to those animals not receiving SRBC. The IPC group was also administered a daily dose of 20 mg/kg/day of cyclophosphamide via single intraperitoneal (i.p.) injection for four consecutive days prior to necropsy. On the day of necropsy (males—Day 91 and females—Day 92), all rats were euthanized via CO2 and the spleens were removed, weighed, and three transverse sections (approximately 2 mm thick) were fixed and processed for standard hematoxylin and eosin (H&E) staining. A single cell suspension was prepared from the remaining spleen portions. Diluted spleen cells were mixed with washed SRBC (same lot used for immunization), guinea pig complement and warm agar with diethylaminoethyl -dextran. The plaques formed as a result of complement- mediated lysis of the SRBC from antibodies secreted by the plasma cells (differentiated B cells) were counted.
The number of animals used in the 90-day study for neurotoxicity and immunotoxicity assessment matches the number of animals needed for current guidelines (from OPPTS 870.6200 Neurotoxicity screening battery: “(2) Number of animals. At least 10 males and 10 females should be used in each dose and control group for behavioral testing” and from OPPTS 870.7800: “(iv) Numbers. (A) At least eight animals should be included in each dose and control group. The number of animals tested should yield sufficient statistical power to detect a twenty percent change based upon the interanimal variation which may be encountered in these assays”. There were only five animals used for the immunotoxicity-positive control group. However, this lower number of animals was not a concern for the statistical power of the test since the positive controls have a greater response which was found to be different from the controls by the statistical analysis.
The data generated from this study with regard to neurotoxicity and immunotoxicity endpoints were considered fully reliable and acceptable by the US EPA upon their review, which confirms that this approach is scientifically acceptable from a regulatory perspective.
Toxicokinetics. Using the most relevant route of administration has favorable implications for 3Rs and also enables generation of more appropriate data, given the expectation that the most significant route of human exposure to the active substances contained in a PPP will be oral, via low levels of residues in food commodities. Rat and rabbit developmental toxicity studies have historically been conducted using the gavage route of administration, which is acceptable under the test guidelines for developmental toxicity. The gavage route can produce irrelevant acute toxicity characteristic of bolus dosing due to the rapid increase in blood concentration of a chemical reaching maximum circulating concentration (Cmax) within a short period of time (tmax). Dietary administration in developmental toxicity studies can provide a more relevant route of administration when considering potential human exposure (i.e., to low levels of residues in food commodities) and reduce the oscillating daily pharmacokinetic profile characterized by short-lived Cmax, and low circulating concentration (Cmin) values that gavage can create, ensuring consistent exposure across all critical periods of fetal development. Gavage and dietary administration of sulfoxaflor in pregnant New Zealand white rabbits has been compared. In probe studies, sulfoxaflor was administered at 0, 10, 15, 20, or 30 mg/kg/day by gavage or 0, 500, or 1000 ppm (0, 20, or 33.3 mg/kg/day) in feed on gestation days (GD) 7–28. Gavage doses of 20 or 30 mg/kg/day exceeded an MTD, while maternal toxicity was similar between gavage at 15 mg/kg/day and diet at 1000 ppm, the identified MTDs. Plasma concentrations of sulfoxaflor in the dietary groups were constant, compared to a three-fold difference between Cmax and Cmin by gavage, while the diurnal systemic dose (diurnal area under the plasma concentration time curve, AUC24h) of the test material was greater than two-fold higher for dietary versus gavage at the respective MTDs (). presents both probe and definitive data; the definitive study illustrated the same TK patterns as seen in the probe study, upon which dosing decisions were taken. Based on these findings from the probe study, the dietary route was chosen for the definitive study at 0, 30, 150, or 750 ppm (0, 5.6, 20.0, or 33.3 mg/kg/day). Animals in the 750 ppm dose group had decreased body weight (12%; GD, 7–28) and body weight gain (55%; GD, 7–13), relative to controls. There was no treatment-related maternal toxicity in the 30 or 150 ppm dose groups and no treatment-related developmental toxicity in any dose group, resulting in maternal and developmental toxicity no-observed-adverse-effect levels (NOAEL) of 150 ppm and 750 ppm (the highest level tested), respectively. The daily systemic dose of sulfoxaflor on GD 27–28 was dose-proportional to the dose-corrected AUC24h values of 18–19 μg sulfoxaflor/h/kg− 1 for animals in the 30, 150, and 750 ppm dose groups. In comparison with gavage administration, in respect to the MTD, the dietary route allowed a more than two-fold greater dose level, regarding both external and systemic doses, with the dietary route providing stable exposure throughout all stages of fetal development.
Table 3. Comparison of the diurnal systemic dose of sulfoxaflor to rabbits administered via bolus oral gavage with that via the dietary route.
In summary, it is difficult to quantify the 3Rs-centric impact the sulfoxaflor toxicologic program had. Although significant numbers of animals were saved for certain specific studies (e.g., 120 rats saved by the integrated 90-day rat study design), sulfoxaflor also had complex toxicologic issues which required MOA/HRF programs (CitationEllis-Hutchings et al. 2014, CitationLeBaron et al. 2014, CitationRasoulpour et al. 2014), which undoubtedly increased the animal numbers used in total compared to active substances with less complex toxicological profiles. However, certainly, animal use was refined wherever possible and all MoA experiments were conducted with in vitro models wherever applicable.
Integration of Toxicokinetics
Integration of TK analysis into all toxicity studies follows principles articulated by the ILSI/HESI-ACSA and new EU Directives (1107/2009 EC). The ACSA project included industry, government, academia and non-governmental organization (NGO) scientists. Their ‘base set’ of principles included an integrated approach to evaluate systemic toxicity, life-stage effects and kinetics, and dose-level selection based on TK (CitationBarton et al. 2006, CitationCarmichael et al. 2006, CitationCooper et al. 2006, CitationDoe et al. 2006).
The integration of TK into PPP toxicity testing programs is a new concept, as described by CitationCreton et al. (2009): “Comprehensive integration of TK into development of a new active substance is a new paradigm that requires commitment from the beginning of the extensive toxicology program. This presents new challenges to this business sector as toxicokinetics has never been an integral part of the toxicology research development program. In addition to a new mindset for the toxicologists, the challenges include when, how and how often blood samples need to be taken and the extent to which analyses extend beyond the simplest of investigations, such as the Cmax of parent molecule in plasma. However, toxicokinetic data can be helpful in several key ways, including study design, dose level selection, comparison of toxicity across species, tentative initial information on potential mode-of-action and even relevance of levels of substances that may one day be detected in humans.”
This approach was utilized for sulfoxaflor whereby TK analysis was performed in most toxicity studies, generating comparative blood and urine TK/metabolism data across dose levels, sexes, study durations, species (rat, mouse, dog, and rabbit), strains, and life stages. Although integration of TK into toxicity studies for the pharmaceutical industry has been a commonplace for many years, this approach is novel in the world of PPP.
In order to better understand the relationship between the systemic dose and the observed toxicity, TK analysis was incorporated in all sub-chronic (4 and 13 week), chronic (12, 18 and 24 month) and developmental toxicity studies. The TK data were used to estimate the daily systemic exposure/dose, measured by the diurnal area under the plasma concentration time curve (AUC24h) (see CitationSaghir et al. 2006, Citation2011 for detail), and urinary elimination of the biomarker(s) across doses and studies. This was part of our commitment to improve the toxicological study design, reduce the use of animals in testing and better understand test material fate in animals at steady-state exposure and the impact of three main factors: (1) prolongation of the exposure; (2) age; and (3) life stage. TK data were used as a guide for the selection of doses for subsequent studies. Doses above which nonlinear kinetics (non-dose-proportional increase in AUC24h) were observed, an indication of stress altering the biological processes of detoxification/activation (CitationOECD 2010—Draft Guidance Document No. 116), were considered not relevant to human exposure scenarios (based upon estimations of human exposure via dietary intake of crop residues). These innovative approaches represent a new paradigm in pesticide regulatory toxicology testing with primary drivers being animal welfare and sound science leading to more informed human risk assessment. Sulfoxaflor was the first active substance in which this approach was followed by this company.
Integration of TK analysis into the sulfoxaflor toxicology testing program was initiated by conducting a probe ADME study in one male and one female rat early in the testing program (the TK parameters were later verified in the regulatory mandated ADME study, ). Sulfoxaflor was rapidly and completely absorbed from the gastrointestinal (GI) tract, well distributed to tissues and rapidly eliminated via urine with almost no metabolism, which made it an ideal for TK investigations. Oral absorption of sulfoxaflor was > 92% in rat () and ≥ 87% in mouse (). Urinary excretion of sulfoxaflor was 86–97% of the administered dose occurring within 48 h post-dosing, biliary elimination accounted for 6–9% of the dose. The elimination t½ of sulfoxaflor from plasma was 4–9 h, 15–35 h, and 17–28 h in the rat, female rabbit and dog short-term repeat-dose studies, respectively ().
Table 4. Toxicokinetic profile of sulfoxaflor in rats.
Table 5. Summary of the excretion of the bolus oral dose used for the estimation of the absorption of sulfoxaflor in mice.
Table 6. Comparative toxicokinetics of sulfoxaflor across doses and species.
As previously discussed, prior to conducting rodent 4-week studies, modified palatability-probe studies were conducted to determine TK in addition to palatability and toxicity. The departure from TK linearity is considered to be the kinetically derived maximum dose (KMD) and can be used, either alone or in conjunction with the conventional MTD, in the selection of high doses for subsequent longer-term studies (CitationSaghir et al. 2012, Citation2013). After 4 weeks of dietary exposure, the plasma AUC24h of sulfoxaflor was proportional across doses in rat (). In mice, after 4 weeks of dietary exposure, the plasma AUC24h was proportional across doses as expected in females (). However, the AUC24h became non-dose proportional (i.e., non-linear) at the two high doses in males, most likely due to saturation of absorption from the GI tract. The TK nonlinearity along with conventional toxicological endpoints was used to select the highest dose for the male mouse for the 13-week toxicity study.
Table 7. Toxicokinetics of sulfoxaflor across doses, species and life stages.
The AUC24h in rats after 13 weeks of exposure remained dose-proportional (); however, in mice AUC24h became significantly nonlinear: > 92 mg/kg/day in males (saturation of elimination) and ≥ 227 mg/kg/day in females (saturation of absorption; , more detail can be found in the study by CitationSaghir et al. 2012). The nonlinear-systemic doses in mice were consistent with the observed biochemical and histopathological changes (). These include a plateau of response at the two higher doses in females, corresponding to a plateau of AUC24h and significantly higher in males due to supralinear AUC24h at the highest dose in the 13-week study. Urinary elimination of sulfoxaflor was less than expected based on the dose levels, probably due to saturation of elimination of the systemic dose from kidneys (male) and saturation of absorption from the GI tract (female), both resulting in less than dose-proportional clearance (data not shown). The limit doses selected for the subsequent 18-month mouse carcinogenicity study were based upon KMD nonlinearity, and was 750 ppm for male (expected, ˜ 65 mg/kg/day) and 1500 ppm for female (expected, ˜ 130 mg/kg/day) based on KMD nonlinearity. Selection of the KMD based on the nonlinearity of the diurnal systemic dose for the long-term carcinogenicity studies resulted in a dose-proportional sulfoxaflor AUC24h in rats and mice after 12 months of dietary exposure ().
Table 8. Comparison of the expected and the observed liver effects in 28- and 90-day mouse studies.
In dogs, the 4-week study was used to determine whether oral gavage was the best route of administration. During the 13-week dog study, absorption of the gavaged sulfoxaflor was most likely saturated in both male and female dogs, which was apparent from the less than dose-proportional increase in AUC24h (). Although the gavage dose to dogs (1–10 mg/kg/day) was much lower than that of other species, for example, rats ingested as high as 151 mg/kg/day of sulfoxaflor, the AUC24h in dogs was higher ( and ). This was apparently due to much slower elimination of sulfoxaflor by dogs compared to that by other species; the plasma elimination half-life in dogs after oral gavage was 22 ± 5 h, which was 4.4-fold longer than rats after a single oral gavage of 4–111 mg/kg ( and ). The slow elimination of sulfoxaflor by dogs resulted in the average (13-, 26-, and 52-week data) dose-corrected AUC24h of 27 ± 5 μg h ml−1, whereas the average (4-, 13-, and 52-week data) for rats and mice was 10 ± 2 and 5 ± 10 μg h ml−1, respectively ( and ). Due to slow clearance, the dose-corrected AUC24h in dogs remained higher than in all developmental and reproductive toxicity studies (, and ).
In all species, females were generally more efficient in clearing sulfoxaflor from their system than the males, which was apparent from the lower diurnal systemic dose of sulfoxaflor in females, even though in most cases they consumed more test material-fortified diet per kg/body weight than males ().
The integrated approach, including TK to determine the fate of sulfoxaflor at different life stages in rat and rabbit developmental/reproductive toxicity studies, allowed a better understanding of the possible reason(s)/mode of observed toxicity, especially at kinetically nonlinear doses. The AUC24h of sulfoxaflor in rabbits exposed to diet was linear up to ˜ 32 mg/kg/day dose and became nonlinear (supra-linear; saturation of elimination) at the highest dose of 35 mg/kg/day (). Sulfoxaflor readily crossed the placenta in rats and was detected in fetal rat blood at 31–92% of the levels detected in dam blood, depending upon the life stage of the developing fetus (). Similarly, rat dams were able to eliminate the systemic burden of sulfoxaflor through milk resulting in blood levels in nursing pups that exceeded dam levels (). These data effectively demonstrated that exposure of sulfoxaflor was adequate during all stages of development, from conception to adulthood: transplacentally during in utero development, through milk during nursing and through diet after weaning in the two-generation reproductive toxicity study.
Integrated MoA studies
The addition of prospective MoA endpoints within repeat-dose toxicity studies as well as inclusion of proactive, specific MoA studies as an integral part of the PPP development program, has numerous potential advantages. The approach taken for sulfoxaflor, for example, was to identify the primary target organ as early as possible in the testing program in palatability studies. The liver was identified as the primary target organ (based on organ weight increase), and therefore in subsequent studies, it was valuable to take liver samples for possible MoA analysis.
It was discussed previously that by fully utilizing each animal in a study, it is possible to reduce and refine animal usage. A specific example of this is the sulfoxaflor 28-day mouse study where the traditional study design of five male and five female mice per dose level was supplemented with satellite groups of three male and three female CD-1 mice at 0 and the mid-dose level for 3 days (termination on Day 4). On Day 4, non-fasted mice (0 and 1500 ppm, 3/sex) were anesthetized with isoflurane and euthanized, and liver samples were collected and weighed. Samples of liver were taken from both lateral and medial lobes (left and right) and fixed in 10% buffered neutral formalin for possible future histologic evaluation. A piece of the remaining tissue was stored in RNALater® and the rest of the liver was flash-frozen in liquid nitrogen for possible analysis of gene expression and liver enzyme activity. The data collected from these satellite groups facilitated the generation of MoA data from a study commonly undertaken as part of the standard testing regime.
In each study protocol, the aim was to reduce and refine animal usage. For example, early in the toxicological program (in the first repeat-dose dietary administration study, the mouse palatability study), the liver was identified as the primary target organ with a CAR/PXR-mediated MoA the likely cause (CitationLeBaron et al. 2013, Citation2014). shows how these data can be used in a MoA/HRF analysis. Therefore, in each subsequent toxicity study, liver samples were taken and stored for potential MoA analysis. Some of these liver samples were used to confirm that a MoA similar to that proposed in mice (i.e., CAR/PXR [pregnane X receptor] activation) likely was also occurring in rats.
Table 9. Sulfoxaflor: temporal and dose response and reversibility for increased relative liver weights rats.
This approach allowed data to be generated from different species (rat, mouse) and from a wide range of dose levels (25–2000 ppm) and timepoints (3-, 7-, 28- and 90 days, 2 years), which was extremely useful when potential MoAs were evaluated using the MoA/HRF in which identifying key events and conducting a dose and temporal relationship analysis, are very important components of the process. Furthermore, this integrated approach to MoA testing was also very useful when addressing another key component of the MoA/HRF analysis—consideration, elimination, and exclusion of other possible MoAs for the effect seen. Direct data were obtained to eliminate other nuclear receptor-mediated MoAs for liver tumors in rodents (PPAR-α [peroxisome proliferator–activated receptor alpha] and AhR [aryl hydrocarbon receptor] nuclear receptor–mediated modes of action), and this was achieved without conducting specific additional studies to test the other possible MoAs. Indeed, an interesting point to note is that the majority of the liver MoA work for sulfoxaflor was conducted in a proactive manner (i.e., whilst the carcinogenicity studies were ongoing and before an increase in the incidence of liver tumors was actually seen) and is described in detail in the study by CitationLeBaron et al. (2013).
Another example of incorporating MoA analysis within standard toxicity studies is from contingency sampling from the rat developmental toxicity study. Dietary administration of 1000 ppm of sulfoxaflor to Crl/CD(SD) rats during gestation has been previously shown to cause neonatal pup death (CitationRasoulpour et al. 2012, CitationEllis-Hutchings et al. 2014). In order to determine whether morphological alterations (e.g., increased collagen deposition) in any region of the lungs were present and related to pup death, one fetus/sex from five control and four 1000-ppm litters (18 samples total) from the definitive developmental toxicity study were collected and preserved in neutral, phosphate-buffered 10% formalin. This MoA seemed possible based upon a lack of in utero mortality in a developmental toxicity study, and no signs of difficult deliveries (dystocia), but increased neonatal mortality observed shortly after birth. Sections from these preserved lung tissues were processed such that each slide contained sections of the trachea, bronchi, bronchioles, and alveoli. Slides were stained with hematoxylin and eosin, and evaluated for histopathological changes. Tissues were archived with the developmental toxicity study.
There were no sulfoxaflor-induced lesions in the trachea, bronchi, bronchioles, or alveoli in any of the treated fetuses examined, and no treatment-related increases in collagen deposition around the airways or alveolar walls or any other changes. This enabled elimination of morphologic abnormalities in the trachea or within the lungs as a potential MoA for this effect. Apart from the reproductive toxicity study, there are only a few regulatory studies where the survival of neonatal rats is monitored; therefore, subsequent studies, which used additional animals, were required to examine and describe the proposed MoA. Ultimately, in vitro experiments were used to conclusively verify this MoA and its lack of relevance to humans (CitationEllis-Hutchings et al. 2014).
Conclusions
Challenges to toxicology-testing requirements and individual study designs provide opportunities for novel approaches, which are science-driven study modifications to generate more robust, and relevant, data while focusing on animal welfare. Innovations are being incorporated into the toxicity testing of new PPPs. In this example, these include integrated TK as well as neurotoxicity, immunotoxicity, and MoA techniques.
Incorporation of TK in all regulatory mandated short- and long-term toxicity studies is supportive of the concept that the relevance of toxicity observed only at kinetically nonlinear doses to estimating potential risk associated with much lower worst-case human-exposure scenarios can be questioned. This is specifically true for PPP active substances, as expected human exposure through environment/residue in food is usually in ng/kg/day range and does not usually necessitate testing such high doses. The proper use of TK data in designing studies and selecting doses would tremendously reduce the number of extraneous experiments (e.g., to define MoA for an effect that occurs only at kinetically saturated doses), and consequently liberate resources for other studies while reducing unnecessary animal use. The use of TK in the toxicity testing of non-pharmaceuticals (e.g., PPPs) has been promoted in the ILSI/HESI-ACSA project publications (CitationBarton et al. 2006, CitationCarmichael et al. 2006, CitationCooper et al. 2006, CitationDoe et al. 2006). Similarly, regulatory agencies are incorporating the generation of TK data in their guidelines (e.g., CitationEC, 2009; CitationECHA, 2008; and CitationOECD 2008, Citation2009, Citation2010). Both the US Environmental Protection Agency and Health Canada's Pest Management Regulatory Agency (PMRA) have accepted the approach of selecting the highest dose at, or slightly above, the point of departure (KMD) from linearity for an extended one-generation toxicity study (CitationU.S. EPA, 2008; 2011; CitationHealth Canada PMRA, 2011). In this case, the dose was half of that suggested by the traditional MTD approach based on body weight loss, liver weight increases, or pathological change(s) in target organ(s).
Additionally, these more robust TK data will also be useful in providing a perspective on human biomonitoring data (blood/urine levels) by enabling a comparison to be made with animal NOAEL/LOAEL blood/urine concentrations of biomarker(s) and allowing more meaningful correlations to be made at the expected extremely low human exposure (CitationBoobis et al. 2006, Citation2008, CitationRhomberg et al. 2007, CitationSaghir et al. 2006, CitationSlikker et al. 2004a, Citation2004b). It is possible that this approach will also facilitate the ‘biomonitoring equivalents’ (BE) approach pioneered by CitationHays and Aylward (2009).
Integration of MoA components into a toxicity study can:
provide mechanistic information early (versus investigations at the end of a research program) giving a significant time saving and, potentially, no delay to regulatory submission while conducting reactive MoA experiments;
establish causality and thus better forecast the likelihood of observing additional apical effects in longer-term studies (e.g., carcinogenesis) and their potential relevance, and hence regulatory impact, to humans;
aid study design and dose selection: refine high dose level and reduce likelihood and mitigate impact of irrelevant high-dose effects;
fully utilize animals in each study, thus reducing and refining animal usage; and
help adopt a flexible approach for further testing and decision-making
The sulfoxaflor toxicology program presented unique opportunities to employ innovative approaches in PPP toxicity testing. This supplement also contains papers which describe in more detail the approach taken to establish the MoA and HRF related to toxicities elicited by sulfoxaflor in the mammalian toxicology studies:
developmental toxicity in rats mediated via the fetal muscle nAChR (CitationEllis-Hutchings et al. 2014);
liver tumors in rodents mediated via CAR/PXR (CitationLeBaron et al. 2014); and
Leydig cell tumors in Fischer 344 rats (CitationRasoulpour et al. 2014)
In the future, with advocacy by RISK21 (CitationEmbry et al. 2014) towards more emphasis being placed upon problem formulation, where the user defines the issue and degree of concern and the exposure information required to make a decision is compiled and evaluated before toxicity testing is initiated, the approaches described herein could help obtain key data early, understand MoA, and thus understand potential regulatory impact of effects before chronic studies are initiated, and ultimately make human health risk assessments more realistic and relevant to actual human exposure scenarios.
Acknowledgments
Many thanks to those who made this publication possible: Robert Ellis-Hutchings, Matthew LeBaron, Dave Geter, Michael Woolhiser, Shakil Saghir, Valerie Marshall, Jennifer Murray, Amanda Andrus, Carol Zablotny, Amy Clark, and Keith Brooks.
Declaration of interest
The authors are employed by The Dow Chemical Company, the developer and producer of sulfoxaflor. The authors have sole responsibility for the writing and content of the paper.
References
- Babcock JM, Gerwick CB, Huang JX, Loso MR, Nakamura G, Nolting SP, et al. (2011). Biological characterization of sulfoxaflor, a novel insecticide. Pest Manag Sci, 67, 328–34.
- Barton HA, Pastoor TP, Baetcke K, Chambers JE, Diliberto J, Doerrer NG, et al. (2006). The acquisition and application of absorption, distribution, metabolism, and excretion (ADME) data in agricultural chemical safety assessments. Crit Rev Toxicol, 36, 9–35.
- Boobis AR, Cohen SM, Dellarco V, McGregor D, Meek ME, Vickers C, et al. (2006). IPCS framework for analyzing the relevance of a cancer mode of action for humans. Crit Rev Toxicol, 36, 781–92.
- Boobis AR, Doe JE, Heinrich-Hirsch B, Meek ME, Munn S, Ruchirawat M, et al. (2008). IPCS framework for analyzing the relevance of a noncancer mode of action for humans. Crit Rev Toxicol, 38, 87–96.
- Carmichael NG, Barton HA, Boobis AR, Cooper RL, Dellarco VL, Doerrer NG, et al. (2006). Agricultural chemical safety assessment: a multisector approach to the modernization of human safety requirements. Crit Rev Toxicol, 36, 1–7.
- Cooper RL, Lamb JC, Barlow SM, Bentley K, Brady AM, Doerrer NG, et al. (2006). A tiered approach to life stages testing for agricultural chemical safety assessment. Crit Rev Toxicol, 36, 69–98.
- Council of Canadian Academies. (2012). Integrating Emerging Technologies into Chemical Safety Assessment: The Expert Panel on the Integrated Testing of Pesticides. ISBN 978–1-926558–38-7, Ottawa, Canada: Council of Canadian Academics.
- Creton S, Billington R, Davies W, Dent MP, Hawksworth GM, Parry S, Travis KZ. (2009). Application of toxicokinetics to improve chemical risk assessment: implications for the use of animals. Regul Tox Pharmacol, 55, 291–9.
- Doe JE, Boobis AR, Blacker A, Dellarco V, Doerrer NG, Franklin C, et al. (2006). A tiered approach to systemic toxicity testing for agricultural chemical safety assessment. Crit Rev Toxicol, 36, 37–68.
- EC (European Commission). (July. (2009)). Guidance to Regulation (EC) No 1272/2008 on Classification, Labelling and Packaging of substances and mixtures.
- ECHA. (2008). Guidance on information requirements and chemical safety assessment. Guidance on toxicokinetics. European Chemicals Agency, Helsinki. Chapter .R7.12.
- Ellis-Hutchings RG, Rasoulpour RJ, Terry C, Carney EW, Billington R. (2014). Human relevance evaluation of a nAChR-medicated fetal muscle contracture mode of action induced by sulfoxaflor. Crit Rev Toxicol, 44, 45–62.
- Embry MR, Bachman A, Bell D, Boobis A, Cohen SM, Dellarco M, et al. (2014). The Risk Assessment in the 21st Century (RISK21): Roadmap and Matrix. Crit Rev Toxicol, in press.
- Hasiwa N, Bailey J, Clausing P, Daneshian M, Eileraas M, Farkas S, et al. (2011). Critical evaluation of the use of dogs in biomedical research and testing in Europe. ALTEX, 28, 326–40.
- Hays SM, Aylward LL. (2009). Using biomonitoring equivalents to interpret human biomonitoring data in a public health risk context. J Appl Toxicol, 29, 275–88.
- Health Canada, Pest Management Regulatory Agency (2011). 2,4-D (DXA) herbicide. - Enhanced One-Generation Reproduction Study Health Canada, Pest Management Regulatory Agency, Ottawa, Ontario.
- Kobel W, Fegert I, Billington R, Lewis R, Betley K, Bomann W, et al. (2010). A 1-year toxicity study in dogs is no longer a scientifically justifiable core data requirement for the safety assessment of pesticides. Crit Rev Toxicol, 40, 1–15.
- Ladics GS, Smith C, Heaps K, Elliott GS, Slone TW, Loveless SE. (1995). Possible incorporation of an immunotoxicological functional assay for assessing humoral immunity for hazard identification purposes in rats on standard toxicology study. Toxicology, 96, 225–38.
- Laviano A, Meguid M, Gleason J, Yang ZJ, Renvyle T. (1996). Comparison of long-term feeding pattern between male and female Fischer 344 rats: influence of estrous cycle. Am J Physiol, 270 (Regulatory Integrative Comp. Physiol. 39), R413–R419.
- LeBaron MJ, Gollapudi B, Terry C, Billington R, Rasoulpour RJ. (2014). Human relevance framework for rodent liver tumors induced by the insecticide sulfoxaflor. Crit Rev Toxicol, 44, 15–24.
- LeBaron MJ, Geter DR, Rasoulpour RJ, Gollapudi BB, Thomas J, Murray J, et al. (2013). An integrated approach for prospectively investigating a mode-of-action for rodent liver effects. Toxicol Appl Pharmacol, 270, 164–73.
- Longhurst C, Babcock JM, Denholm I, Gorman K, Thomas JD, Sparks TC. (2012). Cross-resistance relationships of the sulfoximine insecticide sulfoxaflor with neonicotinoids and other insecticides in the whiteflies Bemisa tabaci and Trialeurodes vaporariorium. Pest Manag Sci, 69, 809–13.
- Meek ME, Bucher JR, Cohen SM, Dellarco V, Hill RN, Lehman-McKeeman LD, et al. (2003). A framework for human relevance analysis of information on carcinogenic modes of action. Crit Rev Toxicol, 33, 591–653.
- NC3Rs. (2011). National Centre for the Replacement, Refinement and Reduction of Animals in Research. Available at: http://www.nc3rs.org.uk/page.asp?id = 7. Last Accessed on: 13th June 2013.
- OECD. (2002). OECD Series on Testing and Assessment Number 32 and OECD Series on Pesticides Number 10: Guidance Notes for Analysis and Evaluation of Repeat-Dose Toxicity Studies. ENV/JM/MPM(2000)18.
- OECD. (2008). Guideline for the testing of chemicals - draft proposal for a revised TG 417: Toxicokinetics, Paris, France. November 2008.
- OECD. (2009). Guideline for the testing of chemicals - draft proposal for an extended one-generation reproductive toxicity study, Paris, France. October 2009.
- OECD. (2010). Draft guidance document No. 116 on the design and conduct of chronic toxicity and carcinogenicity studies, supporting TG 451, 452, 453, Paris, France. April 2010.
- Rasoulpour RJ, Ellis-Hutchings RG, Terry C, Millar NS, Zablotny CL, Gibb A, et al. (2012). A novel mode-of-action mediated by the fetal muscle nicotinic acetylcholine receptor resulting in developmental toxicity in rats. Toxicol Sci, 127, 522–34.
- Rasoulpour RJ, Terry C, LeBaron MJ, Stebbins K, Ellis-Hutchings RJ, Billington R. (2014). Mode-of-action/human relevance framework for rat Leydig cell tumors mediated by sulfoxaflor. Crit Rev Toxicol, 44, 25–44.
- Rhomberg LR, Baetcke K, Blancato J, Bus J, Cohen S, Conolly R, et al. (2007). Issues in the design and interpretation of chronic toxicity and carcinogenicity studies in rodents: approaches to dose selection. Crit Rev Toxicol, 37, 729–837.
- Russell WMS, Burch RL. (1959). The Principles of Humane Experimental Technique. London: Methuen, ISBN: 0900767782.
- Saghir SA, Marty MS, Zablotny CL, Passage JK, Perala AW, Bus JS, et al. (2013). Life-stage-, sex- and dose-dependent dietary toxicokinetics and relationship to toxicity of 2,4-dichlorophenoxyacetic acid (2,4-D) in rats: implications for toxicity test dose selection, design and interpretation. Toxicol Sci, 136, 294–307.
- Saghir SA, Mendrala AL, Bartels MJ, Day SJ, Hansen SC, Sushynski JM, Bus JM. (2006). Strategies to assess systemic exposure of chemicals in subchronic/chronic diet and drinking water studies. Toxicol Appl Pharmacol, 211, 245–60.
- Saghir SA, Rick DL, Clark AJ, Staley JL, Bartels MJ, Terry C, Billington R. (2011). Integration of toxicokinetics into guideline toxicity studies of a new agrochemical. The Toxicologist, 120, 2107.
- Saghir SA, Bartels MJ, Rick DL, McCoy AT, Rasoulpour RJ, Ellis-Hutchings RG, et al. (2012). Assessment of diurnal systemic dose of agrochemicals in regulatory toxicity testing – an integrated approach without additional animal use. Regul Toxicol Pharmacol, 63, 321–32.
- Seed J, Carney EW, Corley RA, Crofton KM, DeSesso JM, Foster PM, et al. (2005). Overview: using mode of action and life stage information to evaluate the human relevance of animal toxicity data. Crit Rev Toxicol, 35, 664–72.
- Slikker W Jr, Andersen ME, Bogdanffy MS, Bus JS, Cohen SD, Conolly RB, et al. (2004a). Dose-dependent transitions in mechanisms of toxicity: case studies. Toxicol Appl Pharmacol, 201, 226–94.
- Slikker W Jr, Andersen ME, Bogdanffy MS, Bus JS, Cohen SD, Conolly RB, et al. (2004b). Dose-dependent transitions in mechanisms of toxicity. Toxicol Appl Pharmacol, 201, 203–25.
- Sonich-Mullin C, Fielder R, Wiltse J, Baetcke K, Dempsey J, Fenner-Crisp P, et al. (2001). IPCS conceptual framework for evaluating a mode of action for chemical carcinogenesis. Regul Toxicol Pharmacol, 34, 146–52.
- U.S. Environmental Protection Agency (2008). 2,4-D. Expedited review. Pharmacokinetic/Range-Finding Studies for Extended F1 1-Generation Reproduction Toxicity Study. Decision No. 395649, Office of Prevention, Pesticides and Toxic Substances, United States Environmental Protection Agency, Washington, D.C.
- U.S. Environmental Protection Agency (2010). 2,4-D: review of extended 1-generation repoduction study and dose-range-finding and pharmacokinetic titration studies. Decision No. 431951, Office of Chemical Safety and Pollution Prevention, United States Environmental Protection Agency, Washington, D.C.
- Watson GB, Loso MR, Babcock JM, Hasler JM, Letherer TJ, Young CD, et al. (2011). Novel nicotinic action of the sulfoximine insecticide sulfoxaflor. Insec Biochem Mol Biol, 41, 432–9.
- Zhu Y, Loso MR, Watson GB, Sparks TC, Rogers RB, Huang JX, et al. (2011). Discovery and characterization of sulfoxaflor, a novel insecticide targeting sap-feeding pests. J Agric Food Chem, 59, 2950–7.