
Abstract
Proteoliposomes have been widely used for in vitro studies of membrane fusion mediated by synaptic proteins. Initially, such studies were made with large unsynchronized ensembles of vesicles. Such ensemble assays limited the insights into the SNARE-mediated fusion mechanism that could be obtained from them. Single particle microscopy experiments can alleviate many of these limitations but they pose significant technical challenges. Here we summarize various approaches that have enabled studies of fusion mediated by SNAREs and other synaptic proteins at a single-particle level. Currently available methods are described and their advantages and limitations are discussed.
Introduction
Membrane fusion is a fundamental cellular process by which two initially distinct membranes merge their hydrophobic cores, resulting in one interconnected structure. When complete fusion occurs, involving both outer and inner leaflets of two membranes, a pore is formed allowing the mixing of the two compartments originally separated by those membranes. Many cellular processes including hormone and neurotransmitter release, protein secretion, egg fertilization, and viral entry into host cells rely on membrane fusion. For fusion processes involving multi-compartment eukaryotic cells, a family of proteins called the soluble N-ethylmaleimide-sensitive factor (NSF) attachment protein receptors (SNAREs) play a critical role in membrane fusion along with other proteins (Jahn & Fasshauer, Citation2012; Südhof, Citation2013; Südhof & Rothman, Citation2009). For example, neurotransmitter release is triggered by Ca2+ influx upon an action potential, leading to fusion of synaptic vesicles with the active zone. It is mediated by a number of synaptic proteins and their complexes, including the neuronal SNAREs (syntaxin, SNAP-25, and synaptobrevin), synaptotagmin, complexin, Munc13, and Munc18 (Jahn & Fasshauer, Citation2012; Südhof & Rothman, Citation2009). Disassembly of SNARE complexes is facilitated by the ATPase NSF along with SNAP proteins; such disassembly is involved in recycling SNARE proteins as well as setting the stage for fusion-competent trans SNARE complexes (Ma et al., Citation2013). Among all biological membrane fusion processes, the fusion of synaptic vesicles with the active zone is unique in that it is fast (sub-millisecond timescale) and tightly regulated by Ca2+.
As a complement to studies in neuronal cultures, in vitro or cell-free assays are powerful tools for discovery since they allow observations and manipulations that are not possible in vivo. As a particularly relevant example for this review, it was a cell-free assay of Golgi prepared from CHO cells that had been infected with vesicular stomatitis virus, that led ultimately to the discovery of NSF and, subsequently, SNAP and SNARE proteins (Balch et al., Citation1984).
Cell free or in vitro assays involve components that are extracted from live cells or that are entirely synthetically produced. Here we review in vitro approaches for studying SNARE-mediated membrane fusion that use synaptic proteins that are expressed in suitable hosts, purified, and reconstituted in proteoliposomes. For a review of studies of other fusion processes, including viral fusion, the reader is referred to the review by Otterstrom & Oijen (Citation2013). We will review both ensemble and single-particle approaches that have been developed, and the challenges that had to be overcome in developing these assays. We will also discuss the strengths and weaknesses of the various approaches.
Ensemble vesicle fusion assays
To our knowledge, the first ensemble fusion assay using fluorescence was reported by Struck et al. (Citation1981) who employed fluorescent lipids to study vesicle–vesicle and vesicle–cell fusion. In this assay, lipid-anchored pairs of fluorophores, N-(7-nitro-2,1,3-benzoxadiazole-4-yl)-phosphatidylethanolamine (NBD-PE) and N-(lissamine rhodamine B sulfonyl)-phosphatidylethanolamine (Rh-PE), are incorporated to one class of lipid vesicles. The fluorescence energy transfer (FRET) from NBD to rhodamine dyes leads to a quenching of the NBD fluorescence intensity. When fluorescent vesicles fuse with other non-fluorescent vesicles or cells, the concentrations of both NBD-PE and Rh-PE in the combined bilayer are reduced since lipids from both classes of vesicles exchange (this is called “lipid mixing”). This dilution leads to dequenching and a consequent increase in NBD fluorescence ().
Figure 1. Ensemble vesicle fusion assays. (A) Vesicle–vesicle fusion assay using fluorescently labeled lipid analogs (Struck et al., Citation1981). Vesicles containing a high concentration of NBD-PE and Rh-PE are characterized by quenching of NBD fluorescence due to FRET between NBD and rhodamine. Upon fusion to unlabeled vesicles the concentration of NBD and rhodamine decreases, resulting in less FRET efficiency and thus a consequent increase in NBD fluorescence (dequenching). (B) Application of the ensemble lipid-mixing vesicle fusion assay to proteoliposomes containing SNARE proteins (Weber et al., Citation1998). NBD-PE and Rh-PE containing vesicles reconstituted with v-SNARES (synaptobrevin shown in blue) fuse with unlabeled vesicle containing t-SNAREs (syntaxin shown in red and SNAP-25 shown in green), resulting in the dequenching of NBD fluorescence.
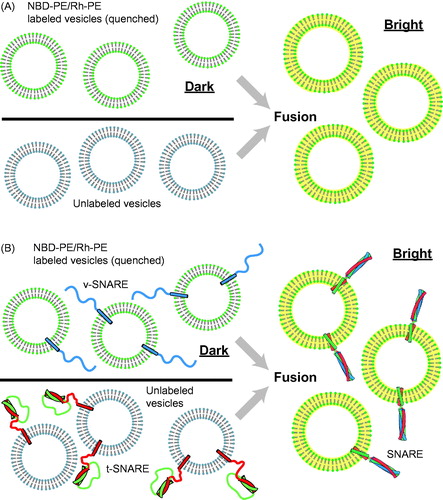
The Struck, Hoekstra & Pagano assay was used by Rothman and colleagues to demonstrate that neuronal SNARE proteins are fusogens (Weber et al., Citation1998). Neuronal SNAREs were reconstituted into different classes of small unilamellar vesicles. One class of vesicles contained synaptobrevin/VAMP (also referred to as a v-SNAREs), and the other class contained syntaxin and SNAP-25 (also referred to collectively as t-SNAREs). The v-SNARE vesicles contained a quenched mixture of NBD-PE and Rh-PE, whereas the t-SNARE vesicles were unlabeled ().
The ensemble lipid-mixing assay by Weber et al. became very popular tool in studies of SNARE-mediated fusion. Unfortunately, this assay cannot distinguish between hemifusion and complete fusion since lipid mixing is necessary but not sufficient for complete fusion. In other words, lipid mixing can occur without or with significantly delayed “content mixing” (the exchange or release of aqueous content). For example, neuronal SNAREs alone do not produce much content mixing, although they readily promote lipid mixing (Diao et al., Citation2012; Kyoung et al., Citation2011), influenza virus-induced fusion content mixing occurs seconds after initial lipid mixing (Floyd et al., Citation2008), and content mixing occurs with minute delay after lipid mixing in vacuolar fusion (Jun & Wickner, Citation2007).
Since inner leaflet mixing should be a stronger indicator for full fusion, a commonly used method to measure inner leaflet mixing involves reduction of the outward facing fluorophores by dithionite (Bhalla et al., Citation2006; Lu et al., Citation2005; Mima et al., Citation2008; Xu et al., Citation2005): for example, this method was used to identify the existence of hemifused states of vesicles with reconstituted SNAREs in ensemble lipid-mixing assays (Hernandez et al., Citation2012). However, a study of DNA-mediated proteoliposome fusion with lipid and small content indicators revealed that lipid mixing of both outer and inner leaflets can occur without content mixing (Chan et al., Citation2009). Indeed, apparent lipid mixing may arise from lipid flip-flop without fusion (Bai &Pagano, Citation1997), or from lipid-dye transfer between adjacent membranes (Ohki et al., Citation1998). These studies also call into question lipid-mixing assays that solely rely on inner-leaflet lipid-mixing indicators alone to assess complete fusion. In a recent study, it was discovered that the disposition of fluorescent lipids with respect to SNAREs had a striking effect: lipid-dequenching is asymmetric with respect to which SNARE protein is present in two proteoliposome classes (Zick & Wickner, Citation2014). For all these reasons, any fusion assay (ensemble or single-particle) that depends solely on lipid-dyes is prone to significant artifacts.
In an attempt to overcome the shortcomings of lipid-mixing assays, they were combined with FRET measurements involving dye pairs that were attached to the luminal side of syntaxin and synaptobrevin (Schuette et al., Citation2004) such that high FRET indicates SNARE complex formation. The idea is that high FRET efficiency between syntaxin and synaptobrevin labels would only be possible during membrane merger since they are initially in different membranes. However, it may be possible that the luminal sides of SNARE proteins may get close to each other without formation of a stable fusion pore. Thus, while useful, this approach is not a substitute for directly monitoring content mixing.
Content mixing measurements are difficult to achieve for ensemble-based assays because of a number of technical hurdles, discussed in more detail below. A first attempt to establish a content mixing assay for SNARE-mediated fusion was reported in 1999 with distinct DNA strands in both vesicle populations (Nickel et al., Citation1999); one strand was 32P-labeled, whereas the other had biotin fused to it. After mixing for an extended period, the sample was lysed, and the content was analyzed by streptavidin pull-down and measuring 32P radioactivity. Unfortunately, in this assay, the content mixing signal was measured after vesicles were lysed by detergent (Triton X-100), leaving the possibility that docked, but unfused vesicles could contribute to the final readout. Moreover, this assay is not suitable for measuring fast fusion kinetics.
Subsequently, interacting domains of SNAREs fused to N-terminal signal sequences were expressed and targeted to mammalian (COS or HeLa) cell surfaces (Hu et al., Citation2003). Clearly, this is strictly speaking not an in vitro assay, but we mentioned it here for completeness. T- and v-SNAREs were expressed in separate cell populations and then projected from the respective cell surfaces. To assess content mixing, a color-mixing assay was utilized where the cytoplasm of one class of cells was labeled with red fluorescent protein (RFP) with a nuclear export signal, and the other class labeled with cyan-colored fluorescent (CFP) protein with a nuclear localization signal. Upon fusion, merged cells have RFP in the cytoplasm that surrounds one or more CFP-labeled nuclei. Unfortunately, the achieved fusion processes were relatively inefficient and slow (10 min time scale) and this cell-based system is therefore unsuitable to study fast Ca2+ triggered fusion (Giraudo et al., Citation2009).
A different ensemble content mixing assay was originally developed for studies of fusion of phospholipid vesicles (Wilschut et al., Citation1980). It uses Tb3+ ions in one class of vesicles, and DPA buffer in another class; upon complex formation, fluorescence occurs. This assay revealed that neuronal SNAREs alone produce little content mixing at low protein to lipid ratios, but that addition of PEG increases lipid mixing, content mixing, but also vesicle leakage (Dennison et al., Citation2006). More recently, Ma et al. (Citation2013) utilized self-quenched sulfurhodamine B in one vesicle population based on one of the single vesicle content-mixing assays described below (Kyoung et al., Citation2011).
A potential problem with utilizing content markers in ensemble assays is that the resulting signal can be affected by leakage of vesicles. For example, when self-quenched dyes are used, even a small population of leaking vesicles can produce a “false positive” signal that can be misinterpreted as fusion. Thus, the absence of leakage must be tested in bulk content-mixing assays, for example by including self-quenched dyes in both classes of vesicles (Yu et al., Citation2013). We also caution that using Ca2+ ions (Shi et al., Citation2012) as a content mixing marker is problematic due to potential leakiness of membranes for small ions, especially near a fusion stalk (Zucchi & Zick, Citation2011). It should also be noted that an osmotic imbalance produced by soluble content markers may affect fusion kinetics (Malinin et al., Citation2002).
A fundamental problem with any ensemble fusion assays is the inability to distinguish between docking and fusion (Cypionka et al., Citation2009); this problem exists regardless of lipid or content indicators, or both, are used. For example, if an added factor results in an increase of docking, it will also result in more hemifusion/fusion, so the assay will indicate increased fusion activity. However, the same happens when the factor increases the intrinsic fusion probability (after docking), without affecting the docking step itself.
Single particle fusion assays
To overcome the shortcomings of ensemble fusion assays, single particle microscopy methods with either lipid or content-mixing indicators, or both, have been developed by several groups. Single particle fusion assays allow direct observation of the fusion process in real time and uncover information that is otherwise hidden by ensemble averaging. Moreover, single particle assays can be combined with single-molecule FRET to study protein–protein interactions at different stages of the fusion process (Brunger et al., Citation2009).
The development of single particle fusion assays was extremely challenging. Indeed, one of us (A.T.B.) initiated a project jointly with the group of Steven Chu in 1999 to develop such an assay. It took many years to eventually develop an assay that achieved Ca2+-dependent triggering with reconstituted synaptic proteins on a millisecond time scale (Diao et al., Citation2012; Kyoung et al., Citation2011; Lai et al., Citation2014).
Single-vesicle bilayer fusion assays
To mimic the geometry of the plasma membrane, the first attempts to develop a single particle fusion assay involved freely diffusing vesicles and a supported bilayer (Bowen et al., Citation2004; Fix et al., Citation2004; Liu et al., Citation2005) (). Among these attempts, the first true content-mixing assay was reported in 2004 (Bowen et al., Citation2004) (). It used a soluble dye (calcein) that was incorporated into “free” vesicles at a sufficiently high concentration that made the dye self-quenched. This particular content marker had been used in studies of protein-free phospholipids and planar phospholipids membranes (Niles, Citation1987). Synaptobrevin molecules were reconstituted into the free vesicles at a reasonably low protein-to-lipid ratio (10–30 synaptobrevin molecules per vesicle with a 50 nm diameter) and labeled with organic dyes (Cy3 or Cy5) (Bowen et al., Citation2004). Syntaxin molecules were reconstituted into the supported bilayer at a concentration of 200 molecules/μm2. Simultaneous monitoring of the fluorescence from the labeled synaptobrevin molecules and the content dye allowed discrimination of docking (indicated by the appearance of synaptobrevin-dye fluorescence), fusion (indicated by a sudden appearance of content dye fluorescence, followed by a slow decay), and bursting (indicated by a sudden appearance of content dye fluorescence followed by rapid disappearance).
Figure 2. Single-vesicle bilayer fusion assays. (A) Lipid labeled (Fix et al., Citation2004) or (B) content labeled vesicles (Bowen et al., Citation2004) are monitored as they fuse to a lipid bilayer formed on a glass imaging surface. Fusion is indicated by the sudden appearance of fluorescence at the bilayer surface followed by a slow decay as the molecules diffuse. Reconstituted proteins are synaptobrevin (blue), syntaxin (red), and SNAP-25 (green). (C) To minimize the possible influence of the glass surface on the mobility of the lipid bilayer, a method with tethered membrane patches has been devised (Rawle et al., Citation2011).
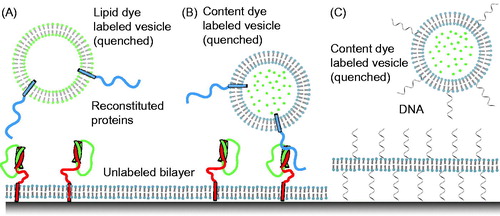
These experiments revealed that as little as one synaptobrevin molecule is sufficient for docking (Bowen et al., Citation2004). However, neuronal SNARE-mediated membrane fusion was relatively rare, and often proceeded seconds after docking ( in Bowen et al., Citation2004). Surprisingly, fusion was apparently triggered by laser-light induced heating or photobleaching-induced radical formation, and fusion was independent of the presence of SNAP-25 (i.e. they only required syntaxin and synaptobrevin). It should be noted, however, that the experimental setup prevented measurement of events right after adding the free vesicles to the supported bilayer because the system was initially equilibrated to establish single-molecule conditions and to avoid non-specific liposome binding. It is therefore possible that initial fusion events might have also occurred, albeit unobservable. We would like to stress that SNAP-25 was required to promote the formation of a stable, parallel interaction between syntaxin and synaptobrevin, as expected for SNARE complex formation involving fully functional SNARE proteins. It should be noted that a subsequent experiment with a similar setup by a different group yielded mostly vesicle bursting events for unknown reasons (Wang et al., Citation2009).
In a different approach, a similar geometry was used (i.e. a deposited bilayer with reconstituted syntaxin/SNAP-25 molecules and freely diffusing vesicles with reconstituted synaptovrevin molecules) (Fix et al., Citation2004). Lipid mixing was monitored using quenched fluorescence of the lipid dye pairs, (7-nitrobenz-2-oxa-1,3-diazole)-1,2-dipalmitoyl-sn-glycero-3-phosphatidylethanolamine (NBD-DPPE) and N-(lyssamine Rhodamine B sulfonyl)-1,2-dipalmitoyl-sn–glycerol-3-phosphatidyl–ethanolamine (Rh-DPPE), in the free vesicles (). Since no content-mixing indicator was used, this experiment allowed the monitoring of docking, undocking, and lipid mixing, but did not allow for the discrimination between hemifusion and complete fusion. Hemifusion/fusion events were inferred from dequenching and subsequent decay of the fluorescence of the lipid dyes owing to diffusion within the deposited bilayer. Surprisingly, the probability of lipid mixing was greatly enhanced in the presence of divalent cations (Mg2+ or Ca2+), in contrast to subsequent experiments with a similar geometry and with labeled lipids (Liu et al., Citation2005). Similar to the studies by Bowen et al. (Citation2004), the docking and lipid mixing results by (Liu et al., Citation2005) were independent of the inclusion of SNAP-25.
These three initial studies with single-vesicle/bilayer geometry (Bowen et al., Citation2004; Fix et al., Citation2004; Liu et al., Citation2005) produced ambiguous results: some of the experiments indicated laser-induced heating as a trigger for content mixing; and docking, lipid mixing, and fusion did not require SNAP-25 or was enhanced by divalent cations. On the other hand, stable SNARE complex formation did require the presence of SNAP-25 (Bowen et al., Citation2004). These inconsistent results were probably caused by the limited mobility of the reconstituted syntaxin molecules in the supported bilayers (Bowen et al., Citation2004). In turn, this reduced mobility was related to the simple method used to generate the supported bilayer: direct condensation of proteoliposomes on a quartz or glass surface.
In an effort to increase the mobility of the supported bilayer, more sophisticated methods involving supported bilayers have been recently developed. In one method, the supported bilayer was separated from the glass surface by a polyethylene glycol (PEG) “brush” of 4 nm height, consisting of PEG molecules linked to phosphoethanolamine (PE) (Karatekin & Rothman, Citation2012; Karatekin et al., Citation2010). The supported bilayer was then formed by condensing vesicles with reconstituted t-SNAREs (syntaxin and SNAP-25), PEG-PE, and a fluorescent-labeled lipid on a quartz surface. The free vesicles included reconstituted synaptobrevin protein molecules and fluorescent lipid labels. The use of fluorescent lipid labels allowed the monitoring of surface coverage of the supported bilayer, as well as docking and lipid mixing of the free vesicles. Both docking and lipid mixing was dependent on the presence of all three neuronal SNAREs, i.e. SNAP-25, syntaxin, and synaptobrevin.
In a second supported bilayer method, the supported bilayer was formed by a two-step process (Domanska et al., Citation2009; Kiessling et al., Citation2010). First, the first leaflet of the bilayer was formed by Langmuir–Blodgett transfer. Second, proteoliposomes were condensed to this monolayer. This method ensures that membrane proteins are properly inserted and thus prevent up-side-down proteins that adhere to the glass or quartz surface and thus may have reduced mobility of properly inserted syntaxin molecules as well (Bowen et al., Citation2004). To monitor docking and lipid mixing, the membranes of the “free” synaptobrevin containing vesicles were labeled with N-(lyssamine rhodamine B sulfonyl)-1,2-dioleoyl-sn-3-phosphatidylehanolamine (Rh-DOPE); in independent experiments, docking was also monitored by using dye-labeled synaptobrevin liposomes. Results from this assay also showed that docking and hemifusion/fusion are dependent on the presence of all three neuronal SNAREs (syntaxin, synaptobrevin, and SNAP-25), and are efficient at ambient temperature and zero Ca2+ concentration. Moreover, experiments with vesicles that were purified from brain and labeled with Rh-DOPE produced similar results (Kiessling et al., Citation2013).
The two methods described above did not utilize a content mixing indicator, so they do not discriminate between hemifusion and complete fusion, and are prone to potential artifacts caused by lipid dyes as discussed above. Moreover, the two methods used supported bilayers that are attached to a surface. It is possible that this coupling may affect the fusion probability or kinetics of fusion pore formation by constraining the membrane and preventing the necessary expansion upon fusion with a vesicle that adds additional lipids to the supported bilayer. In an attempt to overcome this problem, a method has been developed that tethers lipid bilayer membrane patches on solid surfaces (Chung et al., Citation2009). They were formed by rapture of giant-unilamellar vesicles that contain single-strand DNA-lipid hybrid molecules to surfaces with immobilized anti-sense DNA. The tethered membrane patches exhibit high lipid mobility. More recently, this method was combined with a content-mixing fusion assay (Rawle et al., Citation2011). Single unilamellar vesicles were labeled with self-quenched soluble dye (calcein), and docking and fusion were induced by the presence of DNA–lipid hybrid molecules in both vesicles and membrane patches (). This proof-of-principle study shows that the tethered membrane patch might be a good compromise that combines the best features of both single vesicle–vesicle and vesicle–support bilayer approaches. However, it remains to be seen if this assay is applicable to studying synaptic protein-mediated fusion since the presence of DNA interferes with the action of the Ca2+ trigger synaptotagmin.
Single vesicle–vesicle lipid-mixing assays with tethered vesicles
In order to avoid the potential problems with supported bilayers, an alternative approach is to immobilize one class of vesicles on a passivated surface and then to add another class of “free” vesicles to the surface-immobilized vesicles. In a pioneering study, “acceptor” fluorophore (DiD) labeled v-vesicles were immobilized on a PEG-polymer passivated imaging surface () (Yoon et al., Citation2006): tethering was facilitated by biotin-PEG molecules on a PEG-coated glass surface and biotin-PE lipids in the acceptor v-vesicles. Upon addition of neutravidin, the v-vesicles were immobilized. Free donor fluorophore (DiI) labeled t-vesicles were subsequently added and lipid mixing between t- and v-vesicles was measured using total internal reflection (TIR) microscopy (). Notably, the calculated FRET efficiency values from docked v- and t-vesicle pairs (FRET < 0.25) and hemi-fused/fused vesicle pairs (FRET > 0.6) were clearly distinguishable. Docking was observed as a sudden appearance of the fluorescence signal at the imaging surface. Intermediate states of hemifusion/fusion were observed upon donor vesicle docking. The effect of surface tethering of acceptor vesicles on lipid mixing was tested and was shown to be small (see Figure S7 in Yoon et al., Citation2006). Although this single vesicle–vesicle lipid-mixing experiment circumvents one of the deficiencies of the ensemble lipid-mixing assay by distinguishing docking and hemifusion/fusion, it does not discern hemifusion from complete fusion and it may be prone to artifacts caused by lipid-dyes as discussed above.
Figure 3. Single vesicle–vesicle lipid-mixing assays with tethered vesicles. (A) “Acceptor” vesicles containing v-SNAREs (synaptobrevin shown in blue) and labeled with DiD (red phospholipid head groups) and biotin (yellow) are immobilized to a PEG-biotin coated surface through linkage with neutravidin (navy blue). Donor vesicles containing t-SNAREs (syntaxin shown in red and SNAP-25 shown in green) and labeled with DiI (green phospholipids) are added and fusion is measured by measuring FRET using TIR microscopy (Yoon et al., Citation2006). (B) Synaptotagmin (light purple) was added in the lipid-mixing study by Lee et al. (Citation2010).
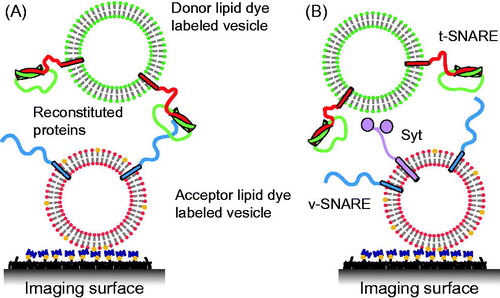
In a follow-up study, this assay was used to study the effect of synaptotagmin on single vesicle-vesicle lipid mixing (Lee et al., Citation2010) (). However, in that experiment a paradoxical decrease of Ca2+-triggered lipid mixing was observed upon increasing the Ca2+ concentration from 10 to 100 μM. It should be noted that this observation has not been re-produced by other groups with different assays. Their observation may be related to the possibility of synaptotagmin C2 cis-interactions with its own membrane when Ca2+ is present prior to docking of vesicles (Vennekate et al., Citation2012), and thereby reducing synaptotagmin trans-interactions with the acceptor vesicles.
Single vesicle–vesicle lipid-mixing assays with freely diffusing vesicles
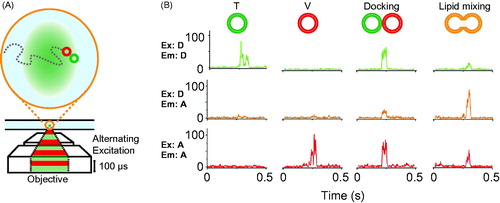
It is desirable to test the potential effect of immobilization of vesicles on a surface on the fusion assay. In order to accomplish this, an alternative method was developed in order to observe single “free” vesicle species diffusing in and out of a small confocal volume (femtoliter) using alternating-laser excitation with donor- and acceptor-dye excitation lasers (ALEX) (Choi et al., Citation2013; Kim et al., Citation2012) (). As before, t- and v-vesicles were labeled with donor and acceptor lipid dyes. Characteristic fluorescent bursts discriminate between a single vesicle, a pair of docked vesicles, or a hemifused/fused vesicle (). The small confocal volume used in these experiments ensures that, on average, only a single vesicle is observed in the excitation volume at any given time. However, the time-resolution for measuring kinetics with the ALEX method is at best ∼ 30 s since a significant number of vesicles need to be observed in the excitation volume in order to obtain sufficient statistics of the populations of undocked, docked, and hemifused/fused vesicles, respectively (Kim et al., Citation2012).
Figure 4. Single vesicle–vesicle lipid-mixing assays with freely diffusing vesicles. (A) Schematic representation of the ALEX method (Choi et al., Citation2013). Dilute mixtures of DiI labeled t-vesicles (donor) are mixed with DiD labeled v-vesicles (acceptor) and imaged using confocal microscopy with alternating laser excitation. The green area represents the confocal detection volume. (B) Representative data collected on individual vesicles diffusing through the confocal volume. Three data channels are recorded as shown: donor emission when directly excited (Ex: D, Em: D); acceptor emission when directly excited (Ex: A, Em: A); and acceptor emission resulting from FRET when the donor is excited (Ex: D, Em: A). The vesicles can be distinguished by their representative fluorescence signals as: t-vesicles only, emission of donor when directly excited, but no acceptor signal; v-vesicle only, no donor signal and emission of acceptor only when directly excited; docked vesicles, emission of both donor and acceptor when excited with the respective excitation sources but no FRET; and fused vesicles resulting in FRET (emission of both donor and acceptor when excited with the green laser). Reproduced with permission from Choi et al. (Citation2013).
Another method to study docking and hemifusion/fusion between freely diffusing vesicles is fluorescence cross-correlation spectroscopy (FCCS) combined with donor fluorescence lifetime analysis (Cypionka et al., Citation2009; Vennekate et al., Citation2012). Similar to the ALEX experiments, donor and acceptor labeled vesicles are mixed and then imaged by confocal microscopy. However, both donor and acceptor fluorophores are excited simultaneously. The two distinct fluorescence signals are then monitored separately, and a fluorescence cross-correlation function and fluorescence lifetime are calculated. This method allows determination of the population of single, docked, or hemifused/fused states.
As with the ALEX assay, the time-resolution of the FCCS assay is limited by the requirement that only a few vesicles pass through the excitation volume at any given time; in the work by (Cypionka et al., Citation2009) the time-resolution was limited to ∼ 90 s. To date, both the ALEX and the FCCS assays have only been used with lipid dyes, rather than content dyes. Thus, studies performed with these assays did not discriminate between hemifusion and complete fusion reactions, and may be subject to artifacts of lipid dyes as discussed above.
Single vesicle–vesicle content-mixing assays
In order to discriminate between different stages of fusion including docking, hemifusion, and complete fusion between immobilized and free vesicles, a single-particle assay was developed that utilizes both content and lipid-mixing indicators (Kyoung et al., Citation2011, Citation2013; Lai et al., Citation2014) (). The geometry of the assay is similar to that of Yoon et al. (Citation2006): immobilized t-vesicles (containing syntaxin and SNAP-25) were tethered to a passivated surface by neutravidin–biotin linkages, and then free v-vesicles (containing reconstituted synaptobrevin and synaptotagmin molecules) were added. This assay employed a small fluorescent content probe (sulforhodamine B, with a molecular weight of 558 Da), comparable in size to that of small neurotransmitter molecules. The assay simultaneously monitored lipid and content mixing by labeling the vesicle membrane with a lipid-dye (DiD) in addition to the content dye. Initially, the vesicles were incubated at zero Ca2+, allowing monitoring of spontaneous fusion prior to Ca2+ addition. At a defined time point, Ca2+ buffer was injected. To our knowledge, this assay is the only single vesicle–vesicle assay that starts from a metastable state at zero Ca2+, followed by injection of Ca2+ at a defined time point.
Representative observed fluorescence intensity time traces (red for lipid, green for content) for immediate content mixing, delayed content mixing, and hemifusion only are shown in ; these traces indicate a variety of fusion intermediates in real-time. By simultaneously observing lipid and content mixing in single vesicle pairs, and performing cryo-electron microscopy under the same conditions, two types of membrane contacts were found before Ca2+ addition: hemifusion intermediates and point contact interactions (Diao et al., Citation2012).
Figure 5. Single vesicle–vesicle content/lipid-mixing assays. (A) Setup of the single vesicle–vesicle assay that simultaneously monitors content and lipid mixing and that allows studies of Ca2+ triggered fusion (Diao et al., Citation2012; Kyoung et al., Citation2011, Citation2013; Lai et al., Citation2014). “Acceptor” vesicles containing syntaxin (red) and SNAP-25 (green) are immobilized via biotinylated lipids (yellow) to a glass surface that has been passivated with biotin-PEG and coated with neutravidin (dark purple). “Donor” vesicles containing high concentrations (and thus self-quenched) of sulforhodamine B in the lumen and DiD lipids are reconstituted with synaptobrevin (blue) and synaptogamin (light purple) molecules. The donor vesicles are incubated with the immobilized acceptor vesicles. Fusion is initiated by flowing buffer containing Ca2+ into the imaging chamber. (B) Docking, hemifusion, and complete fusion are monitored by measuring the fluorescence dequenching of the lipid and content dyes that result from dilution. (C) Representative fluorescence intensity time traces that result from fusion of a single donor vesicle to a single acceptor vesicle illustrating the detection of the individual fusion states shown in panel B. (D) A single vesicle content mixing assay with a large content probe, dye-labeled DNA hairpins (Diao et al., Citation2010). Upon fusion unlabeled DNA molecules complimentary to the FRET labeled hairpin bind and destabilize the hairpin leading to a decrease in FRET.
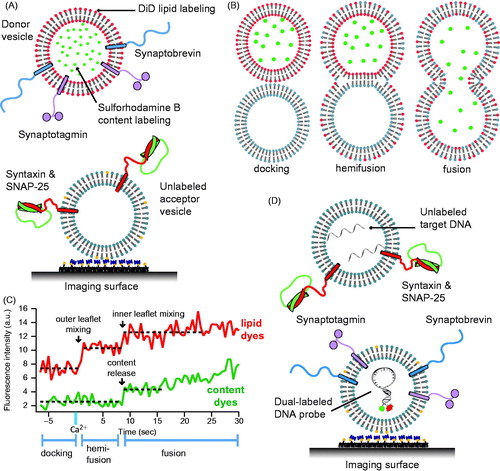
Details of the single vesicle–vesicle content/lipid-mixing assay have been described in reference (Kyoung et al., Citation2013) with important modifications as described in reference (Lai et al., Citation2014): v-vesicles were incubated at a low concentration for a longer period (20–40 min), followed by buffer exchange, and then by Ca2+-injection (originally, v-vesicles were shortly incubated at high concentration, followed by buffer exchange, and subsequent longer incubation period (Kyoung et al., Citation2011). This modification produced more interactions between surface immobilized t-vesicles and free v-vesicles. More recently, we also developed several alternatives to the Ca2+ injection system. In a first variant the cascade-blue dye that was used to indicate the instance of Ca2+ injection was replaced with the soluble dye molecule Cy5 (Lai et al., Citation2014). In a second variant, the time stamp of Ca2+ arrival in the evanescent field was defined by the instance of the first content-mixing event (i.e. stepwise increase of content dye fluorescence intensity) (Diao et al., Citation2012).
A further improvement has been recently made that allows one to monitor both spontaneous and Ca2+ triggered fusion on the same sample by first monitoring fusion events in the absence of Ca2+ right after the initial vesicle association period, followed by Ca2+ injection and continued monitoring of fusion events (Lai et al., Citation2014). Moreover, in order to increase the throughput of the assay and make better use of the vesicle samples, after washing with EGTA containing buffer to remove Ca2+ from the sample chamber, the entire acquisition procedure (vesicle association, spontaneous fusion, and Ca2+-triggered fusion) was repeated up to five times in different imaging areas within the same flow chamber. The higher throughput allows one to collect sufficient numbers of fast Ca2+ triggered fusion events. We are currently investigating how to further improve the efficiency of such triggered events (∼10%, see supporting data in Lai et al., (Citation2014)).
Larger probes have been also used for single vesicle–vesicle content-mixing assays. The single vesicle–vesicle content mixing assay by (Diao et al., Citation2010) () uses a large probe, a Cy3/Cy5 dual-labeled DNA hairpin displaying high FRET was encapsulated inside the surface immobilized v-vesicle (molecular weight 11 kDa). The free t-vesicles contained an unlabeled DNA strand with a sequence complementary to the DNA hairpin. Upon complete fusion the two cavities of vesicles become inter-connected, allowing the two DNA strands to hybridize inducing a large conformational change of the DNA hairpin that can be monitored by a switch from high to low FRET efficiency. However, the assay cannot monitor the formation of initial hemifusion intermediates since it does not include a lipid-mixing indicator. Because of the large size (4 nm in diameter) of the DNA hairpin (Lai et al., Citation2013), this assay will only produce a signal once a large pore has formed.
Important technical considerations
There are many details of fusion assays that may significantly affect the outcome. Here we emphasize some important points that may not be so obvious to the novice.
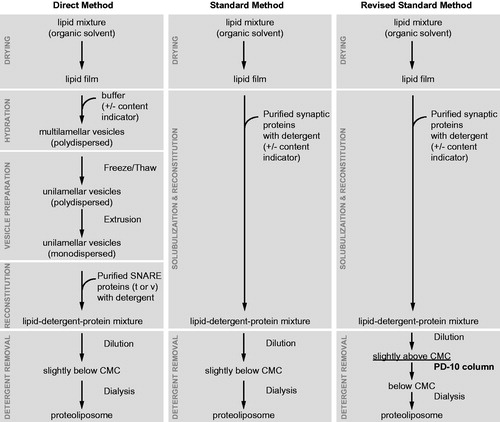
Rizo and co-workers showed that the homogeneity of vesicle size and reconstituted protein density may depend on the method used for reconstitution (Chen et al., Citation2006). The so-called “standard” method consists of detergent co-solubilization of proteins and lipids, whereas the “direct” method consists of the incorporation of detergent solubilized proteins into pre-formed lipid vesicles (). The direct method produces much more homogeneous vesicle size distributions and protein densities than the standard method. However, the modified standard method described by (Kyoung et al., Citation2011, Citation2013) () also produces homogeneous vesicle size distributions and protein number densities. This method promotes the formation of vesicles by diluting the detergent close to the critical micelle concentration rather than lowering it below this point, followed by separation of vesicles from other components on a size exclusion column. The resulting vesicle size and protein density distributions of individual vesicles were found to be homogenous as measured by cryo-EM and single molecule counting experiments, respectively (Kyoung et al., Citation2011).
Figure 6. Proteoliposome reconstitution schemes. Outlined are three methods to create proteoliposomes with reconstituted purified synaptic proteins and various reporter fluorescent dyes. The modified standard method (Kyoung et al., Citation2011, Citation2013) produces vesicles with uniform size and protein distribution. This method has been used for the single vesicle content/lipid-mixing assay described in .
Another important consideration is the directionality of the reconstituted proteins which can be easily tested by a proteolysis assay (Kyoung et al., Citation2011), as well as protein purity and quality, and the absence of any affinity tags (especially His-tags) that may introduce non-specific interactions that are a concern when using single particle methods since they very sensitive to such artifacts.
The surface preparation is critical for experiments involving tethered vesicles. On the one hand, the surface has to provide functional groups for tethering vesicles (e.g. biotin). On the other hand, the surface must not promote non-specific interactions. Tests for uniform surface coverage and non-specific binding are relatively easy to perform (Diao et al., Citation2013; Kyoung et al., Citation2013).
The incorporation of a soluble dye at relatively high concentration may impose stress on the membrane if it is not osmotically balanced by external buffers. Such stresses might introduce leakiness, although in our single vesicle content mixing assay only a very small fraction (less than 0.01%) of leaking vesicles was observed (Kyoung et al., Citation2011). However, the osmotic imbalance may still affect fusion kinetics (Malinin et al., Citation2002).
Outlook
For the single-vesicle lipid and content mixing assays described above, all proteins have to be in the right location and conformation for inducing efficient membrane fusion. Up to now, the proteins used in these assays have been largely unlabeled, limiting the ability to determine the optimum configuration for fast Ca2+-triggered membrane fusion. Thus, in the future, single vesicle content/lipid mixing fusion assays should be combined with single molecule observations in order to probe protein–protein interactions among the fusion proteins in the pre-fusion and post-fusion stages. This requires high spatial and temporal resolution in order to monitor these interactions simultaneously during the fusion processes.
The single vesicle–vesicle content/lipid mixing fusion assay allowed characterization of the kinetics and mechanism of membrane fusion with a reconstituted minimal set of synaptic proteins that mimic Ca2+-triggered synaptic vesicle fusion (Diao et al., Citation2012; Kyoung et al., Citation2011; Lai et al., Citation2014). Moreover, our assay has allowed us to characterize the role of the accessory protein complexin (Lai et al., Citation2014). Other protein factors of the membrane fusion machinery (such as Munc13, Munc18, and NSF/SNAPs) are now being included in our studies (Zhang et al., Citation2015). As single-particle and single-molecule techniques continue to develop and improve, we expect to develop even more powerful fusion assays, and, ultimately, to perform real-time measurements in live neurons.
Acknowledgments
We thank Steven Chu for a longstanding collaboration that led to the development of some of the assays discussed in this review, and Mark Bowen, Keith Weninger, and Jose Rizo for critical reading.
Declaration of interest
This work was supported by the Howard Hughes Medical Institute and the U.S. National Institutes of Health (grant R37-MH63105 to ATB).
References
- Bai J, Pagano RE. (1997). Measurement of spontaneous transfer and transbilayer movement of BODIPY-labeled lipids in lipid vesicles. Biochemistry 36:8840–8
- Balch WE, Dunphy WG, Braell Wa, et al. (1984). Reconstitution of the transport of protein between successive compartments of the golgi measured by the coupled incorporation of N-acetylglucosamine. Cell 39:405–16
- Bhalla A, Chicka MC, Tucker WC, et al. (2006). Ca(2+)-synaptotagmin directly regulates t-SNARE function during reconstituted membrane fusion. Nat Struct Mol Biol 13:323–30
- Bowen ME, Weninger K, Brunger AT, et al. (2004). Single molecule observation of liposome-bilayer fusion thermally induced by soluble N-ethyl maleimide sensitive-factor attachment protein receptors (SNAREs). Biophys J 87:3569–84
- Brunger AT, Weninger K, Bowen M, et al. (2009). Single-molecule studies of the neuronal SNARE fusion machinery. Annu Rev Biochem 78:903–28
- Chan Y-HM, van Lengerich B, Boxer SG. (2009). Effects of linker sequences on vesicle fusion mediated by lipid-anchored DNA oligonucleotides. Proc Natl Acad Sci USA 106:979–84
- Chen X, Araç D, Wang T-M, et al. (2006). SNARE-mediated lipid mixing depends on the physical state of the vesicles. Biophys J 90:2062–74
- Choi B-K, Choi M-G, Kim J-Y, et al. (2013). Large α-synuclein oligomers inhibit neuronal SNARE-mediated vesicle docking. Proc Natl Acad Sci USA 110:4087–92
- Chung M, Lowe RD, Chan Y-HM, et al. (2009). DNA-tethered membranes formed by giant vesicle rupture. J Struct Biol 168:190–9
- Cypionka A, Stein A, Hernandez JM, et al. (2009). Discrimination between docking and fusion of liposomes reconstituted with neuronal SNARE-proteins using FCS. Proc Natl Acad Sci USA 106:18575–80
- Dennison S, Bowen M, Brunger A, et al. (2006). Neuronal SNAREs do not trigger fusion between synthetic membranes but do promote PEG-mediated membrane fusion. Biophys J 90:1661–75
- Diao J, Su Z, Ishitsuka Y, et al. (2010). A single-vesicle content mixing assay for SNARE-mediated membrane fusion. Nat Commun 1:54. doi:10.1038/ncomms1054
- Diao J, Grob P, Cipriano DJ, et al. (2012). Synaptic proteins promote calcium-triggered fast transition from point contact to full fusion. Elife 1:e00109
- Diao J, Cipriano DJ, Zhao M, et al. (2013). Complexin-1 enhances the on-rate of vesicle docking via simultaneous SNARE and membrane interactions. J Am Chem Soc 135:15274–7
- Domanska MK, Kiessling V, Stein A, et al. (2009). Single vesicle millisecond fusion kinetics reveals number of SNARE complexes optimal for fast SNARE-mediated membrane fusion. J Biol Chem 284:32158–66
- Fix M, Melia TJ, Jaiswal JK, et al. (2004). Imaging single membrane fusion events mediated by SNARE proteins. Proc Natl Acad Sci USA 101:7311–16
- Floyd DL, Ragains JR, Skehel JJ, et al. (2008). Single-particle kinetics of influenza virus membrane fusion. Proc Natl Acad Sci USA 105:15382–7
- Giraudo CG, Garcia-Diaz A, Eng WS, et al. (2009). Alternative zippering as an on-off switch for SNARE-mediated fusion. Science 323:512–16
- Hernandez JM, Stein A, Behrmann E, et al. (2012). Membrane fusion intermediates via directional and full assembly of the SNARE complex. Science 336:1581–4
- Hu C, Ahmed M, Melia TJ, et al. (2003). Fusion of cells by flipped SNAREs. Science 300:1745–9
- Jahn R, Fasshauer D. (2012). Molecular machines governing exocytosis of synaptic vesicles. Nature 490:201–7
- Jun Y, Wickner W. (2007). Assays of vacuole fusion resolve the stages of docking, lipid mixing, and content mixing. Proc Natl Acad Sci USA 104:13010–15
- Karatekin E, Rothman JE. (2012). Fusion of single proteoliposomes with planar, cushioned bilayers in microfluidic flow cells. Nat Protoc 7:903–20
- Karatekin E, Di Giovanni J, Iborra C, et al. (2010). A fast, single-vesicle fusion assay mimics physiological SNARE requirements. Proc Natl Acad Sci USA 107:3517–21
- Kiessling V, Domanska MK, Tamm LK. (2010). Single SNARE-mediated vesicle fusion observed in vitro by polarized TIRFM. Biophys J 99:4047–55
- Kiessling V, Ahmed S, Domanska MK, et al. (2013). Rapid fusion of synaptic vesicles with reconstituted target SNARE membranes. Biophys J 104:1950–8
- Kim J-Y, Choi B-K, Choi M-G, et al. (2012). Solution single-vesicle assay reveals PIP2-mediated sequential actions of synaptotagmin-1 on SNAREs. EMBO J 31:2144–55
- Kyoung M, Srivastava A, Zhang Y, et al. (2011). In vitro system capable of differentiating fast Ca2+-triggered content mixing from lipid exchange for mechanistic studies of neurotransmitter release. Proc Natl Acad Sci USA 108:E304–13
- Kyoung M, Zhang Y, Diao J, et al. (2013). Studying calcium-triggered vesicle fusion in a single vesicle–vesicle content and lipid-mixing system. Nat Protoc 8:1–16
- Lai Y, Diao J, Liu Y, et al. (2013). Fusion pore formation and expansion induced by Ca2 + and synaptotagmin 1. Proc Natl Acad Sci USA 110:1333–8
- Lai Y, Diao J, Cipriano DJ, et al. (2014). Complexin inhibits spontaneous release and synchronizes Ca2+-triggered synaptic vesicle fusion by distinct mechanisms. Elife 3:1–14
- Lee H-K, Yang Y, Su Z, et al. (2010). Dynamic Ca2+-dependent stimulation of vesicle fusion by membrane-anchored synaptotagmin 1. Science 328:760–3
- Liu T, Tucker WC, Bhalla A, et al. (2005). SNARE-Driven, 25-millisecond vesicle fusion in vitro. Biophys J 89:2458–72
- Lu X, Zhang F, McNew JA, et al. (2005). Membrane fusion induced by neuronal SNAREs transits through hemifusion. J Biol Chem 280:30538–41
- Ma C, Su L, Seven AB, et al. (2013). Reconstitution of the vital functions of Munc18 and Munc13 in neurotransmitter release. Science 339:421–5
- Malinin VS, Frederik P, Lentz BR. (2002). Osmotic and curvature stress affect PEG-induced fusion of lipid vesicles but not mixing of their lipids. Biophys J 82:2090–100
- Mima J, Hickey CM, Xu H, et al. (2008). Reconstituted membrane fusion requires regulatory lipids, SNAREs and synergistic SNARE chaperones. EMBO J 27:2031–42
- Nickel W, Weber T, McNew JA, et al. (1999). Content mixing and membrane integrity during membrane fusion driven by pairing of isolated v-SNAREs and t-SNAREs. Proc Natl Acad Sci USA 96:12571–6
- Niles WD. (1987). Video fluorescence microscopy studies of phospholipid vesicle fusion with a planar phospholipid membrane. Nature of membrane-membrane interactions and detection of release of contents. J Gen Physiol 90:703–35
- Ohki S, Flanagan TD, Hoekstra D. (1998). Probe transfer with and without membrane fusion in a fluorescence fusion assay. Biochemistry 37:7496–503
- Otterstrom J, van Oijen A. (2013). Visualization of membrane fusion, one particle at a time. Biochemistry 53:1654–68
- Rawle RJ, van Lengerich B, Chung M, et al. (2011). Vesicle fusion observed by content transfer across a tethered lipid bilayer. Biophys J 101:L37–9
- Schuette CG, Hatsuzawa K, Margittai M, et al. (2004). Determinants of liposome fusion mediated by synaptic SNARE proteins. Proc Natl Acad Sci USA 101:2858–63
- Shi L, Shen Q-T, Kiel A, et al. (2012). SNARE proteins: one to fuse and three to keep the nascent fusion pore open. Science 335:1355–9
- Struck DK, Hoekstra D, Pagano RE. (1981). Use of resonance energy transfer to monitor membrane fusion. Biochemistry 20:4093–9
- Südhof TC. (2013). Neurotransmitter release: the last millisecond in the life of a synaptic vesicle. Neuron 80:675–90
- Südhof TC, Rothman JE. (2009). Membrane fusion: grappling with SNARE and SM proteins. Science 323:474–7
- Vennekate W, Schröder S, Lin C-C, et al. (2012). Cis- and trans-membrane interactions of synaptotagmin-1. Proc Natl Acad Sci USA 109:11037–42
- Wang T, Smith EA, Chapman ER, et al. (2009). Lipid mixing and content release in single-vesicle, SNARE-driven fusion assay with 1–5 ms resolution. Biophys J 96:4122–31
- Weber T, Zemelman BV, McNew JA, et al. (1998). SNAREpins: minimal machinery for membrane fusion. Cell 92:759–72
- Wilschut J, Düzgüneş N, Fraley R, et al. (1980). Studies on the mechanism of membrane fusion: kinetics of calcium ion induced fusion of phosphatidylserine vesicles followed by a new assay for mixing of aqueous vesicle contents. Biochemistry 19:6011–21
- Xu Y, Zhang F, Su Z, et al. (2005). Hemifusion in SNARE-mediated membrane fusion. Nat Struct Mol Biol 12:417–22
- Yoon T-Y, Okumus B, Zhang F, et al. (2006). Multiple intermediates in SNARE-induced membrane fusion. Proc Natl Acad Sci USA 103:19731–6
- Yu H, Rathore SS, Lopez Ja, et al. (2013). Comparative studies of Munc18c and Munc18-1 reveal conserved and divergent mechanisms of Sec1/Munc18 proteins. Proc Natl Acad Sci USA 110:E3271–80
- Zhang Y, Diao J, Colbert KN, et al. (2015). Munc18a does not alter fusion rates mediated by neuronal SNAREs, synaptotagmin, and complexin. J Biol Chem doi:10.1074/jbc.M114.630772
- Zick M, Wickner WT. (2014). A distinct tethering step is vital for vacuole membrane fusion. Elife 3:e03251
- Zucchi PC, Zick M. (2011). Membrane fusion catalyzed by a Rab, SNAREs, and SNARE chaperones is accompanied by enhanced permeability to small molecules and by lysis. Mol Biol Cell 22:4635–46