Abstract
Chronic lymphocytic leukemia (CLL) is a clinically and biologically heterogeneous disease where the majority of patients have an indolent disease course, while others may experience a far more aggressive disease, treatment failure and poor overall survival. During the last two decades, there has been an intense search to find novel biomarkers that can predict prognosis as well as guide treatment decisions. Two of the most reliable molecular prognostic markers, both of which are offered in routine diagnostics, are the immunoglobulin heavy chain variable (IGHV) gene mutational status and fluorescence in situ hybridization (FISH) detection of prognostically relevant genomic aberrations (e.g. 11q−, 13q−, +12 and 17p−). In addition to these markers, a myriad of additional biomarkers have been postulated as potential prognosticators in CLL, on the protein (e.g. CD38, ZAP70, TCL1), the RNA (e.g. LPL, CLLU1, micro-RNAs) and the genomic (e.g. TP53, NOTCH1, SF3B1 and BIRC3 mutations) level. Efforts are now being made to test these novel markers in larger patient cohorts as well as in prospective trials, with the ultimate goal to combine the “best” markers in a “CLL prognostic index” applicable for the individual patient. Although it is clear that these studies have significantly improved our knowledge regarding both prognostication and the biology of the disease, there is still an immediate need for recognizing biomarkers that can predict therapy response, and efforts should now focus on addressing this pertinent issue. In the present article, we review the extensive literature in the field of prognostic markers in CLL, focus on the most clinically relevant markers and discuss future directions regarding biomarkers in CLL.
Introduction
Considering the clinical heterogeneity among patients with chronic lymphocytic leukemia (CLL), there has been a great need to find novel biomarkers that can aid prognostication and therapy selection. Besides the established clinical staging systems (i.e. Rai and Binet staging) and some laboratory parameters (e.g. lymphocyte doubling time [LDT], serum lactate dehydrogenase [LDH]), many different types of markers have been identified and evaluated as prognostic factors in CLL. These range from general markers that are measured in serum or blood, to protein markers detectable using flow cytometry, as well as specific genetic markers that can be detected through targeted laboratory tests, such as fluorescence in situ hybridization (FISH) and mutation analysis (). In this review, we provide a comprehensive and partly “historical” summary of prognostic factors in CLL and describe the most important markers that, during recent decades, have been suggested for inclusion in clinical evaluation. Very recent markers that were detected through next-generation sequencing techniques, i.e. NOTCH1, SF3B1 and BIRC3, are also included in this review. In the final part, we also discuss the usefulness of prognostic markers in clinical diagnostics, the absolute need for novel predictive markers that can guide therapy decisions, and how these markers ultimately could be combined as a “CLL prognostic index.”
Table I. Overview of prognostic markers in CLL.
Clinical markers in chronic lymphocytic leukemia
Clinical staging systems
Two clinical staging systems were independently developed by Rai and Binet to facilitate stratification of patients with CLL into different risk groups, and both are currently used in clinics for disease prognostication [Citation1,Citation2]. These staging systems have now been applied for prognostication for more than 30 years, and are based on physical examination and standardized blood tests. Specifically, the detection of signs of disease other than lymphocytosis, such as lymphadenopathy, hepato-/splenomegaly, anemia and thrombocytopenia, aid in the staging of patients into risk groups, i.e. 0–IV for Rai and A–C for Binet staging. Both staging systems have proved useful for estimating outcome, since patients with CLL in Binet stage A or Rai stage 0 have a long overall survival (OS), with an expected median survival exceeding 10 years [Citation3]. In comparison, patients in Binet stage B and Rai stage I/II show an intermediate median survival of 5–7 years, whereas patients in the high-risk groups, Binet stage C and Rai stage III/IV, have a considerably shorter median survival of less than 3 years [Citation3]. Overall, the Rai and Binet staging systems are good prognostic markers in the sense that they are easily assessed and do not depend on laborious and expensive tests. Nevertheless, the clinical stages are not absolute since patients may progress to a higher disease stage, which makes it problematic to assess the disease course in patients within the low-risk group, who constitute at least 70% of patients with CLL at diagnosis. Another limitation is that the staging systems do not provide any information as to how patients will respond to treatment, and can therefore not be used to direct treatment.
Lymphocyte doubling time
Lymphocyte doubling time is defined as the number of months it takes for the lymphocytes to double in absolute number. Hence, a short LDT is directly related to a high proliferation rate and a more aggressive disease. This marker has an independent prognostic significance, and has been shown to correlate with clinical stage and the level of bone marrow infiltration [Citation4]. A LDT of less than 6 months implies an active disease, which is one of the criteria that should be met in order to initiate treatment [Citation5]. However, since patients with a short LDT may be asymptomatic, this marker should not be used as the only indicator to initiate therapy, and is therefore more useful in assessing disease aggressiveness rather than to predict outcome at an early stage of the disease.
Serum markers
Most available serum markers are not specific for CLL but can be easily measured, and provide some useful prognostic information. One of the most common serum markers is LDH, an enzyme that is increased in CLL (as well as other lymphomas), and where a higher level corresponds to several poor-prognostic features and risk of developing Richter syndrome [Citation6–8]. Similarly, a high β2-microglobulin (B2M) level denotes patients with CLL who belong to a more advanced clinical stage with an increased tumor burden [Citation9,Citation10]. Moreover, patients with Binet stage A showing a high B2M level have a shorter progression-free survival (PFS) compared to patients with stage A with low B2M levels, thus indicating that B2M gives additional prognostic information in patients with a quiescent disease [Citation11]. Another serum marker, thymidine kinase (TK), is elevated in patients with CLL with a more aggressive disease, and is associated with unmutated immunoglobulin heavy chain variable (IGHV) genes, high-risk genomic aberrations and a short LDT [Citation12–14]. In one of the studies investigating TK serum levels in CLL, Di Raimondo et al. showed that the TK level can predict response to fludarabine treatment, since as 80% of patients with a low TK level responded to treatment, whereas only 45% of patients with a high TK level responded to treatment [Citation12]. Nevertheless, despite the fact that serum markers may have a role in CLL prognostication, the advent of novel molecular biomarkers has shifted the focus to factors that can provide more disease-specific information regarding survival and treatment responses.
Genetic markers in chronic lymphocytic leukemia
IGHV gene mutational status and beyond
In two landmark papers published in 1999, Hamblin et al. and Damle et al. reported an association between prognosis and the somatic hypermutation status of the IGHV genes in CLL [Citation15,Citation16]. In both these studies, patients with unmutated IGHV genes (40–50% of patients) displayed a more aggressive disease, high-risk cytogenetics and a poor outcome, while IGHV-mutated genes instead were associated with a more favorable clinical course with long OS. This important subclassification of CLL has now been replicated in many subsequent studies, and the IGHV gene mutational status is currently one of the most commonly used prognostic markers and also widely accepted as one of the most stable and reliable indicators of clinical outcome [Citation17–22]. illustrates the different pathogenic mechanisms that are known to be involved in IGHV-mutated and -unmutated CLL.
Figure 1. Different molecular mechanisms driving IGHV-mutated and unmutated CLL. An illustration of the most important genetic, RNA and protein based prognostic markers in relation to the IGHV gene mutational status. NOTCH1 activating mutations and BIRC3 disrupting mutations lead to enhanced NF-κB signaling and proliferation in IGHV-unmutated CLL. ZAP70 is involved in BcR signaling and its overexpression leads to enhanced proliferation through, e.g. NF-κB signaling. Disruption of TP53 through deletion and/or mutation is more common in IGHV-unmutated CLL and leads to dysfunctional cell cycle arrest resulting in enhanced cell survival. Although the reason for higher LPL and CLLU1 expression in CLL is unknown, these markers are correlated to high risk.
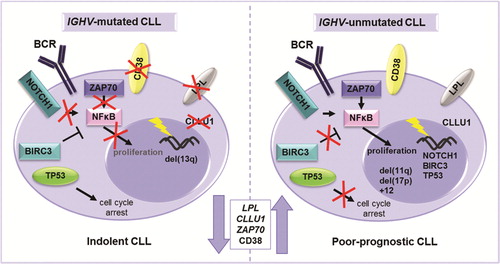
IGHV-mutated cases were originally defined as having less than 98% identity to the corresponding germline sequence, since it was important to distinguish between patients expressing mutated IGHV genes and patients having naturally occurring polymorphisms [Citation16]. Despite the fact that other cut-off values, such as 94% or 95% identity, have been suggested throughout the years, most publications have since established the initial 98% cut-off value to be the best discriminator of clinical outcome, although patients displaying minimally or borderline mutated IGHV genes, i.e. 97–98% germline identity, are to be evaluated with caution [Citation19–23].
That notwithstanding, we reported the first exception to the mutated/unmutated “rule,” where patients with CLL carrying IGHV3-21 genes were commonly IGHV-mutated but still displayed poor clinical outcome [Citation24]. This finding has also been replicated in subsequent studies, and today, cases of CLL with IGHV3-21 rearrangements are thought to constitute a specific subgroup of patients with poor prognosis, independent of IGHV gene mutational status [Citation25–27].
Approximately half of IGHV3-21-expressing patients were shown early on to display almost identical complementarity determining region 3 (CDR3) sequences, the main determinant for antigen specificity, as well as restricted lambda light-chain IGVL3-21 usage [Citation27–29]. This was one of the first observations of B cell receptor (BcR) “stereotypy” among patients with CLL, pointing to a key role for antigen selection in leukemogenesis [Citation30,Citation31]. Since then, numerous subsets with quasi-identical or stereotyped BcRs have been characterized [Citation29,Citation31–33]. In fact, today we know that almost 30% of all patients with CLL display stereotyped BcRs and can be subdivided into 19 major subsets [Citation34]. Intriguingly, patients with CLL within certain stereotyped subsets have also been shown to have similar clinical and biological features [Citation33,Citation35]. These include, for instance, stereotyped subset 1 patients (IGHV1/5/7 clan genes combined with IGKV1-39/IGKV1D-39 genes) who all carry unmutated IGHV genes and display a particularly poor prognosis, and stereotyped subset 4 cases (IGHV4-34/IGKV2-30) who all show mutated IGHV genes, young age at diagnosis and IgG-switched disease, and are rarely in need of treatment [Citation33]. Furthermore, some studies have indicated that only IGHV3-21 patients with stereotyped BcRs (now denoted as subset 2) are associated with a poor outcome; however, this has not been confirmed by others, who observed an equally poor prognosis in stereotyped and non-stereotyped subset 2 patients [Citation26,Citation27,Citation33,Citation35,Citation36]. In the coming years, large, multicenter studies will hopefully reveal the full impact of stereotypy on prognosis and outcome in CLL.
Genetic landscape in chronic lymphocytic leukemia
For many years it has been well established that the genomic landscape in CLL is characterized by certain recurrent genomic alterations, such as deletions of 11q, 13q, 17p and trisomy 12, which also have immediate prognostic impact for patients [Citation37–40]. Importantly, patients with deletions of 17p and/or TP53 mutations have the most aggressive phenotype, and these are the only markers that currently are recommended to direct treatment decisions [Citation5,Citation41]. Other structural aberrations with prognostic significance involve translocations and genomic complexity, even though their presence is fairly low in CLL [Citation42–46]. In recent times, the advent of next-generation sequencing (NGS) techniques has helped uncover novel genetic mutations in, for example, NOTCH1, SF3B1 and BIRC3 [Citation47–51]. In fact, mutations in these genes have recently been suggested to be included in the prognostic evaluation, which currently involves the known recurrent aberrations and TP53 mutations [Citation39,Citation52]. In the following section, the biological function of genetic markers and their relation to prognosis are briefly described.
Known recurrent genomic aberrations
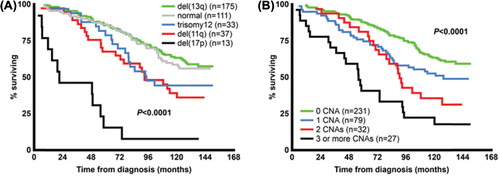
FISH is the gold-standard method applied in clinic diagnostics for detection of the known recurrent genomic aberrations. As mentioned above, these aberrations include deletions of 11q, 13q, 17p and trisomy 12, and provide important information regarding patient outcome when classified according to the hierarchal model [] initially proposed by Döhner et al. [Citation37,Citation53]. The application of FISH or microarrays will detect the known recurrent aberrations at a frequency of up to 80–90% at diagnosis, and patients without any of these aberrations appear to have an intermediate to good prognosis [Citation37,Citation39,Citation54].
Figure 2. Overall survival in patients according to known recurrent aberrations (A) and in relation to increasing genomic complexity (B) in a population-based Scandinavian CLL cohort.
Deletion of 11q
The deletion of 11q is most often monoallelic and carried by 10–17% of patients with CLL [Citation37,Citation54]. The minimal deleted region (MDR), which is 2–3 Mbp in size and located to 11q22.3-q23.1, is known to encode several tumor suppressor genes including ATM [Citation55,Citation56]. This gene plays an important role in cell cycle regulation by activating p53 and augmenting the DNA damage response. Of importance, studies focusing on ATM in CLL have shown that up to a third of patients carrying del(11q) have somatic or germline ATM mutations [Citation57,Citation58]. Recently, mutations in BIRC3, which is encoded on 11q22.2, were detected particularly in advanced CLL stages with fludarabine refractoriness [Citation50]. Since 11q deletion does not always result in mono- or biallelic silencing of ATM, an additional plausible candidate gene is BIRC3, which may play a role in patients carrying del(11q), as suggested in a recent review by Rossi et al. [Citation59].
The deletion of 11q is associated with younger age at diagnosis and an inferior outcome [Citation37]. Moreover, it has also been shown to correlate with unmutated IGHV genes and lymphadenopathy [Citation60]. Nevertheless, even if patients with deletion of 11q have a poor survival, clinical trials have shown that a combination of fludarabine, cyclophosphamide and rituximab (FCR) increases the therapy response and survival time compared to treatment with fludarabine and cyclophosphamide alone [Citation61]. Hence, 11q deletion could now be considered as a new predictive marker, at least for fit patients who tolerate the FCR regimen.
Deletion of 13q
Deletion of 13q is the most frequently occurring genomic alteration in CLL at diagnosis, presented at a frequency of 35–45% as the sole abnormality [Citation37,Citation54]. This deletion is associated with the most favorable prognosis when compared to deletions of 11q, 17p and trisomy 12 [Citation37,Citation54]. Deletions of 13q vary markedly in size, with losses ranging from ˜300 kbp up to > 70 Mbp [Citation54,Citation62,Citation63]. The MDR is located at 13q14, and several genes encoded in this region, e.g. DLEU1, DLEU2, TRIM13, RB1 and the microRNAs miR15a/16-1, have been suggested as candidate genes [Citation64–67]. One of the documented biological functions of miR15a/16-1 is down-regulation of the anti-apoptotic gene BCL2 through post-translational mRNA repression, which may lead to an increased anti-apoptotic resistance [Citation68].
Interestingly, it has been shown that the size of the 13q deletion is associated with outcome, since patients with CLL with larger aberrations have a shorter time to treatment (TTT) and OS, indicating that several genes included in the deletion have an effect on the disease course [Citation54,Citation63,Citation69,Citation70]. Moreover, Van Dyke et al. investigated the prognostic significance of 13q deletions and found that patients with homozygous and heterozygous deletions had a similar time to first treatment (TFT) and OS, however that patients with a larger del(13q) clone had a poorer TFT compared to those with a smaller del(13q) clone [Citation71]. This observation most probably reflects a greater tumor cell growth potential and an increased risk of becoming aggressive in patients with a larger del(13q) clone [Citation71].
Deletion of 17p and TP53 mutations
The deletion of 17p is detected at a frequency of 3–7% at diagnosis [Citation37,Citation54]. The 17p deletion often involves the entire p-arm, but some losses are focused to the 17p13.1 region, which encodes the TP53 gene among several other genes. This gene is a key regulator of the cell cycle, since it induces cell cycle arrest and promotes DNA repair or apoptosis when the cell has accumulated DNA damage [Citation72]. As expected, genomic complexity is commonly detected in patients with del(17p), and this is most probably due to the fact that these patients have lost their cell repair mechanism and more easily acquire additional aberrations [Citation43,Citation54,Citation73,Citation74]. CLL patients with a del(17p) commonly present other poor-prognostic markers such as unmutated IGHV genes and high expression of CD38 and ZAP70 [Citation19,Citation43].
Importantly, mutations of the TP53 gene have been detected in patients with CLL with or without a concomitant del(17p), although mutations in patients without the 17p deletion are infrequent at diagnosis [Citation75–77]. Several studies performed by Zenz et al. have focused on TP53 mutations in CLL, and have provided a better understanding of these mutations and their consequences in this disease [Citation77–80]. For instance, it is now known that TP53 deletions/mutations increase in progressive stages and accumulate in chemorefractory patients [Citation40,Citation54,Citation81,Citation82]. In fact, del(17p) and TP53 mutations are thought to represent approximately 40% of the cases that are resistant to treatment [Citation79]. Evidently, patients with CLL with TP53 mutation and/or deletion have the worst outcome, with an aggressive disease course and short OS () in combination with a poor response to treatment, as shown by several recent clinical trials [Citation61,Citation83–85]. In addition, more than a third of all cases that develop Richter transformation carry a deletion of 17p [Citation86,Citation87]. For these reasons, it is now suggested that detection of TP53 mutations should be included in the genetic screening of patients with CLL (at least exons 4–9), in order to identify patients who may be resistant to conventional therapy [Citation41]. Instead, these patients should be given alternative treatments, such as allogeneic stem cell transplant or alemtuzumab combined with high-dose steroids [Citation88].
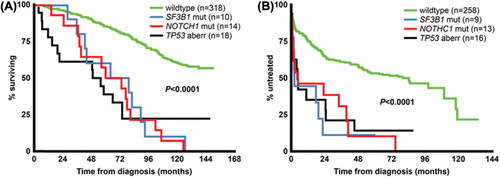
Figure 3. Prognostic impact of TP53 aberrations, NOTCH1 and SF3B1 mutations on overall survival and time to treatment in newly diagnosed CLL. For analysis of overall survival (A), six cases with concurrent del(17p) and NOTCH1 or SF3B1 mutations were included in the “TP53 aberration” subgroup, whereas for time to treatment analysis (B), five cases included in the “TP53 aberration” subgroup also displayed NOTCH1 or SF3B1 mutations. There were no statistically significant differences when comparing the TP53 aberration subgroup to either NOTCH1 or SF3B1 mutation subgroups for either overall survival or time to treatment.
Trisomy 12
Since large aberrations are readily detected with cytogenetics, trisomy 12 was one of the first aberrations considered as recurrent and associated with prognostic information in CLL [Citation53,Citation89]. This aberration is detected in 11–16% of patients at diagnosis and is associated with an intermediate prognosis [Citation37,Citation41,Citation53,Citation54,Citation89]. Moreover, patients with this trisomy have a good response to treatment, and the aberration therefore does not increase at relapse or in refractory patient groups. Trisomy 12 has been connected with concurrent trisomy of chromosomes 18 and 19 [Citation54,Citation90–92], and the combination of +12, +18 and +19 has been shown in IgG-switched CLL [Citation93]. Interestingly, some patients with CLL with trisomy 12 and IgG-switched disease also express stereotyped IGHV4-39/IGKV1(D)-39 BcRs [Citation94] and a concurrent t(14;19)(q32;q13), involving the IGH locus and the BCL3 gene. All these findings point to a stepwise acquisition of genomic aberrations during leukemogenesis, at least for certain subgroups [Citation95,Citation96].
Translocations
In CLL, translocations are found in up to 20–30%, although some studies have included a high number (40%) of pretreated patients, which may explain the frequent occurrence of these aberrations [Citation43,Citation45]. Both balanced and unbalanced translocations are detected in CLL, and the fusions recurrently involve the IGH gene on 14q32 [e.g. t(14;19)(q32;q13) involving juxtaposition of the BCL3 gene to the IGH gene promoter], and less commonly, the IGL loci on 2p11 and 22q11 [Citation43,Citation45,Citation96]. Moreover, 13q14, 11q13, 18q21 and 19q13 have been detected as recurrent partners in translocations to the IG gene loci [Citation43,Citation45]. The presence of translocations may also provide prognostic information in CLL, since it has been shown that patients with these structural aberrations have a shorter OS and treatment-free survival (TFS) [Citation45]. For instance, Van Den Neste et al. showed that chromosomal translocations, and in particular those that are unbalanced, predict treatment failure, TFS and OS in patients treated with cladribine [Citation46].
Genomic complexity
At diagnosis only a small proportion of patients with CLL have a high number of genomic aberrations, a finding which is termed genomic complexity, and specifically includes patients carrying ≥ 3 deletions and/or gains [Citation42]. Genomic aberrations may accumulate during the course of the disease and lead to a more complex karyotype, thus being more commonly detected in patients prior to treatment initiation or at relapse. For instance, Van Den Neste et al. studied patients with CLL treated with cladribine and detected genomic complexity in 20% of patients, who had a shorter TFS and OS compared to patients with fewer aberrations [Citation46]. Of importance, it has been shown that genomic complexity is associated with deletions of 11q and 17p involving ATM and TP53, respectively [Citation42,Citation43]. Naturally, a loss of function of these genes will lead to dysfunctional cell-cycle and impaired DNA-repair mechanisms, hence promoting a higher genomic complexity. It has also been shown that patients with a high number of genomic aberrations often have unmutated IGHV genes, are CD38 positive and have short telomeres [Citation43,Citation97]. Moreover, Kujawski et al. demonstrated that genomic complexity is an independent risk marker for disease progression, as patients with ≥ 3 aberrations had a shorter time to first and second treatment [Citation44]. Correspondingly, we showed that patients with a high number of large gains or losses (> 5 Mbp) had a shorter TTT and OS [] [Citation42]. Recently, another form of genomic complexity, termed chromothripsis, was found in 5% of patients with CLL by application of high-resolution single nucleotide polymorphism (SNP) arrays [Citation98]. Chromothripsis is defined as massive genomic rearrangement within a single chromosome, and is believed to increase during a single cell division [Citation99]. This phenomenon was linked to unmutated IGHV genes, poor-prognostic aberrations and a worse outcome in terms of PFS and OS [Citation98]. Taken together, it is likely that a high number of aberrations acquired sequentially or during one single event marks a similar adverse effect on patient outcome.
Novel gene mutations
NOTCH1
NOTCH1, which is encoded on chromosome 9q34.3, was the first gene to be recurrently detected with mutations by the application of NGS in two independent studies of CLL [Citation47,Citation48]. In the study by Puente et al., mutations of this gene were identified in 12% of cases of CLL, whereas Fabbri and co-workers presented a lower frequency of 8.3% [Citation47,Citation48]. Notably, we recently investigated the presence of NOTCH1 mutations at diagnosis in a population-based cohort and found a lower mutation frequency (4.7%) than previously reported in CLL, which probably reflects the unselective nature of our patient cohort [Citation52].
Mutations of NOTCH1 are most often detected within the intracellular proline, glutamic acid, serine and threonine (PEST) domain, and generate a premature stop codon that results in a C-terminal deficient, constitutively active NOTCH1 [Citation47,Citation48]. This activation supports the nuclear factor κB (NF-κB) pathway through interaction with the IKK complex [Citation100]. In B cells, NOTCH1 stimulation has been shown to participate in the formation of terminally differentiated antibody-secreting plasma cells [Citation101]. Enhanced NOTCH1 signaling has also been demonstrated to increase cell survival and apoptosis resistance in CLL cells [Citation102]. Moreover, NOTCH1 mutations are associated with unmutated IGHV genes and are more frequently detected in patients carrying trisomy 12 [Citation103–107]. However, in a recent study including CLL samples taken at diagnosis, we could not confirm the correlation of NOTCH1 mutations and trisomy 12 [Citation52]. Importantly, these patients appear to have a considerably shorter survival compared to patients with NOTCH1 wild-type [] and additionally display an increased risk of developing Richter syndrome [Citation47,Citation48,Citation52,Citation106,Citation108,Citation109].
SF3B1
The SF3B1 gene on chromosome 2q33.1 encodes a protein that is included in the spliceosome machinery, important for the pre-processing of mRNA prior to protein synthesis [Citation110]. It is involved in alternative splicing, which gives rise to different splice variants that form protein variants encoded by the same gene. Two recent exome sequencing studies detected SF3B1 mutations in 10–15% of cases of CLL [Citation49,Citation51]. Still, a considerably lower SF3B1 mutation frequency (3.6%) was detected in our investigation of newly diagnosed patients with CLL [Citation52]. The mutations in SF3B1 are detected at several hotspot exons in the C-terminal HEAT motifs [Citation49,Citation51]. Of note, Wang et al. showed that patients with CLL with mutations within this gene have an aberrant splicing of SF3B1 target mRNA, such as BRD2 and RIOK3 [Citation51]. Moreover, patients with SF3B1 mutations have a considerably shorter survival [] and are more likely to progress, compared to patients without SF3B1 mutations [Citation49,Citation51,Citation111]. However, in contrast to NOTCH1 mutations, SF3B1 disruption does not appear to increase the risk of developing Richter syndrome [Citation108]. The presence of SF3B1 mutations is associated with deletions of 11q or ATM mutations, hence suggesting that these aberrations have a cooperative effect in CLL pathogenesis [Citation51]. Very recently, we observed a strikingly higher proportion (44%) of SF3B1 gene mutations in poor-prognostic subset 2 versus other stereotyped subsets with adverse prognosis (0–5%), alluding to subset-biased acquisition of mutations during leukemogenesis [Citation163].
BIRC3
The baculoviral IAP repeat containing 3 (BIRC3) gene is encoded on chromosome 11q22.2, and has recently been suggested as an alternative candidate gene to ATM in patients with 11q deletions [Citation50]. This gene encodes a member of the inhibitor of apoptosis protein (IAP) family, which negatively regulates the non-canonical NF-κB pathway through inhibition of the NF-κB inducing kinase MAP3K14. Disruption of BIRC3 through mutations or deletions has recently been detected in splenic marginal zone lymphoma and CLL [Citation50,Citation112]. Mutation of BIRC3 leads to aberrant interaction and degradation of MAP3K14, contributing to a constitutive activation of non-canonical NF-κB signaling [Citation50]. Aberrant activation of the NF-κB pathway has been previously documented in CLL [Citation113,Citation114], and the detection of disruptive BIRC3 mutations, as well as activating mutations of MYD88, has provided an additional explanation of the constitutive NF-κB activation in CLL [Citation48,Citation50,Citation51].
Importantly, Rossi et al. detected BIRC3 mutations at a frequency of 4% at diagnosis, but at a much higher frequency (24%) in fludarabine-refractory CLL [Citation50]. Interestingly, these mutations were absent in patients responding to fludarabine treatment, neither were they found in patients with monoclonal B cell lymphocytosis (MBL) [Citation50]. Moreover, the same study showed that the BIRC3 mutations were only found in chemorefractory patients with wild-type TP53, and that patients with either of these gene mutations had an equally poor outcome [Citation50]. Since it is estimated that TP53 mutations account for approximately 40% of the fludarabine refractoriness in CLL, BIRC3 mutations may represent a large fraction of the remaining 60% therapy-resistant cases [Citation79]. Hence, evaluation of both TP53 and BIRC3 mutations would therefore be valuable to include in the routine analysis of patients with CLL in order to detect prospective fludarabine non-responsive cases [Citation39,Citation50].
Protein and RNA-based markers in chronic lymphocytic leukemia
Extensive efforts have been made to find surrogate markers that can substitute for the laborious analysis of the IGHV gene mutational status. This has led to a plethora of markers that have been identified at the protein as well as the RNA level. Indeed, some of these molecules are good surrogate markers for the IGHV gene mutation status, even though a number of the markers that were originally suggested have in subsequent studies been shown to have an independent prognostic value. Because of the abundance of molecules suggested as prognostic markers, it is impossible to describe all here, and we therefore focus on discussing the most relevant and evaluated ones.
CD38
The transmembrane glycoprotein CD38 is expressed by cells of hematopoietic origin, with a high expression in activated B and T cells as well as natural killer and dendritic cells [Citation115]. CD38 has several functions, and can act either as a surface receptor by interacting with PECAM1 (CD31), or as an enzyme, regulating the intracellular calcium level [Citation115]. In CLL, CD38 expression was initially determined to correlate with the IGHV gene mutational status, with a high expression in IGHV-unmutated CLL, and was therefore proposed as a surrogate marker for the more technically complex evaluation of the mutation status [Citation15]. However, further investigation of CD38 showed that the correlation with IGHV mutation was incomplete, and that CD38 instead could function as an independent prognostic marker [Citation19,Citation116,Citation117]. Nevertheless, several obstacles have augmented the use of CD38 as a prognostic maker, namely: (i) the fact that CD38 expression may vary during the course of the disease, (ii) the heterogeneity of CD38 expressing CLL cells within a blood sample and (iii) the lack of a clearly defined cut-off value for the definition of CD38 positivity [Citation116,Citation118]. Indeed, the cut-off value for CD38 expression has varied considerably between studies (from 5% up to 30%), which makes it problematic to compare the attained results. On the other hand, CD38, which is present on the cell surface of CLL cells as well as other hematological malignant cells, has been proposed as a therapeutic target of monoclonal antibodies. Recently, the combination of lenalidomide with the anti-CD38 monoclonal antibody daratumumab was shown to be effective and complementary in treatment of multiple myeloma, and may thus have an analogous effect in CLL treatment [Citation119].
ZAP70
ZAP70 encodes the ζ-chain associated protein kinase 70 kDa, which is a tyrosine kinase normally expressed by natural killer cells and T cells [Citation120]. ZAP70 has a central role in T lymphocytes and is involved in cell migration, apoptosis, T cell receptor signaling and T cell activation [Citation121]. Moreover, both malignant and normal B cells express ZAP70 at various differentiation stages [Citation122]. The protein is also known to play a role in BcR signaling and it is recruited to the BcR signaling complex upon antigen binding, with a similar mechanism to that of the structurally homologous SYK protein [Citation123,Citation124]. In CLL, the presence of high ZAP70 expression was initially identified by Rosenwald et al. in a gene-expression study comparing IGHV-mutated and IGHV-unmutated cases [Citation125]. This study revealed an overexpression of ZAP70 in patients with unmutated IGHV genes, and was consequently suggested as a surrogate marker for the IGHV mutational status. Several studies validated this finding and reported that ZAP70 predicted an unfavorable disease course in terms of disease progression and OS [Citation126–128]. Nevertheless, subsequent studies focusing on the relationship between ZAP70 expression and IGHV mutational status showed discordant results. For instance, patients with IGHV3-21 gene usage were shown to have high ZAP70 expression regardless of IGHV mutational status, whereas cases with del(11q) and del(17p) may display low ZAP70 expression despite having unmutated IGHV genes [Citation129]. Since ZAP70 is an intracellular protein, which requires cell fixation and permeabilization in order to be stained and analyzed, the application of flow cytometry for ZAP70 detection is not as simple as CD38 determination. This complicating factor has limited the application of flow cytometry for ZAP70 detection, although international efforts initiated by the European Research Initiative on CLL (ERIC) have led to a proposal for a standardized assay (www.ericll.org). On the other hand, mRNA expression of ZAP70 has also been correlated to prognosis, and this finding has opened up the possibility of quantitative mRNA measurement, which might be more reliable compared to flow cytometry measurements.
LPL
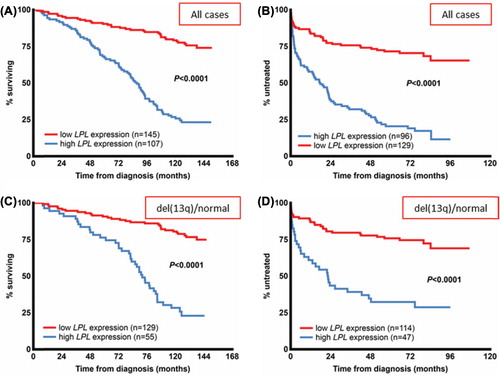
Lipoproteinlipase (LPL) is an enzyme that plays a central role in lipid metabolism by hydrolyzing circulating triacylglycerides into free fatty acids and monoacylglycerol, and which is highly expressed in muscle, adipose tissue and mammary glands [Citation130]. In addition, LPL has been suggested to play a role in monocyte and macrophage cell adhesion, and this may also be the pathological function in CLL cells, as they are devoid of LPL catalytic activity [Citation131,Citation132]. Two gene expression studies performed in 2001 showed that LPL was one of the most differently expressed genes when comparing CLL samples with and without IGHV gene mutations [Citation125,Citation133]. Subsequent studies revealed that a high LPL level correlates to a poor outcome and is associated with poor-prognostic markers such as CD38 and ZAP70 expression, a short LDT and high-risk genomic aberrations [Citation132,Citation134–139]. Moreover, it has also been shown that OS, TTT, event-free survival and TFS can be predicted by LPL expression alone, or in combination with the expression of ADAM29 [Citation132,Citation134–139] When we investigated LPL expression among other RNA based prognostic markers, i.e. ZAP70, CLLU1, TCL1A and MCL1, the former was shown to be the strongest predictor of prognosis among these factors [ and ] [Citation135]. Notably, our comparative study showed that LPL could distinguish patients with otherwise good prognostic markers such as mutated IGHV genes, Binet stage A, favorable cytogenetic changes and absence of CD38 expression [ and ] [Citation135]. This finding implies that LPL can aid the prognostic evaluation, especially in low-risk patients at diagnosis.
Figure 4. LPL expression status and clinical outcome in CLL. Overall survival (A) and time to treatment (B) according to LPL expression status in a newly diagnosed population-based CLL cohort. (C, D) Patients with good cytogenetic markers, i.e. absence of the known recurrent genomic aberrations or presence of del(13q) from the same cohort. In the patient group with good-risk cytogenetic markers, overall survival (C) and time to treatment (D) are stratified by LPL expression status. The expression cut-off for LPL was defined using receiver operating characteristic (ROC) curve analysis and median survival.
LPL appears to be the most reliable RNA-based prognostic marker to date, and the fact that it can be measured directly in peripheral blood, without the requirement for cell sorting such as for ZAP70 analysis, makes it a promising marker for clinical use [Citation140]. Still, additional studies should investigate its robustness and stability over time, and LPL measurement must to be standardized before it can be introduced in the clinic.
CLLU1
In 2006, Buhl et al. identified a gene on chromosome 12q22 that was uniquely expressed in CLL cells [Citation141]. A high expression of this gene, which was named CLL upregulated gene 1 (CLLU1), could not be linked to trisomy 12, since these two features were unrelated [Citation141]. CLLU1 was initially shown as highly expressed in poor-prognostic patients with unmutated IGHV genes [Citation141]. In addition, high CLLU1 expression has been shown in patients with poor-prognostic genomic aberrations, an advanced disease stage and ZAP70 expression [Citation135,Citation142–144]. These studies also revealed that CLLU1 can predict OS, TTT and TFS [Citation135,Citation142–144]. Recently, a study of 515 patients with CLL included in the Lymphoma Research Foundation (LRF) CLL4 randomized trial showed that high CLLU1 expression correlated with poor-prognostic markers such as unmutated IGHV genes and high ZAP70 and CD38 expression [Citation145]. However, it did not hold as an independent predictor of OS, nor did it predict response to treatment [Citation145]. A similar result was shown in a study performed by our research group, where CLLU1 expression did not independently predict OS upon multivariate analysis [Citation135]. Instead, it has been suggested that CLLU1 can be used as a prognostic marker in patients younger than 70 years with favorable prognostic markers, to identify patients who are likely to require treatment [Citation143]. Nevertheless, CLLU1 has been proposed as a marker for minimal residual disease, since it is a sensitive marker for detecting low levels of circulating residual cells in the blood after therapy [Citation146].
MicroRNA
Several microRNA (miRNA) expression studies focusing on CLL have revealed differences in expression levels between distinct prognostic subgroups [Citation147–149]. In the pivotal study, Calin et al. evaluated the expression of 190 miRNAs in 94 patients with CLL and established a signature of 13 miRNAs that could discriminate between ZAP70 positive/IGHV-unmutated CLL with poor prognosis and ZAP70 negative/IGHV-mutated CLL with an indolent disease [Citation147]. Subsequent studies of global miRNA expression have confirmed some of the primary findings made by Calin et al., although with incomplete overlap, suggesting that methodological differences and variations in patient material introduce discordant results [Citation148,Citation149]. Additionally, a number of studies have shown that specific miRNAs can be used as prognostic markers, e.g. miR-21, miR-29, mir-34a, mir-181b and miR-223 [Citation79,Citation150–154]. Through fingerprinting of miRNAs in patients with CLL with del(17p), Rossi et al. identified that up-regulation of miR-21 predicted a worse outcome in terms of a shorter OS, and that down-regulation of mir-181b was associated with therapy refractoriness [Citation151]. The biological role of mir-181b has been shown to involve down-regulation of BCL2 and TCL1, of which the latter is also regulated by miR-29 [Citation150,Citation155]. Another interesting finding is that mir-34a, which is a direct transcriptional target of the p53 pathway, is down-regulated in patients with del(17p) and/or TP53 mutation, and has a reduced expression in patients with fludarabine-refractory CLL [Citation79,Citation153]. Furthermore, Stamatopolous et al. showed a predictive role for miR-29c and mir-223 in the assessment of OS and TFS [Citation152]. In addition, several other studies on miR-223 have shown that down-regulation of this miRNA can predict OS, TFS and PFS, and it has therefore been suggested as a promising prognostic marker [Citation152,Citation154]. Nonetheless, since the miRNAs are involved in post-transcriptional mRNA regulation, these small RNA molecules may have a higher degree of expression level variations over time, and may thus be problematic to standardize and introduce in a clinical setting.
Toward a prognostic index?
Considering the significant disease heterogeneity and the wealth of prognostic markers that have been implicated in CLL during recent years, several attempts have recently been made to construct a model that contains several clinically relevant prognostic biomarkers. In the following sections we review some of these models and their pros and cons.
Haferlach and colleagues proposed a new scoring system for predicting OS and TTT, in which the following parameters were included: age at diagnosis (≥ 65 years), white blood cell count (WBC) (≥ 20 × 109/L), IGHV mutational status, TP53 deletion/mutation, translocations involving the IGH locus and number of chromosomal aberrations evaluated through cytogenetics [Citation156]. The patients (n = 349) were placed in a favorable, intermediate or unfavorable prognostic group based on scores assigned to each evaluated parameter. A high WBC count, old age at diagnosis and unmutated IGHV genes contributed with one point each, while a TP53 deletion, translocations involving the IGH locus and genomic complexity would each add two points to the total score [Citation156]. Using this model, 77% of all cases were evaluated as having favorable prognosis (< 4 points), while 5% were scored as unfavorable, with a prognostic index ≥ 6. Since conventional cytogenetics for detection of chromosomal aberrations and translocations is not commonly performed on CLL samples, the applicability of this model in clinical routine might be problematic and limited, at least in the current setting.
Wierda et al. constructed a nomogram or alignment chart to evaluate the probability of 2- and 4-year TFS in patients with CLL [Citation157]. In their model, points were assigned for lymph node characteristics, including the number of enlarged lymph nodes and the size of the largest lymph node in the neck. Other parameters used for the creation of a prognostic score included serum LDH levels stratified by IGHV mutation status, and the presence of cytogenetic aberrations according to the hierarchal model [Citation37]. This multivariate model may be useful for identifying patients who require early therapy; however, it has not yet been tested for other important clinical endpoints such as OS and in independent cohorts.
Focusing on early stage CLL, Pepper and colleagues showed in a large study of newly diagnosed patients with CLL (n = 1154) that only LDT, IGHV mutation status, CD38 and age at diagnosis were independent prognostic factors for TFT and OS, whereas neither FISH nor ZAP70 held as independent markers at diagnosis [Citation158]. Therefore, the authors went on to propose a diagnostic work-up for patients with Binet stage A, in which a combination of CD38 expression, IGHV mutational status and LDT could be analyzed and which would be sufficient to monitor patients for the first few years. On the other hand, CLL-FISH should only be carried out at disease progression to guide treatment decisions, which is in line with recent guidelines recommending FISH analysis to be performed before initiation of therapy rather than at diagnosis [Citation159,Citation160].
In a very recent publication, Rossi et al. proposed an updated hierarchical classification for prediction of OS by integrating mutational and cytogenetic analysis in CLL [Citation39]. Here, 1273 patients with CLL were assigned to one of four CLL risk groups based on the presence of: (i) TP53 and/or BIRC3 abnormalities (high risk), (ii) NOTCH1 and/or SF3B1 mutations and/or del(11q) (intermediate risk), (iii) trisomy 12 or normal cytogenetics (low risk) and (iv) del(13q) (very low risk). According to the authors, this new model displayed superior prognostication accuracy compared to the “old” hierarchical classification as proposed by Döhner et al. [Citation37]. Additionally, through time-dependent analysis, this model was found to maintain prognostic power at any time from diagnosis, making this classification system particularly interesting. Conversely to this finding, we reported a similarly poor outcome in patients carrying NOTCH1, SF3B1 and TP53 mutations () [Citation52]. Nevertheless, these results implicate the importance of including the mutational analysis of these novel genes, in addition to detection of the known recurrent aberrations, since the presence of any of the mutations may reclassify the patients into a higher risk-group. Therefore, it will be important to include NOTCH1, SF3B1 and BIRC3 in forthcoming clinical trials to evaluate their impact on outcome in CLL. Since these genes are involved in many key signaling pathways that lead to an aggravation of the disease, they may also represent novel therapeutic targets in CLL.
Concluding remarks and future directions
Many different biomarkers have been suggested as novel prognostic factors in CLL, and some of these markers have been outlined in the previous sections. Importantly, for all prognostic markers that aspire to be applied in clinical routine diagnostics, it is essential that: (i) each potential biomarker is validated in independent larger patient materials as well as in prospective studies, preferentially in clinical trials to investigate its relation to a given treatment; (ii) each potential biomarker is compared to currently established prognostic markers using Cox regression analysis to test whether the marker under investigation displays independent prognostic power; and (iii) the stability of a given marker is investigated in longitudinal studies as well as how this marker is influenced by treatment. Apropos the latter point, it is now well known that some of the currently applied prognostic markers, i.e. presence or absence of the known recurrent genomic aberrations, may change over time through clonal evolution, and it is therefore more important to assess the presence of these aberrations before initiation of treatment as well as at treatment failure or relapse [Citation39,Citation54,Citation161,Citation162]. Contrary to this, the IGHV mutational status is very stable and will never change during the course of the disease, and this marker can hence be measured at any time point [Citation15–17].
Despite this abundance of prognostic markers in CLL, one of the most important tasks right now is to identify biomarkers that can act as predictive markers in terms of directing treatment. Today, the only markers influencing treatment decisions are TP53 mutations and/or deletions, and to some extent 11q deletions. However, the recent finding that other novel mutations, such as BIRC3 mutations, which accumulate in chemorefractory patients may in the near future qualify as predictive markers of treatment failure should this marker be validated in independent cohorts [Citation50]. In the coming years, it is thus very important that the most promising novel markers are always included in clinical trials, as also has been proposed in the international CLL guidelines [Citation5], so that they can be evaluated regarding their predictive capacity in relation to the different treatment protocols applied.
In the end, many of the suggested markers will never reach the stage where they can be included in routine diagnostics. Nevertheless, it is still essential to investigate the biological role of such prognostic markers, since their presence or absence may provide important clues for a better understanding of the underlying pathogenesis of CLL, which ultimately may enable the design of novel treatment options. For instance, the detection of BIRC3, NOTCH1 and SF3B1 mutations has not only identified new prognostic subsets, but also added new insights into different dysregulated pathways involved in the pathobiology of CLL, such as enhanced NF-κB signaling (BIRC3, NOTCH1) as well as deregulation of the spliceosome machinery (SF3B1) [Citation47–51]. Moreover, the finding that several miRNAs are deregulated in CLL, and that these small molecules can provide a link to the deregulation of many other genes, is very important for understanding the complex network that is operating in these tumor cells, although the miRNAs may not be the most applicable prognostic indicators in the clinic.
Probably we will see further attempts in the near future to construct a “CLL prognostic index” taking into account the most important biomarkers. From our perspective, the recently proposed model integrating cytogenetic and molecular analysis appears particularly attractive, since it will detect patients with CLL with high-risk genetic aberrations who are in direct need of alternative treatment. Definitely, we foresee that large collaborative efforts, probably including up to 5000–10 000 patients, are now needed to be able to reach solid conclusions about which markers to investigate, at what time point and in relation to which other marker.
Supplementary Material
Download Zip (2.4 MB)Potential conflict of interest:
Disclosure forms provided by the authors are available with the full text of this article at www.informahealthcare.com/lal.
This research was supported by the Nordic Cancer Union, the Swedish Cancer Society, the Swedish Research Council and the Lion's Cancer Research Foundation, Uppsala.
References
- Binet JL, Auquier A, Dighiero G, et al. A new prognostic classification of chronic lymphocytic leukemia derived from a multivariate survival analysis. Cancer 1981;48:198–206.
- Rai KR, Sawitsky A, Cronkite EP, et al. Clinical staging of chronic lymphocytic leukemia. Blood 1975;46:219–234.
- Hamblin TJ. Prognostic markers in chronic lymphocytic leukaemia. Best Pract Res Clin Haematol 2007;20:455–468.
- Montserrat E, Sanchez-Bisono J, Vinolas N, et al. Lymphocyte doubling time in chronic lymphocytic leukaemia: analysis of its prognostic significance. Br J Haematol 1986;62:567–575.
- Hallek M, Cheson BD, Catovsky D, et al. Guidelines for the diagnosis and treatment of chronic lymphocytic leukemia: a report from the International Workshop on Chronic Lymphocytic Leukemia updating the National Cancer Institute-Working Group 1996 guidelines. Blood 2008;111:5446–5456.
- Fasola G, Fanin R, Gherlinzoni F, et al. Serum LDH concentration in non-Hodgkin's lymphomas. Relationship to histologic type, tumor mass, and presentation features. Acta Haematol 1984;72:231–238.
- Lee JS, Dixon DO, Kantarjian HM, et al. Prognosis of chronic lymphocytic leukemia: a multivariate regression analysis of 325 untreated patients. Blood 1987;69:929–936.
- Rossi D, Cerri M, Capello D, et al. Biological and clinical risk factors of chronic lymphocytic leukaemia transformation to Richter syndrome. Br J Haematol 2008;142:202–215.
- Hallek M, Wanders L, Ostwald M, et al. Serum beta(2)-microglobulin and serum thymidine kinase are independent predictors of progression-free survival in chronic lymphocytic leukemia and immunocytoma. Leuk Lymphoma 1996;22:439–447.
- Tsimberidou AM, Wen S, O’Brien S, et al. Assessment of chronic lymphocytic leukemia and small lymphocytic lymphoma by absolute lymphocyte counts in 2,126 patients: 20 years of experience at the University of Texas M.D. Anderson Cancer Center. J Clin Oncol 2007;25:4648–4656.
- Gentile M, Cutrona G, Neri A, Molica S, Ferrarini M, Morabito F. Predictive value of beta2-microglobulin (beta2-m) levels in chronic lymphocytic leukemia since Binet A stages. Haematologica 2009; 94:887–888.
- Di Raimondo F, Giustolisi R, Lerner S, et al. Retrospective study of the prognostic role of serum thymidine kinase level in CLL patients with active disease treated with fludarabine. Ann Oncol 2001;12:621–625.
- Magnac C, Porcher R, Davi F, et al. Predictive value of serum thymidine kinase level for Ig-V mutational status in B-CLL. Leukemia 2003;17:133–137.
- Matthews C, Catherwood MA, Morris TC, et al. Serum TK levels in CLL identify Binet stage A patients within biologically defined prognostic subgroups most likely to undergo disease progression. Eur J Haematol 2006;77:309–317.
- Damle RN, Wasil T, Fais F, et al. Ig V gene mutation status and CD38 expression as novel prognostic indicators in chronic lymphocytic leukemia. Blood 1999;94:1840–1847.
- Hamblin TJ, Davis Z, Gardiner A, et al. Unmutated Ig V(H) genes are associated with a more aggressive form of chronic lymphocytic leukemia. Blood 1999;94:1848–1854.
- Ghia P, Stamatopoulos K, Belessi C, et al. ERIC recommendations on IGHV gene mutational status analysis in chronic lymphocytic leukemia. Leukemia 2007;21:1–3.
- Langerak AW, Davi F, Ghia P, et al. Immunoglobulin sequence analysis and prognostication in CLL: guidelines from the ERIC review board for reliable interpretation of problematic cases. Leukemia 2011;25:979–984.
- Krober A, Seiler T, Benner A, et al. V(H) mutation status, CD38 expression level, genomic aberrations, and survival in chronic lymphocytic leukemia. Blood 2002;100:1410–1416.
- Lin K, Sherrington PD, Dennis M, et al. Relationship between p53 dysfunction, CD38 expression, and IgV(H) mutation in chronic lymphocytic leukemia. Blood 2002;100:1404–1409.
- Oscier DG, Gardiner AC, Mould SJ, et al. Multivariate analysis of prognostic factors in CLL: clinical stage, IGVH gene mutational status, and loss or mutation of the p53 gene are independent prognostic factors. Blood 2002;100:1177–1184.
- Tobin G, Thunberg U, Laurell A, et al. Patients with chronic lymphocytic leukemia with mutated VH genes presenting with Binet stage B or C form a subgroup with a poor outcome. Haematologica 2005;90:465–469.
- Hamblin TJ, Davis ZA, Oscier DG. Determination of how many immunoglobulin variable region heavy chain mutations are allowable in unmutated chronic lymphocytic leukaemia - long-term follow up of patients with different percentages of mutations. Br J Haematol 2008;140:320–323.
- Tobin G, Thunberg U, Johnson A, et al. Somatically mutated Ig V(H)3-21 genes characterize a new subset of chronic lymphocytic leukemia. Blood 2002;99:2262–2264.
- Bomben R, Dal Bo M, Capello D, et al. Comprehensive characterization of IGHV3-21-expressing B-cell chronic lymphocytic leukemia: an Italian multicenter study. Blood 2007;109:2989–2998.
- Ghia EM, Jain S, Widhopf GF 2nd, et al. Use of IGHV3-21 in chronic lymphocytic leukemia is associated with high-risk disease and reflects antigen-driven, post-germinal center leukemogenic selection. Blood 2008;111:5101–5108.
- Thorselius M, Krober A, Murray F, et al. Strikingly homologous immunoglobulin gene rearrangements and poor outcome in VH3-21-using chronic lymphocytic leukemia patients independent of geographic origin and mutational status. Blood 2006;107:2889–2894.
- Tobin G, Thunberg U, Johnson A, et al. Chronic lymphocytic leukemias utilizing the VH3-21 gene display highly restricted Vlambda2-14 gene use and homologous CDR3s: implicating recognition of a common antigen epitope. Blood 2003;101:4952–4957.
- Tobin G, Thunberg U, Karlsson K, et al. Subsets with restricted immunoglobulin gene rearrangement features indicate a role for antigen selection in the development of chronic lymphocytic leukemia. Blood 2004;104:2879–2885.
- Chiorazzi N, Rai KR, Ferrarini M. Chronic lymphocytic leukemia. N Engl J Med 2005;352:804–815.
- Rosen A, Murray F, Evaldsson C, Rosenquist R. Antigens in chronic lymphocytic leukemia–implications for cell origin and leukemogenesis. Semin Cancer Biol 2010;20:400–409.
- Messmer BT, Albesiano E, Efremov DG, et al. Multiple distinct sets of stereotyped antigen receptors indicate a role for antigen in promoting chronic lymphocytic leukemia. J Exp Med 2004;200:519–525.
- Stamatopoulos K, Belessi C, Moreno C, et al. Over 20% of patients with chronic lymphocytic leukemia carry stereotyped receptors: pathogenetic implications and clinical correlations. Blood 2007;109: 259–270.
- Agathangelidis A, Darzentas N, Hadzidimitriou A, et al. Stereotyped B-cell receptors in one-third of chronic lymphocytic leukemia: a molecular classification with implications for targeted therapies. Blood 2012;119:4467–4475.
- Bomben R, Dal Bo M, Capello D, et al. Molecular and clinical features of chronic lymphocytic leukaemia with stereotyped B cell receptors: results from an Italian multicentre study. Br J Haematol 2009;144:492–506.
- Ghia P, Stamatopoulos K, Belessi C, et al. Geographic patterns and pathogenetic implications of IGHV gene usage in chronic lymphocytic leukemia: the lesson of the IGHV3-21 gene. Blood 2005; 105:1678–1685.
- Dohner H, Stilgenbauer S, Benner A, et al. Genomic aberrations and survival in chronic lymphocytic leukemia. N Engl J Med 2000; 343:1910–1916.
- Gunnarsson R, Mansouri L, Rosenquist R. Exploring the genetic landscape in chronic lymphocytic leukemia using high-resolution technologies. Leuk Lymphoma 2013 Feb 28. [Epub ahead of print]
- Rossi D, Rasi S, Spina V, et al. Integrated mutational and cytogenetic analysis identifies new prognostic subgroups in chronic lymphocytic leukemia. Blood 2013;121:1403–1412.
- Zenz T, Mertens D, Kuppers R, et al. From pathogenesis to treatment of chronic lymphocytic leukaemia. Nat Rev Cancer 2010;10:37–50.
- Pospisilova S, Gonzalez D, Malcikova J, et al. ERIC recommendations on TP53 mutation analysis in chronic lymphocytic leukemia. Leukemia 2012;26:1458–1461.
- Gunnarsson R, Isaksson A, Mansouri M, et al. Large but not small copy-number alterations correlate to high-risk genomic aberrations and survival in chronic lymphocytic leukemia: a high-resolution genomic screening of newly diagnosed patients. Leukemia 2010;24:211–215.
- Haferlach C, Dicker F, Schnittger S, et al. Comprehensive genetic characterization of CLL: a study on 506 cases analysed with chromosome banding analysis, interphase FISH, IgV(H) status and immunophenotyping. Leukemia 2007;21:2442–2451.
- Kujawski L, Ouillette P, Erba H, et al. Genomic complexity identifies patients with aggressive chronic lymphocytic leukemia. Blood 2008;112:1993–2003.
- Mayr C, Speicher MR, Kofler DM, et al. Chromosomal translocations are associated with poor prognosis in chronic lymphocytic leukemia. Blood 2006;107:742–751.
- Van Den Neste E, Robin V, Francart J, et al. Chromosomal translocations independently predict treatment failure, treatment-free survival and overall survival in B-cell chronic lymphocytic leukemia patients treated with cladribine. Leukemia 2007;21:1715–1722.
- Fabbri G, Rasi S, Rossi D, et al. Analysis of the chronic lymphocytic leukemia coding genome: role of NOTCH1 mutational activation. J Exp Med 2011;208:1389–1401.
- Puente XS, Pinyol M, Quesada V, et al. Whole-genome sequencing identifies recurrent mutations in chronic lymphocytic leukaemia. Nature 2011;475:101–105.
- Quesada V, Conde L, Villamor N, et al. Exome sequencing identifies recurrent mutations of the splicing factor SF3B1 gene in chronic lymphocytic leukemia. Nat Genet 2012;44:47–52.
- Rossi D, Fangazio M, Rasi S, et al. Disruption of BIRC3 associates with fludarabine chemorefractoriness in TP53 wild-type chronic lymphocytic leukemia. Blood 2012;119:2854–2862.
- Wang L, Lawrence MS, Wan Y, et al. SF3B1 and other novel cancer genes in chronic lymphocytic leukemia. N Engl J Med 2011; 365:2497–2506.
- Mansouri L, Cahill N, Gunnarsson R, et al. NOTCH1 and SF3B1 mutations can be added to the hierarchical prognostic classification in chronic lymphocytic leukemia. Leukemia 2013;27:512–514.
- Juliusson G, Oscier DG, Fitchett M, et al. Prognostic subgroups in B-cell chronic lymphocytic leukemia defined by specific chromosomal abnormalities. N Engl J Med 1990;323:720–724.
- Gunnarsson R, Mansouri L, Isaksson A, et al. Array-based genomic screening at diagnosis and during follow-up in chronic lymphocytic leukemia. Haematologica 2011;96:1161–1169.
- Stilgenbauer S, Liebisch P, James MR, et al. Molecular cytogenetic delineation of a novel critical genomic region in chromosome bands 11q22.3-923.1 in lymphoproliferative disorders. Proc Natl Acad Sci USA 1996;93:11837–11841.
- Taylor AM, Metcalfe JA, Thick J, et al. Leukemia and lymphoma in ataxia telangiectasia. Blood 1996;87:423–438.
- Austen B, Powell JE, Alvi A, et al. Mutations in the ATM gene lead to impaired overall and treatment-free survival that is independent of IGVH mutation status in patients with B-CLL. Blood 2005;106:3175–3182.
- Bullrich F, Rasio D, Kitada S, et al. ATM mutations in B-cell chronic lymphocytic leukemia. Cancer Res 1999;59:24–27.
- Rossi D, Fangazio M, Gaidano G. The spectrum of genetic defects in chronic lymphocytic leukemia. Mediterr J Hematol Infect Dis 2012;4:e2012076.
- Dohner H, Stilgenbauer S, James MR, et al. 11q deletions identify a new subset of B-cell chronic lymphocytic leukemia characterized by extensive nodal involvement and inferior prognosis.Blood 1997;89:2516–2522.
- Hallek M, Fischer K, Fingerle-Rowson G, et al. Addition of rituximab to fludarabine and cyclophosphamide in patients with chronic lymphocytic leukaemia: a randomised, open-label, phase 3 trial. Lancet 2010;376:1164–1174.
- Mosca L, Fabris S, Lionetti M, et al. Integrative genomics analyses reveal molecularly distinct subgroups of B-cell chronic lymphocytic leukemia patients with 13q14 deletion. Clin Cancer Res 2010;16:5641–5653.
- Ouillette P, Erba H, Kujawski L, et al. Integrated genomic profiling of chronic lymphocytic leukemia identifies subtypes of deletion 13q14. Cancer Res 2008;68:1012–1021.
- Liu Y, Corcoran M, Rasool O, et al. Cloning of two candidate tumor suppressor genes within a 10 kb region on chromosome 13q14, frequently deleted in chronic lymphocytic leukemia. Oncogene 1997;15:2463–2473.
- Liu Y, Grander D, Soderhall S, et al. Retinoblastoma gene deletions in B-cell chronic lymphocytic leukemia. Genes Chromosomes Cancer 1992;4:250–256.
- Liu Y, Szekely L, Grander D, et al. Chronic lymphocytic leukemia cells with allelic deletions at 13q14 commonly have one intact RB1 gene: evidence for a role of an adjacent locus. Proc Natl Acad Sci USA 1993;90:8697–8701.
- Calin GA, Dumitru CD, Shimizu M, et al. Frequent deletions and down-regulation of micro-RNA genes miR15 and miR16 at 13q14 in chronic lymphocytic leukemia. Proc Natl Acad Sci USA 2002;99:15524–15529.
- Cimmino A, Calin GA, Fabbri M, et al. miR-15 and miR-16 induce apoptosis by targeting BCL2. Proc Natl Acad Sci USA 2005;102: 13944–13949.
- Mian M, Rinaldi A, Mensah AA, et al. Del(13q14.3) length matters: an integrated analysis of genomic, fluorescence in situ hybridization and clinical data in 169 chronic lymphocytic leukaemia patients with 13q deletion alone or a normal karyotype. Hematol Oncol 2012;30:46–49.
- Parker H, Rose-Zerilli MJ, Parker A, et al. 13q deletion anatomy and disease progression in patients with chronic lymphocytic leukemia. Leukemia 2011;25:489–497.
- Van Dyke DL, Shanafelt TD, Call TG, et al. A comprehensive evaluation of the prognostic significance of 13q deletions in patients with B-chronic lymphocytic leukaemia. Br J Haematol 2010;148:544–550.
- Xu-Monette ZY, Medeiros LJ, Li Y, et al. Dysfunction of the TP53 tumor suppressor gene in lymphoid malignancies. Blood 2012;119:3668–3683.
- Dicker F, Herholz H, Schnittger S, et al. The detection of TP53 mutations in chronic lymphocytic leukemia independently predicts rapid disease progression and is highly correlated with a complex aberrant karyotype. Leukemia 2009;23:117–124.
- Forconi F, Rinaldi A, Kwee I, et al. Genome-wide DNA analysis identifies recurrent imbalances predicting outcome in chronic lymphocytic leukaemia with 17p deletion. Br J Haematol 2008;143:532–536.
- Malcikova J, Smardova J, Rocnova L, et al. Monoallelic and biallelic inactivation of TP53 gene in chronic lymphocytic leukemia: selection, impact on survival, and response to DNA damage. Blood 2009;114:5307–5314.
- Zainuddin N, Murray F, Kanduri M, et al. TP53 mutations are infrequent in newly diagnosed chronic lymphocytic leukemia. Leuk Res 2011;35:272–274.
- Zenz T, Krober A, Scherer K, et al. Monoallelic TP53 inactivation is associated with poor prognosis in chronic lymphocytic leukemia: results from a detailed genetic characterization with long-term follow-up. Blood 2008;112:3322–3329.
- Zenz T, Eichhorst B, Busch R, et al. TP53 mutation and survival in chronic lymphocytic leukemia. J Clin Oncol 2010;28:4473–4479.
- Zenz T, Habe S, Denzel T, et al. Detailed analysis of p53 pathway defects in fludarabine-refractory chronic lymphocytic leukemia (CLL): dissecting the contribution of 17p deletion, TP53 mutation, p53-p21 dysfunction, and miR34a in a prospective clinical trial. Blood 2009; 114:2589–2597.
- Zenz T, Vollmer D, Trbusek M, et al. TP53 mutation profile in chronic lymphocytic leukemia: evidence for a disease specific profile from a comprehensive analysis of 268 mutations. Leukemia 2010;24:2072–2079.
- Lozanski G, Heerema NA, Flinn IW, et al. Alemtuzumab is an effective therapy for chronic lymphocytic leukemia with p53 mutations and deletions. Blood 2004;103:3278–3281.
- Stilgenbauer S, Zenz T, Winkler D, et al. Subcutaneous alemtuzumab in fludarabine-refractory chronic lymphocytic leukemia: clinical results and prognostic marker analyses from the CLL2H study of the German Chronic Lymphocytic Leukemia Study Group. J Clin Oncol 2009;27:3994–4001.
- Catovsky D, Richards S, Matutes E, et al. Assessment of fludarabine plus cyclophosphamide for patients with chronic lymphocytic leukaemia (the LRF CLL4 Trial): a randomised controlled trial. Lancet 2007;370:230–239.
- Grever MR, Lucas DM, Dewald GW, et al. Comprehensive assessment of genetic and molecular features predicting outcome in patients with chronic lymphocytic leukemia: results from the US Intergroup Phase III Trial E2997. J Clin Oncol 2007;25:799–804.
- Stilgenbauer S, Eichhorst BF, Busch R, et al. Biologic and clinical markers for outcome after fludarabine (F) or F plus cyclophosphamide (FC) - comprehensive analysis of the CLL4 trial of the GCLLSG. Blood 2008;112(Suppl. 1): Abstract 2089.
- Dohner H, Fischer K, Bentz M, et al. p53 gene deletion predicts for poor survival and non-response to therapy with purine analogs in chronic B-cell leukemias. Blood 1995;85:1580–1589.
- Fenaux P, Preudhomme C, Lai JL, et al. Mutations of the p53 gene in B-cell chronic lymphocytic leukemia: a report on 39 cases with cytogenetic analysis. Leukemia 1992;6:246–250.
- Pettitt AR, Matutes E, Oscier D. Alemtuzumab in combination with high-dose methylprednisolone is a logical, feasible and highly active therapeutic regimen in chronic lymphocytic leukaemia patients with p53 defects. Leukemia 2006;20:1441–1445.
- Juliusson G, Robert KH, Ost A, et al. Prognostic information from cytogenetic analysis in chronic B-lymphocytic leukemia and leukemic immunocytoma. Blood 1985;65:134–141.
- Juliusson G, Merup M. Cytogenetics in chronic lymphocytic leukemia. Semin Oncol 1998;25:19–26.
- Schwaenen C, Nessling M, Wessendorf S, et al. Automated array-based genomic profiling in chronic lymphocytic leukemia: development of a clinical tool and discovery of recurrent genomic alterations. Proc Natl Acad Sci USA 2004;101:1039–1044.
- Sellmann L, Gesk S, Walter C, et al. Trisomy 19 is associated with trisomy 12 and mutated IGHV genes in B-chronic lymphocytic leukaemia. Br J Haematol 2007;138:217–220.
- Ibbotson R, Athanasiadou A, Sutton LA, et al. Coexistence of trisomies of chromosomes 12 and 19 in chronic lymphocytic leukemia occurs exclusively in the rare IgG-positive variant. Leukemia 2012;26:170–172.
- Athanasiadou A, Stamatopoulos K, Gaitatzi M, et al. Recurrent cytogenetic findings in subsets of patients with chronic lymphocytic leukemia expressing IgG-switched stereotyped immunoglobulins. Haematologica 2008;93:473–474.
- Chapiro E, Radford-Weiss I, Bastard C, et al. The most frequent t(14;19)(q32;q13)-positive B-cell malignancy corresponds to an aggressive subgroup of atypical chronic lymphocytic leukemia. Leukemia 2008;22:2123–2127.
- Martin-Subero JI, Ibbotson R, Klapper W, et al. A comprehensive genetic and histopathologic analysis identifies two subgroups of B-cell malignancies carrying a t(14;19)(q32;q13) or variant BCL3-translocation. Leukemia 2007;21:1532–1544.
- Roos G, Krober A, Grabowski P, et al. Short telomeres are associated with genetic complexity, high-risk genomic aberrations, and short survival in chronic lymphocytic leukemia. Blood 2008;111:2246–2252.
- Edelmann J, Holzmann K, Miller F, et al. High-resolution genomic profiling of chronic lymphocytic leukemia reveals new recurrent genomic alterations. Blood 2012;120:4783–4794.
- Stephens PJ, Greenman CD, Fu B, et al. Massive genomic rearrangement acquired in a single catastrophic event during cancer development. Cell 2011;144:27–40.
- Vilimas T, Mascarenhas J, Palomero T, et al. Targeting the NF-kappaB signaling pathway in Notch1-induced T-cell leukemia. Nat Med 2007;13:70–77.
- Santos MA, Sarmento LM, Rebelo M, et al. Notch1 engagement by Delta-like-1 promotes differentiation of B lymphocytes to antibody-secreting cells. Proc Natl Acad Sci USA 2007;104:15454–15459.
- Rosati E, Sabatini R, Rampino G, et al. Constitutively activated Notch signaling is involved in survival and apoptosis resistance of B-CLL cells. Blood 2009;113:856–865.
- Del Giudice I, Rossi D, Chiaretti S, et al. NOTCH1 mutations in + 12 chronic lymphocytic leukemia (CLL) confer an unfavorable prognosis, induce a distinctive transcriptional profiling and refine the intermediate prognosis of + 12 CLL. Haematologica 2012;97:437–441.
- Balatti V, Bottoni A, Palamarchuk A, et al. NOTCH1 mutations in CLL associated with trisomy 12. Blood 2012;119:329–331.
- Oscier DG, Rose-Zerilli MJ, Winkelmann N, et al. The clinical significance of NOTCH1 and SF3B1 mutations in the UK LRF CLL4 trial. Blood 2013;121:468–475.
- Rossi D, Rasi S, Fabbri G, et al. Mutations of NOTCH1 are an independent predictor of survival in chronic lymphocytic leukemia. Blood 2012;119:521–529.
- Villamor N, Conde L, Martinez-Trillos A, et al. NOTCH1 mutations identify a genetic subgroup of chronic lymphocytic leukemia patients with high risk of transformation and poor outcome. Leukemia 2012 Dec 6. [Epub ahead of print]
- Rossi D, Rasi S, Spina V, et al. Different impact of NOTCH1 and SF3B1 mutations on the risk of chronic lymphocytic leukemia transformation to Richter syndrome. Br J Haematol 2012;158:426–429.
- Sportoletti P, Baldoni S, Cavalli L, et al. NOTCH1 PEST domain mutation is an adverse prognostic factor in B-CLL. Br J Haematol 2010;151:404–406.
- Wahl MC, Will CL, Luhrmann R. The spliceosome: design principles of a dynamic RNP machine. Cell 2009;136:701–718.
- Rossi D, Bruscaggin A, Spina V, et al. Mutations of the SF3B1 splicing factor in chronic lymphocytic leukemia: association with progression and fludarabine-refractoriness. Blood 2011;118:6904–6908.
- Rossi D, Deaglio S, Dominguez-Sola D, et al. Alteration of BIRC3 and multiple other NF-kappaB pathway genes in splenic marginal zone lymphoma. Blood 2011;118:4930–4934.
- Buggins AG, Pepper C, Patten PE, et al. Interaction with vascular endothelium enhances survival in primary chronic lymphocytic leukemia cells via NF-kappaB activation and de novo gene transcription. Cancer Res 2010;70:7523–7533.
- Herishanu Y, Perez-Galan P, Liu D, et al. The lymph node microenvironment promotes B-cell receptor signaling, NF-kappaB activation, and tumor proliferation in chronic lymphocytic leukemia. Blood 2011;117:563–574.
- Malavasi F, Deaglio S, Funaro A, et al. Evolution and function of the ADP ribosyl cyclase/CD38 gene family in physiology and pathology. Physiol Rev 2008;88:841–886.
- Hamblin TJ, Orchard JA, Ibbotson RE, et al. CD38 expression and immunoglobulin variable region mutations are independent prognostic variables in chronic lymphocytic leukemia, but CD38 expression may vary during the course of the disease. Blood 2002;99:1023–1029.
- Thunberg U, Johnson A, Roos G, et al. CD38 expression is a poor predictor for VH gene mutational status and prognosis in chronic lymphocytic leukemia. Blood 2001;97:1892–1894.
- Ghia P, Guida G, Stella S, et al. The pattern of CD38 expression defines a distinct subset of chronic lymphocytic leukemia (CLL) patients at risk of disease progression. Blood 2003;101:1262–1269.
- van der Veer MS, de Weers M, van Kessel B, et al. Towards effective immunotherapy of myeloma: enhanced elimination of myeloma cells by combination of lenalidomide with the human CD38 monoclonal antibody daratumumab. Haematologica 2011;96:284–290.
- Chan AC, Iwashima M, Turck CW, et al. ZAP-70: a 70 kd protein-tyrosine kinase that associates with the TCR zeta chain. Cell 1992;71:649–662.
- Negishi I, Motoyama N, Nakayama K, et al. Essential role for ZAP-70 in both positive and negative selection of thymocytes. Nature 1995;376:435–438.
- Scielzo C, Camporeale A, Geuna M, et al. ZAP-70 is expressed by normal and malignant human B-cell subsets of different maturational stage. Leukemia 2006;20:689–695.
- Chen L, Widhopf G, Huynh L, et al. Expression of ZAP-70 is associated with increased B-cell receptor signaling in chronic lymphocytic leukemia. Blood 2002;100:4609–4614.
- Efremov DG, Gobessi S, Longo PG. Signaling pathways activated by antigen-receptor engagement in chronic lymphocytic leukemia B-cells. Autoimmun Rev 2007;7:102–108.
- Rosenwald A, Alizadeh AA, Widhopf G, et al. Relation of gene expression phenotype to immunoglobulin mutation genotype in B cell chronic lymphocytic leukemia. J Exp Med 2001;194:1639–1647.
- Crespo M, Bosch F, Villamor N, et al. ZAP-70 expression as a surrogate for immunoglobulin-variable-region mutations in chronic lymphocytic leukemia. N Engl J Med 2003;348:1764–1775.
- Orchard JA, Ibbotson RE, Davis Z, et al. ZAP-70 expression and prognosis in chronic lymphocytic leukaemia. Lancet 2004;363:105–111.
- Wiestner A, Rosenwald A, Barry TS, et al. ZAP-70 expression identifies a chronic lymphocytic leukemia subtype with unmutated immunoglobulin genes, inferior clinical outcome, and distinct gene expression profile. Blood 2003;101:4944–4951.
- Krober A, Bloehdorn J, Hafner S, et al. Additional genetic high-risk features such as 11q deletion, 17p deletion, and V3-21 usage characterize discordance of ZAP-70 and VH mutation status in chronic lymphocytic leukemia. J Clin Oncol 2006;24:969–975.
- Wang CS, Hartsuck J, McConathy WJ. Structure and functional properties of lipoprotein lipase. Biochim Biophys Acta 1992;1123:1–17.
- Mamputu JC, Renier G. Differentiation of human monocytes to monocyte-derived macrophages is associated with increased lipoprotein lipase-induced tumor necrosis factor-alpha expression and production: a process involving cell surface proteoglycans and protein kinase C. Arterioscler Thromb Vasc Biol 1999;19:1405–1411.
- Mansouri M, Sevov M, Fahlgren E, et al. Lipoprotein lipase is differentially expressed in prognostic subsets of chronic lymphocytic leukemia but displays invariably low catalytical activity. Leuk Res 2010;34:301–306.
- Klein U, Tu Y, Stolovitzky GA, et al. Gene expression profiling of B cell chronic lymphocytic leukemia reveals a homogeneous phenotype related to memory B cells. J Exp Med 2001;194:1625–1638.
- Heintel D, Kienle D, Shehata M, et al. High expression of lipoprotein lipase in poor risk B-cell chronic lymphocytic leukemia. Leukemia 2005;19:1216–1223.
- Kaderi MA, Kanduri M, Buhl AM, et al. LPL is the strongest prognostic factor in a comparative analysis of RNA-based markers in early chronic lymphocytic leukemia. Haematologica 2011;96: 1153–1160.
- Kienle D, Benner A, Laufle C, et al. Gene expression factors as predictors of genetic risk and survival in chronic lymphocytic leukemia. Haematologica 2010;95:102–109.
- Oppezzo P, Vasconcelos Y, Settegrana C, et al. The LPL/ADAM29 expression ratio is a novel prognosis indicator in chronic lymphocytic leukemia. Blood 2005;106:650–657.
- Van Bockstaele F, Pede V, Janssens A, et al. Lipoprotein lipase mRNA expression in whole blood is a prognostic marker in B cell chronic lymphocytic leukemia. Clin Chem 2007;53:204–212.
- van't Veer MB, Brooijmans AM, Langerak AW, et al. The predictive value of lipoprotein lipase for survival in chronic lymphocytic leukemia. Haematologica 2006;91:56–63.
- Sevov M, Rosenquist R, Mansouri L. RNA-based markers as prognostic factors in chronic lymphocytic leukemia. Expert Rev Hematol 2012;5:69–79.
- Buhl AM, Jurlander J, Jorgensen FS, et al. Identification of a gene on chromosome 12q22 uniquely overexpressed in chronic lymphocytic leukemia. Blood 2006;107:2904–2911.
- Buhl AM, Jurlander J, Geisler CH, et al. CLLU1 expression levels predict time to initiation of therapy and overall survival in chronic lymphocytic leukemia. Eur J Haematol 2006;76:455–464.
- Josefsson P, Geisler CH, Leffers H, et al. CLLU1 expression analysis adds prognostic information to risk prediction in chronic lymphocytic leukemia. Blood 2007;109:4973–4979.
- Stamatopoulos B, Meuleman N, De Bruyn C, et al. A molecular score by quantitative PCR as a new prognostic tool at diagnosis for chronic lymphocytic leukemia patients. PLoS One 2010;5. pii: e12780.
- Gonzalez D, Else M, Wren D, et al. CLLU1 expression has prognostic value in CLL after first-line therapy in younger patients and those with mutated IGHV genes. Haematologica 2013;98:274–278.
- Buhl AM, James DF, Neuberg D, et al. Analysis of CLLU1 expression levels before and after therapy in patients with chronic lymphocytic leukemia. Eur J Haematol 2011;86:405–411.
- Calin GA, Ferracin M, Cimmino A, et al. A MicroRNA signature associated with prognosis and progression in chronic lymphocytic leukemia. N Engl J Med 2005;353:1793–1801.
- Fulci V, Chiaretti S, Goldoni M, et al. Quantitative technologies establish a novel microRNA profile of chronic lymphocytic leukemia. Blood 2007;109:4944–4951.
- Marton S, Garcia MR, Robello C, et al. Small RNAs analysis in CLL reveals a deregulation of miRNA expression and novel miRNA candidates of putative relevance in CLL pathogenesis. Leukemia 2008;22:330–338.
- Pekarsky Y, Santanam U, Cimmino A, et al. Tcl1 expression in chronic lymphocytic leukemia is regulated by miR-29 and miR-181. Cancer Res 2006;66:11590–11593.
- Rossi S, Shimizu M, Barbarotto E, et al. microRNA fingerprinting of CLL patients with chromosome 17p deletion identify a miR-21 score that stratifies early survival. Blood 2010;116:945–952.
- Stamatopoulos B, Meuleman N, Haibe-Kains B, et al. microRNA-29c and microRNA-223 down-regulation has in vivo significance in chronic lymphocytic leukemia and improves disease risk stratification. Blood 2009;113:5237–5245.
- Zenz T, Mohr J, Eldering E, et al. miR-34a as part of the resistance network in chronic lymphocytic leukemia. Blood 2009;113:3801–3808.
- Zhou K, Yi S, Yu Z, et al. MicroRNA-223 expression is uniformly down-regulated in B cell lymphoproliferative disorders and is associated with poor survival in patients with chronic lymphocytic leukemia. Leuk Lymphoma 2012;53:1155–1161.
- Neilson JR, Zheng GX, Burge CB, et al. Dynamic regulation of miRNA expression in ordered stages of cellular development. Genes Dev 2007;21:578–589.
- Haferlach C, Dicker F, Weiss T, et al. Toward a comprehensive prognostic scoring system in chronic lymphocytic leukemia based on a combination of genetic parameters. Genes Chromosomes Cancer 2010;49:851–859.
- Wierda WG, O’Brien S, Wang X, et al. Multivariable model for time to first treatment in patients with chronic lymphocytic leukemia. J Clin Oncol 2011;29:4088–4095.
- Pepper C, Majid A, Lin TT, et al. Defining the prognosis of early stage chronic lymphocytic leukaemia patients. Br J Haematol 2012;156:499–507.
- Onkologiskt centrum, Södra sjukhusregionen, 2008-2010. National guidelines for investigation and treatment of chronic lymphocytic leukemia. Available from: http://www.oc.gu.se/pdf/KLL_Nationella_Riktlinjer_20060601.pdf
- British Committee for Standards in Haematology. Guidelines on the diagnosis, investigation and management of chronic lymphocytic leukaemia. Available from: http://www.bcshguidelines.com/documents/Revised_CLL_guideline_july_13_v1.pdf
- Shanafelt TD, Witzig TE, Fink SR, et al. Prospective evaluation of clonal evolution during long-term follow-up of patients with untreated early-stage chronic lymphocytic leukemia. J Clin Oncol 2006;24:4634–4641.
- Stilgenbauer S, Sander S, Bullinger L, et al. Clonal evolution in chronic lymphocytic leukemia: acquisition of high-risk genomic aberrations associated with unmutated VH, resistance to therapy, and short survival. Haematologica 2007;92:1242–1245.
- Strefford J, Sutton LA, Baliakas P, Agathangelidis A, Malčíková J, Plevova K, Scarfó L, Davis Z, Stalika E, Cortese D, Cahill N, Bredo Pedersen L, Francia di Celle P, Tzenou T, Geisler C, Panagiotidis P, Langerak AW, Chiorazzi N, Pospisilova S, Oscier D, Davi F, Belessi C, Mansouri L, Ghia P, Stamatopoulos K, Rosenquist R. Distinct patterns of novel gene mutations in poor-prognostic stereotyped subsets of chronic lymphocytic leukemia: the case of SF3B1 and subset #2. Leukemia. 2013; doi: 10.1038/leu.2013.98.