Abstract
Elevated levels of pro-oxidants and various markers of oxidative tissue damage were found in diabetic patients, indicating involvement of oxidative stress in the pathogenesis of diabetes mellitus (DM). On one side, physiological levels of reactive oxygen species (ROS) play an important role in redox signaling of various cells, while on the other, excessive ROS production can jeopardize the integrity and physiological functions of cellular macromolecules, in particular proteins, thus contributing to the pathogenesis of DM. Reactive aldehydes, especially 4-hydroxynonenal (HNE), are considered as second messengers of free radicals that act both as signaling molecules and as cytotoxic products of lipid peroxidation causing long-lasting biological consequences, in particular by covalent modification of macromolecules. Accordingly, the HNE and related reactive aldehydes may play important roles in the pathophysiology of DM, both in the development of the disease and in its progression and complications due to the following: (i) exposure of cells to supraphysiological levels of 4-hydroxyalkenals, (ii) persistent and sustained generation of 4-hydroxyalkenals that progressively affect vulnerable cells that lack an efficient bioactive aldehyde neutralization system, (iii) altered redox signaling influenced by reactive aldehydes, in particular by HNE, and (iv) induction of extracellular generation of similar aldehydes under secondary pathological conditions, such as low-grade inflammation.
Introduction
Millions of people worldwide suffer from diabetes mellitus (DM), a chronic metabolic disease that is characterized by hyperglycemia due to absolute or relative lack of insulin secretion and/or its metabolic activity. Pancreatic islets of Langerhans function as glucose sensors and are responsible for maintaining glucose homeostasis and metabolism. Among the three different types of endocrine cells in these islets, β-cells are responsible for insulin synthesis and secretion. Therefore, the integrity, mass, and function of β-cells are critical to maintain normoglycemia. In response to hyperglycemia, the most important peptide hormone secreted from β-cells is insulin and its exocytosis relies on optimal mitochondrial function [Citation1]. Generally, glucose-stimulated insulin secretion involves entry of glucose to β-cells by glucose transporter 2 (GLUT2) across the membrane, followed by glucokinase-mediated phosphorylation and initiation of glycolysis [Citation2]. Subsequently, adenosine diphosphate (ADP) is converted into adenosine triphosphate (ATP), leading to the closure of ATP-sensitive K+ channels, depolarization of the plasma membrane, and activation of voltage-gated Ca2+ channels, while Ca2+ influx leads to the rise of cytosolic Ca2+ concentration and triggers the exocytosis of insulin [Citation3]. Relative contribution of glucose and lipid products for oxidative catabolism could modulate metabolic profile of mitochondria. Acetyl coenzyme-A is a key player in this modulated pathway, resulting further in Ca2+-evoked exocytosis of insulin [Citation4].
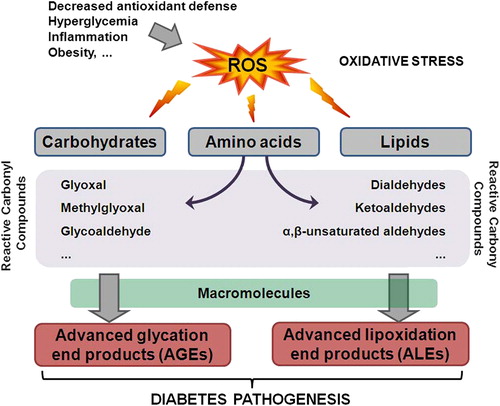
Hitherto, it is established that chronic hyperglycemia and hyperlipidemia are deleterious to the function of β-cells. Under glucose toxicity and lipotoxicity conditions, the fine balance between pro-oxidants and antioxidants in the cells is disturbed and leads to a chronic oxidative stress that subsequently contribute to impaired glucose-induced insulin release [Citation5,Citation6]. Indeed, elevated levels of pro-oxidants and various markers of oxidative tissue damage were found in diabetic patients, indicating important roles of oxidative stress in the pathogenesis of the disease [Citation5] (). Of interest are the findings that oxidative stress and lipid peroxidation are crucial for inducing β-cell dysfunction and death during the progression of DM [Citation7]. This review discusses some aspects on the role of oxidative stress in the onset, progression, and complications of DM.
Figure 1. The role of oxidative stress in diabetes pathogenesis. Decreased antioxidant defense, hyperglycemia, inflammation, and obesity contribute to the ROS increase in the body. Elevated levels of ROS can damage carbohydrates, amino acids, and lipids leading to the formation of RCC. RCC can further react with macromolecules yielding advanced glycation or lipoxidation end products, AGE or ALE, respectively. Both AGE and ALE have important role in the onset and progression of diabetes.
Susceptibility of β-cells to oxidative stress
Pancreatic β-cells are particularly vulnerable to endogenous or exogenous oxidative stress, which contributes to β-cell dysfunction and death [Citation8]. Mitochondria are the main source of intracellular reactive oxygen species (ROS). During the respiratory chain in the inner membrane of mitochondria, reactive superoxide anion (•O2‐) is formed. This radical may be further converted in β-cells into hydrogen peroxide (H2O2) by the action of superoxide dismutase (SOD) [Citation9]. Normally, catalase (CAT) and glutathione peroxidases (GPXs) can neutralize H2O2, However, since β-cells express very low levels of Cu/Zn SOD, Mn SOD, CAT, and GPXs in comparison with the other cells in the body [Citation10], they are more susceptible to oxidative stress. In addition, reactive aldehydes that have longer half-life than the abovementioned radicals are considered as “second messengers of free radicals” involved in β-cell reaction to oxidative stress.
Dual signaling roles of ROS in β-cells
As in the case of other cell types, dynamic changes in the ATP levels and ROS generation by glycolytic and respiratory metabolism represent the key energetic metabolism that makes the fundaments also in the β-cell signaling [Citation11]. At first, ROS were thought to be the by-products of respiratory mitochondria metabolism; however, ROS are currently considered as important players in numerous signaling pathways that regulate a variety of cellular responses [Citation12,Citation13]. For instance, H2O2 is recognized a ubiquitous intracellular messenger and numerous signaling molecules involved in insulin secretion in β-cells have been recognized as its downstream targets. Hence, H2O2 derived from glucose metabolism represents one of the metabolic signals for insulin secretion [Citation11].
Nevertheless, excessive and/or sustained ROS production can jeopardize the integrity and physiological functions of macromolecules and contribute to pathogenesis of DM [Citation14]. High levels of H2O2 have been shown to inactivate mitochondria and alter mitochondria signals for insulin secretion [Citation15]. Indeed, Li and colleagues showed that transient oxidative stress, induced by 200 μM H2O2 for 10 min, induces β-cells dysfunction lasting over days [Citation16]. The main component in cellular defense against ROS toxicity is the transcription factor NF-E2-related factor 2 (Nrf2) that in response to elevated ROS levels induces the transcription of numerous antioxidant enzymes [Citation17]. Although the Nrf2 action is necessary for a proper redox homeostasis, it could also blunt normal ROS signals [Citation17]. Interestingly, excessive activity of antioxidants in β-cells deregulates insulin secretion [Citation18]. Therefore, it is necessary to elucidate the effects of cellular adaptive responses to disturbed redox homeostasis in pathophysiology of DM.
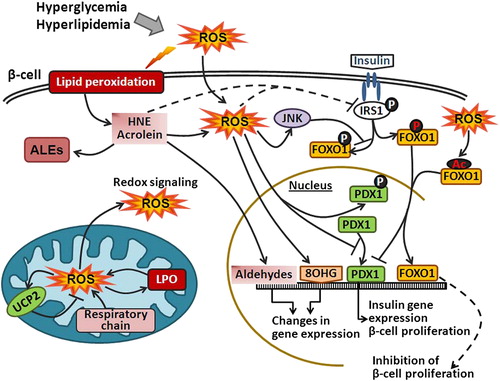
Furthermore, an essential role in the β-cell proliferation, survival, and function has been assigned to the transcriptional factors pancreatic duodenal homeobox 1 (PDX1), forkhead box O (FOXO) and to the uncoupling protein 2 (UCP2) (). The function of these proteins is sensitive to elevated levels of ROS [Citation18]. For example, increasing the levels of ROS accelerates the degradation of PDX1 due to targeted phosphorylation on Ser61 and Ser66 [Citation19]. Oxidative stress also regulates FOXO activity through various post-translational modifications (i.e., phosphorylation, acetylation, and ubiquitination) [Citation20]. It has been shown that elevated ROS levels lead to the activation of FOXO, which upregulates the transcription of antioxidant genes [Citation21]. Similarly, UCP2, which functions as a molecular sensor and suppressor of mitochondria-derived •O2‐, is also considered to be a negative regulator of insulin release [Citation22,Citation23]. Matsuoka and colleagues reported that ROS induced augmented expression of c-Jun in diabetic islets and decrease musculoaponeurotic fibrosarcoma oncogene homolog A (MafA) activity [Citation24]. It has been shown that overexpression of GPX in β-cells can prevent the loss of MafA and protect the cells from chronic hyperglycemia-induced dysfunction [Citation25]. ROS can interfere with this mechanism by inactivating MafA and thus compromises this defense mechanism [Citation26].
Figure 2. Oxidative stress and HNE impair insulin signaling. Hyperglycemia and hyperlipidemia induce ROS generation, which in turn promote lipid peroxidation and formation of reactive aldehydes (e.g. HNE) that can further bind to IRS and impair IRS function. Elevated intracellular ROS can damage DNA and lead to the formation of 8OHG. In addition, ROS can induce UCP2 in mitochondria or inactivate PDX1. Furthermore, activation of IRS1 leads to phosphorylation of FOXO1 transcription factor and FOXO1 nuclear export. However, ROS can either directly induce acetylation of FOXO1 or through JNK phosphorylate FOXO1 at different residues and thus promote FOXO1 nuclear translocation. FOXO1 also competes for the PDX1 promoter and represses the expression of PDX1 that is responsible for β-cell proliferation.
Hyperglycemia-induced oxidative stress
Elevated blood glucose levels contribute to the formation of ROS in various tissues and also contribute to the ‘hyperglycemic/glycemic memory’. The ‘glycemic memory’ is a term coined for the continued progression of tissue damage that persists after abolition of hyperglycemia and attainment of normoglycemia. This phenomenon was partly explained by the persistent epigenetic changes caused by mitochondria-induced ROS production during hyperglycemia. It has been shown that ROS induce long-lasting monomethylation of histone lysine residue H3K4 in the proximal promoter of the nuclear factor kappaB (NF-κB) subunit p65, which consequently leads to sustained increase in p65 gene expression and further in the expression of p65-dependent pro-inflammatory genes. Conversely, hyperglycemia-induced overproduction of ROS can cause demethylation of H3K9, thus reducing the inhibition of p65 gene expression and acting synergistically with other epigenetic changes that might contribute to the onset of diabetes [Citation27,Citation28].
There are four main pathways affected by hyperglycemia-induced overproduction of ROS: (i) activation of isoforms of protein kinase C (PKC), (ii) increased hexosamine pathway flux and augmented flux of glucose and other sugars through polyol pathway, (iii) increased formation of advanced glycation end products (AGEs), and (iv) increased expression of receptor for AGEs (RAGE) and its activating ligands [Citation27,Citation29].
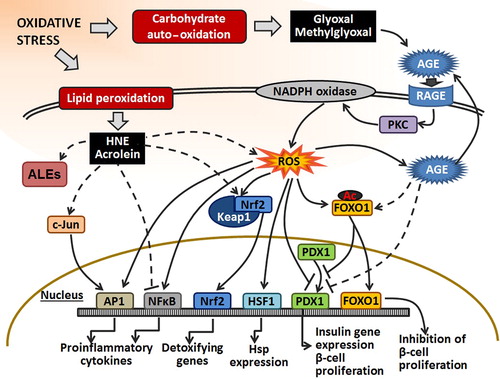
In addition to the above-mentioned direct effects of ROS, equally important are the indirect effects caused by covalent modifications of proteins by reactive carbonyl compounds (RCC). Namely, in contrast to the very short half-life of ROS, which is in the range of nanoseconds and milliseconds, RCC are more stable and with the average half-life of minutes to hours [Citation30]. Due to their higher stability, the non-charged RCC may escape the cells and react with extracellular targets and therefore become more deleterious than ROS. RCC can modify macromolecules and alter cellular signaling pathways (). The RCC are formed by auto-oxidation of carbohydrates, lipids, or amino acids. Some of the RCC that can modify proteins or lipids yielding AGEs are formed by auto-oxidation of carbohydrates and include glyoxal, methylglyoxal, and glycoaldehyde [Citation31]. The crucial mechanism for the role of AGE in altered gene expression was described well before [Citation32]. AGEs bind to the cell surface receptor RAGE, and induce phosphorylation of PKC, with subsequent NADPH oxidase activation that yields to an excessive intracellular ROS formation [Citation33] and activation of key transcription factors, such as NFκB and AP1 that cause multiple pathological changes in gene expression [Citation34,Citation35]. Furthermore, it has been suggested that AGEs might deteriorate β-cell function in patients with long-term hyperglycemia [Citation36]. RAGE signaling pathway has also been implicated in the upregulation of heat shock factor-1 (HSF1) [Citation37], which regulates the transcription of heat shock proteins (HSP) that protect the proteins exposed to non-enzymatic protein modifications. Therefore, available data suggest that HSP might be the essential in preventing insulin resistance in obesity [Citation38].
Figure 3. The effect of RCC on transcription factors. Under oxidative stress, RCC are formed by auto-oxidation of carbohydrates (e.g., glyoxal and methylglyoxal) or by lipid peroxidation (e.g., HNE and acrolein). LPO-derived RCC can react with NFκB and suppress its action. Also, they can indirectly activate AP1 and Nrf2 by phosphorylation of c-Jun or alkylation of Keap1 in the Keap1–Nrf2 complex, respectively. Moreover, they can affect transcription factors indirectly through ROS signal transduction pathways. The effect of RCC, derived from carbohydrate oxidation, on the transcription factors is either achieved through AGE or ROS transduction pathway. AGE can promote acetylation of FOXO1 that can inhibit proliferation of β-cells and insulin gene expression. Also, binding of AGE to AGE receptor (RAGE) stimulates ROS production by PKC and NADPH oxidase. Furthermore, elevated ROS can activate AP1, NFκB, and HSF1, and inhibit PDX1 binding to DNA. Accordingly, they induce pro-inflammatory gene expression, Hsp expression, and inhibit insulin gene expression and β-cell proliferation. Also, ROS can induce dissociation of Nrf2 from Keap1–Nrf2 complex enabling Nrf2 translocation to the nucleus and activation of the expression of detoxifying genes.
The RCC formed by lipid peroxidation of polyunsaturated fatty acids (PUFAs) are hydroperoxides and endoperoxides that further undergo fragmentation and produce a variety of RCC intermediates [Citation30]. The most reactive PUFA-derived RCC are α,β-unsaturated aldehydes, like 4-hydroxyl-trans-2-nonenal (HNE) and acrolein, followed by dialdehydes such as malondialdehyde (MDA) and finally ketoaldehydes such as 4-oxo-trans-2-nonenal (ONE) [Citation39]. Some of these, in particular HNE, can be retained in the membranes due to their lipophilic property; yet, they can also move within the cell and between the cells, even if conjugated with proteins or peptides [Citation40]. Growing evidence suggests that HNE can induce insulin resistance, thus eliciting its important role in DM pathogenesis [Citation41]. Nucleophilic groups in macromolecules, such as aminophospholipids or certain amino acid moieties, are sensitive to RCC resulting in their modification and formation of crosslinks and adducts, and are termed advanced lipoxidation end products (ALEs) [Citation30]. Different ALE adducts like HNE-His and acrolein-Lys can be formed by RCC reaction with nucleophilic sites (i.e., His, Cys, Lys, or Agr) of proteins through Michael addition.
Hitherto, there is a growing body of evidence that PUFA-derived reactive aldehydes have also an important role in cellular signaling, both under physiological and under pathological circumstances [Citation42–44]. PUFA-derived aldehydes can directly alter cellular signaling pathways by interfering with the function of various transcription factors. HNE and acrolein have been shown to inhibit NFκB [Citation45,Citation46] while by phosphorylation of c-Jun they activate AP-1 [Citation45,Citation47]. Both NFκB and AP1 are responsible for the expression of proinflammatory cytokines and contribute to the propagation of diabetic complications. In addition, HNE was shown to alkylate the Keap1, a cytoplasmic inhibitor of Nrf2, and leads to dissociation of Nrf2 from the Keap1–Nrf2 complex, its translocation into the nucleus, and expression of detoxifying genes [Citation48].
Furthermore, PUFA-derived RCC, like MDA, HNE, and acrolein, can react with exocyclic groups of DNA bases yielding various alkylated products. The most commonly modified DNA base is guanine that in the reaction with PUFA-derived RCC can yield eteno adducts such as MDA-dG [Citation30].
Demozay and colleagues showed that exogenously added HNE to 3T3-L1 murine adipocytes caused insulin resistance due to an enhanced degradation of the insulin receptor substrate (IRS)-1/IRS-2 proteins. They have also shown that neutralization of HNE, by expressing fatty aldehyde dehydrogenase in the cells, partially restores the insulin-induced IRS-1 tyrosine phosphorylation and the abnormal metabolic responses that were induced by HNE [Citation49].
Pathophysiological association of HNE with T2DM was also demonstrated by the recent clinical study of relationships among skeletal muscle lipid peroxidation, intramyocellular lipid content, and insulin sensitivity in nine insulin-sensitive and insulin-resistant subjects, as well as in T2DM patients [Citation50]. Namely, HNE-protein adducts were elevated 1.6-fold in T2DM adults when compared with insulin-sensitive adults, whereas insulin-resistant adults showed intermediate levels. Moreover, intramyocellular lipid content was increased by 4.0- and 1.9-fold in T2DM and insulin-resistant subjects, respectively, when compared with insulin sensitive subjects. However, protein carbonyls were not different among groups and did not correlate with other measured variables. Thus, the study has shown that skeletal muscle protein-HNE adducts are related to the severity of insulin resistance in humans and that muscle lipid peroxidation could be involved in the development of insulin resistance [Citation50]. These findings support the overall concepts of pathophysiology of lipid peroxidation based on the signaling effects of HNE and its high affinity to bind to proteins and peptides causing their non-enzymatic modifications of structure and function [Citation51].
Diabetic ketoacidosis (DKA) is a dangerous complication of DM and is associated with decreased levels of serum SOD, in comparison with those of healthy individuals [Citation52]. Furthermore, DKA is also involved in chronic complications of diabetic encephalopathy, which might be related to the vicious circle of oxidative stress associated with a secondary brain damage, as in the case of fetal brain edema in which DKA-in is also associated with increased levels of HNE, 8-hydroxyguanosine (8OHG), and heme-oxygenase-1 (HO-1) in the pyramidal neurons of hippocampus [Citation53]. Similarly, the HNE-protein adducts were shown to accumulate also in endothelial and Schwann cells of the peripheral nerve, neurons, astrocytes and oligodendrocytes of the spinal cord and neurons and glial cells of the dorsal root ganglia [Citation54]. The inhibition of poly(ADP-ribose) polymerase (PARP) may diminish the accumulation of HNE-protein adducts in peripheral nerves, spinal cord, and dorsal rood ganglion neurons indicating important role of PARP and HNE in diabetic peripheral neuropathy [Citation54], while HNE was also shown to upregulate aldose reductase, an enzyme of the polyol pathway related to microvascular complications in DM [Citation55]. Indeed, carbonyl/PUFA pathway might promote localized oxidative stress in tissues sensitive to diabetic damage such as retina and aortic tissue [Citation56]. These data point not only to oxidative stress in general but in particular to the HNE and related aldehydes as important factors of DM pathophysiology.
Relationship between obesity, reactive aldehydes, and DM pathology
Excessive HNE generation has also been suggested to be involved in obesity by promoting fatty acid synthesis and suppressing fatty acid β-oxidation [Citation57], although it is likely that lipid peroxidation itself is not a prerequisite for obesity. Namely, the study carried out on mice fed with a high fat diet (70% energy as fat) has shown that such a diet increased body weight and fat mass development, impaired glycemia and insulinemia, but it decreased malondialdehyde (MDA determined as thiobarbituric acid-reactive substances) content in the liver as well as in epididymal, subcutaneous, and visceral adipose tissues [Citation58]. Thus, lipid peroxidation, at least in the important metabolic organs studied, does not appear to be necessarily related to the development of metabolic disorders associated with diabetes and obesity. In favor of this assumption are findings of the recent in vitro study showing that acute and repeated exposure of adipocytes to physiological concentrations of HNE is sufficient to promote subsequent oxidative stress, impaired adipogenesis; alter the expression of adipokines; and increase lipolytic gene expression and subsequent increase in free fatty acid release [Citation59]. Accordingly, it may be assumed that HNE has diverse effects toward adipocyte homeostasis and adipocyte differentiation, which may be important for the pathogenesis of obesity and metabolic syndrome. Similar findings of diverse effects of HNE in vitro were revealed twenty years ago for the first time, when the aldehyde was found to be not only the cytotoxic by-product of lipid peroxidation but also bifunctional, growth regulating factor interacting with various cytokines [Citation60,Citation61]. Therefore, not only cytotoxic, but also regulatory effects of HNE, known nowadays as a signaling molecule involved in regulation of major cellular activities [Citation13,Citation44] as well as a potent factor modifying structure and function of numerous cellular and extracellular proteins [Citation62], might help understanding the involvement of HNE and related aldehydes in pathophysiology of DM. Of importance is to keep in mind that HNE has, in particular, affinity to bind to the membrane-associated proteins [Citation63], thus allowing its bioactivities to be obtained at the site of its origin as well as remote from the membrane lipids, while non-enzymatic modifications of proteins by HNE (ALE) can be influenced, even reverted by hormetic effects nutriceuticals, especially those with antioxidant capacities [Citation64–66]. These aspects might help understanding some contradictory data in the literature and encourage further necessary studies similar to the one done by Vincent et al. who found that elevated levels of HNE, observed in obese people, could be reduced by exercise and the change of diet [Citation67].
In addition, recently, Shevalye and colleagues have shown that high-calorie/high-fat diet causes systemic and renal oxidative stress and is associated with prediabetic nephropathy. They have observed a 38% increase in HNE adducts in the renal cortex of mice fed with a high-fat diet for 16 weeks [Citation68]. Obesity is also associated with the development of gestational DM in pregnant woman [Citation69]. Maternal hyperglycemia induces oxidative stress and contributes to diabetic embryopathy. Recently, the increased levels of HNE and MDA in embryos of diabetic wild-type mice were described [Citation70]. However, these effects of oxidative stress were successfully mitigated by SOD1 overexpression that blocked maternal hyperglycemia-induced PKC activation [Citation70].
The role 4-Hydroxyalkenals as endogenous activators of the peroxisome proliferator-activated receptor-δ (PPARδ) in diabetes
Peroxisome proliferator-activated receptor-δ (PPARδ) is a ligand-activated transcription factor which dimerizes with the retinoid X receptor (RXR) to form a heterodimer that interacts with the PPAR response element (PPRE) in the promoters of target genes. This complex regulates the transcription of various proteins in adipose tissue, skeletal muscles, skin, cancer cells, and in inflammatory processes [Citation71]. Of particular interest are studies showing that the pharmacological activation of PPARδ could attenuate the metabolic syndrome by ameliorating dyslipidemia, preventing obesity, improving peripheral insulin sensitivity, and ultimately reducing symptoms of non-insulin-dependent diabetes [Citation71–77]. Various saturated and unsaturated fatty acids as well as PUFA-derived mediators have been proposed to function as endogenous activators of PPARδ [Citation71,Citation78–81]. Yet, no consensus on the nature of endogenous activating ligands has yet been reached. However, small synthetic molecular weight activators (e.g., GW501516) and antagonists (e.g., GSK0660) have been developed and tested in a variety of in vitro and in vivo experimental systems [Citation82–86]. Ravnskjaer et al. [Citation87] and Riahi et al. [Citation88] have recently shown that the lipid peroxidation products of arachidonic acid and linoleic acid, namely HNE and 4-hydroxydodecadienal (4-HDDE), are potent activators of PPARδ in pancreatic β-cells and vascular endothelial cells, respectively. Cohen et al. have studied, in detail, the role of HNE in rat islets of Langerhans and in the INS-1E β-cell line, which was derived from rat insulinoma [Citation89]. Two important observations have been reported in this study: first, exogenously added HNE caused dose-dependent apoptosis and cell death and second, stressful conditions, like high glucose levels, altered the turn-over of fatty acids in membrane phospholipids due to the activation of phospholipase A2 and subsequent release of arachidonic and linoleic acids to the cell interior. These two fatty acids were readily peroxidized in the oxidative environment induced by high glucose and fragmented to 4-hydroxyalkenals [Citation90,Citation91]. In INS-1E cells, this process leads to an augmented production of HNE. Yet, the levels of HNE under these conditions were significantly below the cytotoxic range and the cells remained viable and maintained a normal insulin secretory function. Moreover, these conditions augmented glucose-stimulated insulin secretion to levels typically seen in the adaptation phase response of β-cells to hyperglycemia.
Several lines of evidence link HNE-dependent activation of PPARδ to this phenomenon. The use of the PPARδ antagonist GSK0660 or silencing of PPARδ expression in β-cells abolished both high glucose- and HNE-augmenting effects of insulin secretion, whereas the PPARδ agonist GW501516 mimicked them. Moreover, neutralization of endogenously generated HNE with N-acetyl cysteine blocked the stimulatory effect. Finally, HNE and GW501516 induced the expression of a reporter gene (luciferase) in a transactivation assay in INS-1E cells in a PPRE-dependent manner, whereas the antagonist GSK0660 blocked this effect. These findings point to a clear distinction between physiological functions of intracellular HNE and pathophysiological effects of extracellular of HNE in cells. Under mild oxidative stress and accelerated phospholipid turn-over, the generation of 4-hydroxyalkenals is augmented due to the release of precursor PUFAs to the cell interior. The levels of free intracellular 4-hydroxyalkenal in the cells depend on the rate of lipid peroxidation, the degree of covalent interactions with nucleophilic moieties in macromolecules, and the efficacy of the inherent neutralization enzymes (i.e., fatty aldehyde dehydrogenase and glutathione peroxidase) of these bioactive aldehydes in cells [Citation91]. Similarly, small molecular weight antioxidants, and in particular L-carnosine, that covalently bind and sequester 4-hydoroxyakenals greatly contribute to steady-state level of these reactive molecules in the cells as well as they could influence glucose metabolism and scavenge reactive carbonyls. Thus, Sauerhöfer et al. observed correlation of L-carnosine levels with β-cell mass in transgenic mice model mimicking the expression pattern of human carnosinase 1 (hCN1) resulting in the weight loss and glycosuria [Citation92], while Aldini et al. used obese rats to reveal that some of the major biological effects of carnosine, such as prevention of dyslipidemia, hypertension, and renal injury, reflect direct carbonyl scavenging activities of carnosine [Citation93]. However, interactions of carnosine and HNE and related aldehydes have yet to be understood well.
The study on INS-1E cells and freshly isolated rat islets of Langerhans [Citation89] demonstrate that the intracellular levels of HNE generated under glucose-induced oxidative stress are not harmful to the cells. On the contrary, HNE actually functions as an important signaling molecule in the adaptive hypersecretion of insulin from β-cells in response to long-term hyperglycemia. Further support to this contention comes from the study of the effect of high glucose on the expression of glucose transporter-1 (GLUT-1) in vascular endothelial, in which 4-HDDE activates PPARδ, which increases the expression of the protein calreticulin that destabilizes GLUT-1 mRNA and ultimately lowers glucose uptake. It has been proposed that this mechanism equips endothelial cells with an effective natural protective mechanism against deleterious effects of uncontrolled influx of glucose to the cells under hyperglycemic conditions. Yet, it is clear that extended oxidative stressful conditions, which cause sustained generation of 4-hydroxyalkenals, may progressively impair cell structure and function due to the accumulation of stable adducts 4-hydroxyalkenals formed with proteins, phospholipids, and DNA. In addition, the deterioration of cells is accelerated when they are exposed to exogenous high levels of 4-hydroxyalkenals produced in the vicinity of the cells. Vazdar et al. have recently shown that the entry of exogenous HNE to the cytoplasm of cells through membrane phospholipids is very fast and efficient [Citation40]. High levels of HNE have been associated with inflammatory processes [Citation94,Citation95] under pathological conditions such as atherosclerosis or obesity [Citation90,Citation96,Citation97]. Therefore, 4-hydroxyalkenal-mediated cell and organ dysfunction similar to these pathological conditions may result from (i) exposure of cells to supraphysiological levels of 4-hydroxyalkenals, (ii) persistent and sustained generation of 4-hydroxyalkenals that progressively affect vulnerable cells lacking an efficient bioactive aldehyde neutralization and sequestration systems, (iii) altered redox signaling influenced by reactive aldehydes, in particular by HNE, and (iv) extracellular generation of these aldehydes under pathological conditions such as inflammation.
Conclusion
Reactive aldehydes generated under various pathophysiological processes of oxidative stress associated with lipid peroxidation act as second messengers of free radicals and may therefore be of major relevance in development and progression of various metabolic diseases, in particular of DM. However, to elucidate the complete mechanism of PUFA-derived reactive aldehydes in DM pathophysiology, furt her studies are needed.
Declaration of interest
The authors report no declarations of interest. The authors alone are responsible for the content and writing of the paper.
The study was supported by the Croatian Ministry of Science, Education and Sports and by the COST CM1001 Action of the European Union. Shlomo Sasson is the Adolf D. and Horty Storch Chair in Pharmaceutical Sciences, at the Faculty of Medicine, The Hebrew University of Jerusalem, Israel.
References
- Maechler P. Mitochondria as the conductor of metabolic signals for insulin exocytosis in pancreatic beta-cells. Cell Mol Life Sci 2002;59:1803–1818.
- Tarasov A, Dusonchet J, Ashcroft F. Metabolic regulation of the pancreatic beta-cell ATP-sensitive K+ channel: a pas de deux. Diabetes 2004;53:S113–S122.
- Eliasson L, Abdulkader F, Braun M, Galvanovskis J, Hoppa MB, Rorsman P. Novel aspects of the molecular mechanisms controlling insulin secretion. J Physiol 2008;586: 3313–3324.
- Li N, Stojanovski S, Maechler P. Mitochondrial hormesis in pancreatic β cells: does uncoupling protein 2 play a role?. Oxid Med Cell Longev 2012;2012:740849.
- Robertson RP, Harmon J, Tran PO, Poitout V. Beta-cell glucose toxicity, lipotoxicity, and chronic oxidative stress in type 2 diabetes. Diabetes 2004;53:S119–S124.
- Köhnke R, Mei J, Park M, York DA, Erlanson-Albertsson C. Fatty acids and glucose in high concentration down-regulates ATP synthase beta-subunit protein expression in INS-1 cells. Nutr Neurosci 2007;10:273–278.
- Lenzen S. Oxidative stress: the vulnerable beta-cell. Biochem Soc Trans 2008;36:343–347.
- Rhodes C. Type 2 diabetes-a matter of beta -cell life and death?. Science 2005;307:380–384.
- Gurgul E, Lortz S, Tiedge M, Jörns A, Lenzen S. Mitochondrial catalase overexpression protects insulin-producing cells against toxicity of reactive oxygen species and proinflammatory cytokines. Diabetes 2004;53:2271–2280.
- Tiedge M, Lortz S, Munday R, Lenzen S. Complementary action of antioxidant enzymes in the protection of bioengineered insulin-producing RINm5F cells against the toxicity of reactive oxygen species. Diabetes 1998;47:1578–1585.
- Pi J, Bai Y, Zhang Q, Wong V, Floering LM, Daniel K, et al. Reactive oxygen species as a signal in glucose-stimulated insulin secretion. Diabetes 2007;56:1783–1791.
- Zivkovic M, Poljak-Blazi M, Zarkovic K, Mihaljevic D, Schaur RJ, Zarkovic N. Oxidative burst of neutrophils against melanoma B16-F10. Cancer Lett 2007;246:100–108.
- Jaganjac M, Cacev T, Cipak A, Kapitanović S, Gall Troselj K, Zarković N. Even stressed cells are individuals: second messengers of free radicals in pathophysiology of cancer. Croat Med J 2012;53:304–309.
- Evans JL, Goldfine ID, Maddux BA, Grodsky GM. Are oxidative stressactivated signaling pathways mediators of insulin resistance and β-cell dysfunction?. Diabetes 2003;52:1–8.
- Maechler P, Jornot L, Wollheim CB. Hydrogen peroxide alters mitochondrial activation and insulin secretion in pancreatic beta cells. J Biol Chem 1999;274:27905–27913.
- Li N, Brun T, Cnop M, Cunha DA, Eizirik DL, Maechler P. Transient oxidative stress damages mitochondrial machinery inducing persistent beta-cell dysfunction. J Biol Chem 2009; 284:23602–23612.
- Jaganjac M. Possible involvement of granulocyte oxidative burst in Nrf2 signaling in cancer. Indian J Med Res 2010;131:609–616.
- Lei XG, Vatamaniuk MZ. Two tales of antioxidant enzymes on β cells and diabetes. Antioxid Redox Signal 2011;14: 489–503.
- Boucher M-J, Selander L, Carlsson L, Edlund H. Phosphorylation marks IPF1 = PDX1 protein for degradation by glycogen synthase kinase 3-dependent mechanisms. J Biol Chem 2006; 281:6395–6403.
- Ponugoti B, Dong G, Graves DT. Role of forkhead transcription factors in diabetes-induced oxidative stress. Exp Diabetes Res 2012;2012:939751.
- Storz P. Forkhead homeobox type O transcription factors in the responses to oxidative stress. Antioxid Redox Signal 2011;14:593–605.
- Zhang CY, Baffy G, Perret P, Krauss S, Peroni O, Grujic D, et al. Uncoupling protein-2 negatively regulates insulin secretion and is a major link between obesity, beta cell dysfunction, and type 2 diabetes. Cell 2001;105:745–755.
- Krauss S, Zhang CY, Scorrano L, Dalgaard LT, St-Pierre J, Grey ST, Lowell BB. Superoxide-mediated activation of uncoupling protein 2 causes pancreatic beta cell dysfunction. J Clin Invest 2003;112:1831–1842.
- Matsuoka TA, Kaneto H, Miyatsuka T, Yamamoto T, Yamamoto K, Kato K, et al. Regulation of MafA expression in pancreatic beta-cells in db/db mice with diabetes. Diabetes 2010;59:1709–1720.
- Harmon JS, Bogdani M, Parazzoli SD, Mak SS, Oseid EA, Berghmans M, et al. beta-Cell-specific overexpression of glutathione peroxidase preserves intranuclear MafA and reverses diabetes in db/db mice. Endocrinology 2009;150:4855–4862.
- Robertson RP, Harmon JS. Diabetes, glucose toxicity, and oxidative stress: a case of double jeopardy for the pancreatic islet beta cell. Free Radic Biol Med 2006;41:177–184.
- Giacco F, Brownlee M. Oxidative stress and diabetic complications. Circ Res 2010;107:1058–1070.
- Brasacchio D, Okabe J, Tikellis C, Balcerczyk A, George P, Baker EK, et al. Hyperglycemia induces a dynamic cooperativity of histone methylase and demethylase enzymes associated with gene-activating epigenetic marks that coexist on the lysine tail. Diabetes 2009;58:1229–1236.
- Rolo AP, Palmeira CM. Diabetes and mitochondrial function: role of hyperglycemia and oxidative stress. Toxicol Appl Pharmacol 2006;212:167–178.
- Pamplona R. Advanced lipoxidation end-products. Chem Biol Interact 2011;192:14–20.
- Suzuki D, Miyata T. Carbonyl stress in the pathogenesis of diabetic nephropathy. Intern Med 1999;38:309–314.
- Wautier MP, Chappey O, Corda S, Stern DM, Schmidt AM, Wautier JL. Activation of NADPH oxidase by AGE links oxidant stress to altered gene expression via RAGE. Am J Physiol Endocrinol Metab 2001;280:E685–E694.
- Thallas-Bonke V, Thorpe SR, Coughlan MT, Fukami K, Yap FY, Sourris KC, et al. Inhibition of NADPH oxidase prevents advanced glycation end product-mediated damage in diabetic nephropathy through a protein kinase C-alpha- dependent pathway. Diabetes 2008;57:460–469.
- Schramm A, Matusik P, Osmenda G, Guzik TJ. Targeting NADPH oxidases in vascular pharmacology. Vascul Pharmacol 2012;56:216–231.
- Clynes R, Moser B, Yan SF, Ramasamy R, Herold K, Schmidt AM. Receptor for AGE (RAGE): weaving tangled webs within the inflammatory response. Curr Mol Med 2007; 7:743–751.
- Lim M, Park L, Shin G, Hong H, Kang I, Park Y. Induction of apoptosis of Beta cells of the pancreas by advanced glycation end-products, important mediators of chronic complications of diabetes mellitus. Ann N Y Acad Sci 2008;1150:311–315.
- Sangle GV, Zhao R, Mizuno TM, Shen GX. Involvement of RAGE, NADPH oxidase, and Ras/Raf-1 pathway in glycated LDL-induced expression of heat shock factor-1 and plasminogen activator inhibitor-1 in vascular endothelial cells. Endocrinology 2010;151:4455–4466.
- Chung J, Nguyen AK, Henstridge DC, Holmes AG, Chan MH, Mesa JL, et al. HSP72 protects against obesity-induced insulin resistance. Proc Natl Acad Sci U S A 2008;105:1739–1744.
- Esterbauer H, Schaur RJ, Zollner H. Chemistry and biochemistry of 4-hydroxynonenal, malonaldehyde and related aldehydes. Free Radic Biol Med 1991;11:81–128.
- Vazdar M, Jurkiewicz P, Hof M, Jungwirth P, Cwiklik L. Behavior of 4-hydroxynonenal in phospholipid membranes. J Phys Chem B 2012;116:6411–6415.
- Mattson MP. Roles of the lipid peroxidation product 4-hydroxynonenal in obesity, the metabolic syndrome, and associated vascular and neurodegenerative disorders. Exp Gerontol 2009; 44:625–633.
- Jaganjac M, Matijevic T, Cindric M, Cipak A, Mrakovcic L, Gubisch W, Zarkovic N. Induction of CMV-1 promoter by 4-hydroxy-2-nonenal in human embryonic kidney cells. Acta Biochim Pol 2010;57:179–183.
- Jaganjac M, Poljak-Blazi M, Schaur RJ, Zarkovic K, Borovic S, Cipak A, et al. Elevated neutrophil elastase and acrolein-protein adducts are associated with W256 regression. Clin Exp Immunol 2012;170:178–185.
- Zarkovic N. 4-hydroxynonenal as a bioactive marker of pathophysiological processes. Mol Aspects Med 2003;24: 281–291.
- Go YM, Halvey PJ, Hansen JM, Reed M, Pohl J, Jones DP. Reactive aldehyde modification of thioredoxin-1 activates early steps of inflammation and cell adhesion. Am J Pathol 2007;171:1670–1681.
- Page S, Fischer C, Baumgartner B, Haas M, Kreusel U, Loidl G, et al. 4-Hydroxynonenal prevents NF-kappaB activation and tumor necrosis factor expression by inhibiting IkappaB phosphorylation and subsequent proteolysis. J Biol Chem 1999;274:11611–11618.
- Dickinson DA, Forman HJ. Cellular glutathione and thiols metabolism. Biochem Pharmacol 2002;64:1019–1026.
- Grimsrud PA, Xie H, Griffin TJ, Bernlohr DA. Oxidative stress and covalent modification of protein with bioactive aldehydes. J Biol Chem 2008;283:21837–21841.
- Demozay D, Mas JC, Rocchi S, Van Obberghen E. FALDH reverses the deleterious action of oxidative stress induced by lipid peroxidation product 4-hydroxynonenal on insulin signaling in 3T3-L1 adipocytes. Diabetes 2008;57:1216–1226.
- Ingram KH, Hill H, Moellering DR, Hill BG, Lara-Castro C, Newcomer B, et al. Skeletal muscle lipid peroxidation and insulin resistance in humans. J Clin Endocrinol Metab 2012; 97:1182–1186.
- Negre-Salvayre A, Auge N, Ayala V, Basaga H, Boada J, Brenke R, et al. Pathological aspects of lipid peroxidation. Free Radic Res 2010;44:1125–1171.
- Shen XP, Li J, Zou S, Wu HJ, Zhang Y. The relationship between oxidative stress and the levels of serum circulating adhesion molecules in patients with hyperglycemia crises. J Diabetes Complications 2012;26:291–295.
- Hoffman WH, Siedlak SL, Wang Y, Castellani RJ, Smith MA. Oxidative damage is present in the fatal brain edema of diabetic ketoacidosis. Brain Res 2011;1369: 194–202.
- Lupachyk S, Shevalye H, Maksimchyk Y, Drel VR, Obrosova IG. PARP inhibition alleviates diabetes-induced systemic oxidative stress and neural tissue 4-hydroxynonenal adduct accumulation: correlation with peripheral nerve function. Free Radic Biol Med 2011;50:1400–1409.
- Gleissner CA, Sanders JM, Nadler J, Ley K. Upregulation of aldose reductase during foam cell formation as possible link among diabetes, hyperlipidemia, and atherosclerosis. Arterioscler Thromb Vasc Biol 2008;28:1137–1143.
- Pennathur S, Ido Y, Heller JI, Byun J, Danda R, et al. Reactive carbonyls and polyunsaturated fatty acids produce a hydroxyl radical-like species: a potential pathway for oxidative damage of retinal proteins in diabetes. J Biol Chem 2005;280:22706–22714.
- Singh SP, Niemczyk M, Saini D, Awasthi YC, Zimniak L, Zimniak P. Role of the electrophilic lipid peroxidation product 4-hydroxynonenal in the development and maintenance of obesity in mice. Biochemistry 2008;47:3900–3911.
- Sohet FM, Neyrinck AM, Dewulf EM, Bindels LB, Portois L, Malaisse WJ, et al. Lipid peroxidation is not a prerequisite for the development of obesity and diabetes in high-fat-fed mice. Br J Nutr 2009;102:462–469.
- Dasuri K, Ebenezer P, Fernandez-Kim SO, Zhang L, Gao Z, Bruce-Keller AJ, et al. Role of physiological levels of 4-hydroxynonenal on adipocyte biology: implications for obesity and metabolic syndrome. Free Radic Res 2013; 47:8–19.
- Zarkovic N, Ilic Z, Jurin M, Scahur RJ, Puhl H, Esterbauer H. Stimulation of HeLa cell growth by physiological concentrations of 4-hydroxynonenal. Cell Biochem Funct 1993;11: 279–286.
- Zarkovic N, Schaur RJ, Puhl H, Jurin M, Esterbauer H. Mutual dependence of growth modifying effects of 4-hydroxy-nonenal and fetal calf serum in vitro. Free Radic Biol Med 1994;16: 877–884.
- Zarkovic N, Cipak A, Jaganjac M, Borovic S, Zarkovic K. Pathophysiological relevance of aldehydic protein modifications. J Proteomics 2013, in press doi:pii:S1874-3919(13)00066-3.10.1016/j.prot.2013.02.004
- Zivkovic M, Zarkovic K, Skrinjar LJ, Waeg G, Poljak-Blazi M, Borovic S, et al. A new method for detection of HNE-histidine conjugates in rat inflammatory cells. Croat Chem Acta 2005;78:91–98.
- Srividhya R, Zarkovic K, Stroser M, Waeg G, Zarkovic N, Kalaiselvi P. Mitochondrial alterations in aging rat brain: effective role of (2)- epigallo catechin gallate. Int J Develop Neurosci 2009;27:223–231.
- Casos K, Zaragoza MC, Zarkovic N, Zarkovic K, Andrisic L, Portero-Otín M, et al. A fish oil-rich diet reduces vascular oxidative stress in apoE-/- mice. Free Radic Res 2010;44:821–829.
- Augustyniak A, Bartosz G, Cipak A, Duburs G, Horakova L, Luczaj W, et al. Natural and synthetic antioxidants – an updated overview. Free Radic Res 2010;44:1216–1262.
- Vincent HK, Innes KE, Vincent KR. Oxidative stress and potential interventions to reduce oxidative stress in overweight and obesity. Diabetes Obes Metab 2007;9:813–839.
- Shevalye H, Lupachyk S, Watcho P, Stavniichuk R, Khazim K, Abboud HE, Obrosova IG. Prediabetic nephropathy as an early consequence of the high-calorie/high-fat diet: relation to oxidative stress. Endocrinology 2012;153:1152–1161.
- Torloni MR, Betrán AP, Horta BL, Nakamura MU, Atallah AN, Moron AF, Valente O. Prepregnancy BMI and the risk of gestational diabetes: a systematic review of the literature with meta-analysis. Obes Rev 2009;10:194–203.
- Weng H, Li X, Reece EA, Yang P. SOD1 suppresses maternal hyperglycemia-increased iNOS expression and consequent nitrosative stress in diabetic embryopathy. Am J Obstet Gynecol 2012;206:448.e1–7.
- Wagner KD, Wagner N. Peroxisome proliferator-activated receptor beta/delta (PPARbeta/delta) acts as regulator of metabolism linked to multiple cellular functions. Pharmacol Ther 2010;125:423–435.
- Kocalis HE, Turney MK, Printz RL, Laryea GN, Muglia LJ, Davies SS, et al. Neuron-specific deletion of peroxisome proliferator-activated receptor delta (PPARdelta) in mice leads to increased susceptibility to diet-induced obesity. PLoS One 2012;7:e42981.
- Salvado L, Serrano-Marco L, Barroso E, Palomer X, Vazquez-Carrera M. Targeting PPARbeta/delta for the treatment of type 2 diabetes mellitus. Expert Opin Ther Targets 2012;16:209–223.
- Lee CH, Olson P, Hevener A, Mehl I, Chong LW, Olefsky JM, et al. PPARdelta regulates glucose metabolism and insulin sensitivity. Proc Natl Acad Sci U S A 2006;103:3444–3449.
- Tanaka T, Yamamoto J, Iwasaki S, Asaba H, Hamura H, Ikeda Y, et al. Activation of peroxisome proliferator-activated receptor delta induces fatty acid beta-oxidation in skeletal muscle and attenuates metabolic syndrome. Proc Natl Acad Sci U S A 2003;100:15924–15929.
- Wang YX, Lee CH, Tiep S, Yu RT, Ham J, Kang H, Evans RM. Peroxisome-proliferator-activated receptor delta activates fat metabolism to prevent obesity. Cell 2003;113: 159–170.
- Oliver WR Jr, Shenk JL, Snaith MR, Russell CS, Plunket KD, Bodkin NL, et al. A selective peroxisome proliferator- activated receptor delta agonist promotes reverse cholesterol transport. Proc Natl Acad Sci U S A 2001;98:5306–5311.
- Mottillo EP, Bloch AE, Leff T, Granneman JG. Lipolytic products activate peroxisome proliferator-activated receptor (PPAR) alpha and delta in brown adipocytes to match fatty acid oxidation with supply. J Biol Chem 2012;287: 25038–25048.
- Naruhn S, Meissner W, Adhikary T, Kaddatz K, Klein T, Watzer B, et al. 15-hydroxyeicosatetraenoic acid is a preferential peroxisome proliferator-activated receptor beta/delta agonist. Mol Pharmacol 2010;77:171–184.
- Yu Z, Schneider C, Boeglin WE, Brash AR. Epidermal lipoxygenase products of the hepoxilin pathway selectively activate the nuclear receptor PPARalpha. Lipids 2007;42:491–497.
- Xu HE, Lambert MH, Montana VG, Parks DJ, Blanchard SG, Brown PJ, et al. Molecular recognition of fatty acids by peroxisome proliferator-activated receptors. Mol Cell 1999;3: 397–403.
- Lamers C, Schubert-Zsilavecz M, Merk D. Therapeutic modulators of peroxisome proliferator-activated receptors (PPAR): a patent review (2008-present). Expert Opin Ther Pat 2012;22:803–841.
- Maltarollo VG, Honorio KM. Ligand- and structure-based drug design strategies and PPARdelta/alpha selectivity. Chem Biol Drug Des 2012;80:533–544.
- Batista FA, Trivella DB, Bernardes A, Gratieri J, Oliveira PS, Figueira AC, et al. Structural insights into human peroxisome proliferator activated receptor delta (PPAR-delta) selective ligand binding. PLoS One 2012;7:e33643.
- Shearer BG, Steger DJ, Way JM, Stanley TB, Lobe DC, Grillot DA, et al. Identification and characterization of a selective peroxisome proliferator-activated receptor beta/delta (NR1C2) antagonist. Mol Endocrinol 2008;22:523–529.
- Sznaidman ML, Haffner CD, Maloney PR, Fivush A, Chao E, Goreham D, et al. Novel selective small molecule agonists for peroxisome proliferator-activated receptor delta (PPARdelta) – synthesis and biological activity. Bioorg Med Chem Lett 2003;13:1517–1521.
- Ravnskjaer K, Frigerio F, Boergesen M, Nielsen T, Maechler P, Mandrup S. PPARdelta is a fatty acid sensor that enhances mitochondrial oxidation in insulin-secreting cells and protects against fatty acid-induced dysfunction. J Lipid Res 2010;51:1370–1379.
- Riahi Y, Sin-Malia Y, Cohen G, Alpert E, Gruzman A, Eckel J, et al. The natural protective mechanism against hyperglycemia in vascular endothelial cells: roles of the lipid peroxidation product 4-hydroxydodecadienal and peroxisome proliferator- activated receptor delta. Diabetes 2010;59:808–818.
- Cohen G, Riahi Y, Shamni O, Guichardant M, Chatgilialoglu C, Ferreri C, Kaiser N, Sasson S. Role of lipid peroxidation and PPAR-delta in amplifying glucose-stimulated insulin secretion. Diabetes 2011;60:2830–2842.
- Cohen G, Riahi Y, Sasson S. Lipid peroxidation of poly-unsaturated fatty acids in normal and obese adipose tissues. Arch Physiol Biochem 2011;117:131–139.
- Riahi Y, Cohen G, Shamni O, Sasson S. Signaling and cytotoxic functions of 4-hydroxyalkenals. Am J Physiol Endocrinol Metab 2010;299:E879–E886.
- Aldini G, Orioli M, Rossoni G, Savi F, Braidotti P, Vistoli G, et al. The carbonyl scavenger carnosine ameliorates dyslipidaemia and renal function in Zucker obese rats. J Cell Mol Med 2011;15:1339–1354.
- Sauerhöfer S, Yuan G, Braun GS, Deinzer M, Neumaier M, Gretz N, et al. L-carnosine, a substrate of carnosinase-1, influences glucose metabolism. Diabetes 2007;56:2425–32.
- Poli G, Schaur RJ, Siems WG, Leonarduzzi G. 4-hydroxynonenal: a membrane lipid oxidation product of medicinal interest. Med Res Rev 2008;28:569–631.
- Parola M, Bellomo G, Robino G, Barrera G, Dianzani MU. 4-Hydroxynonenal as a biological signal: molecular basis and pathophysiological implications. Antioxid Redox Signal 1999; 1:255–284.
- Usatyuk PV, Natarajan V. Hydroxyalkenals and oxidized phospholipids modulation of endothelial cytoskeleton, focal adhesion and adherens junction proteins in regulating endothelial barrier function. Microvasc Res 2012;83: 45–55.
- Leonarduzzi G, Chiarpotto E, Biasi F, Poli G. 4-Hydroxynonenal and cholesterol oxidation products in atherosclerosis. Mol Nutr Food Res 2005;49:1044–1049.