
Abstract
Background: Methylmalonic aciduria and homocystinuria type C (cblC), a disorder of vitamin B12 (cobalamin) metabolism caused by mutations in the MMACHC gene, presents with many systemic symptoms, including neurological, cognitive, psychiatric, and thromboembolic events. Retinal phenotypes, including maculopathy, pigmentary retinopathy, and optic atrophy are common in early onset form of the disease but are rare in adult onset forms.
Materials and Methods: An adult Hispanic female presented with decreased central vision, bilateral pericentral ring scotomas and bull’s eye-appearing macular lesions at 28 years of age. Her medical history was otherwise unremarkable except for iron deficiency anemia and both urinary tract and kidney infections. Screening of the ABCA4 gene, mutations in which frequently cause bull’s eye maculopathy, was negative. Subsequently, analysis with whole exome sequencing was performed.
Results: Whole exome sequencing discovered compound heterozygous mutations in MMACHC, c.G482A:p.Arg161Gln and c.270_271insA:p.Arg91Lysfs*14, which segregated with the disease in the family. The genetic diagnosis was confirmed by biochemical laboratory testing, showing highly elevated urine methylmalonic acid/creatinine and homocysteine levels, and suggesting disease management with hydroxycobalamin injections and carnitine supplementation.
Conclusions: In summary, a unique case of an adult patient with bull’s eye macular lesions and no clinically relevant systemic symptoms was diagnosed with cblC by genetic screening and follow-up biochemical laboratory tests.
INTRODUCTION
Methylmalonic aciduria and homocystinuria type C (cblC; OMIM #277400) is a recessively inherited disorder of vitamin B12 (cobalamin) metabolism,Citation1 which is caused by mutations in the MMACHC gene.Citation2 Maculopathy (including bull’s eye maculopathy), pigmentary retinopathy, and optic atrophy are all common in early-onset disease.Citation3 Conversely, ocular findings are rare,Citation4,Citation5 in the “late-onset” type, where systemic symptoms become apparent after age 4.Citation1 Although optic atrophy and peripheral retinal pigmentary changes have been noted in late-onset cblC diseaseCitation4,Citation6 a maculopathy has not been described.Citation1 Patients with the late-onset form present with a variety of systemic deficits, including neurological complaints, cognitive decline, psychiatric disturbances, thromboembolic events, and gait disturbances.Citation4,Citation7 Some of the late-onset cblC cases also present retinal changes, but no late-onset cblC case has been described presenting without systemic symptoms but with a retinal degenerative phenotype.Citation1
MATERIALS AND METHODS
Case Report
A 28-year-old Hispanic female () presented in 2006 with decreased central vision and mild photoaversion of approximately two years duration. She reported subjectively normal color vision, night vision and peripheral vision. Her medical history was positive for iron deficiency anemia, occasional urinary tract infections, and recurrent kidney infections in 2002. Best corrected Snellen visual acuities (BCVA) were 20/80 OD and 20/70−2 OS with a low myopic refraction. She correctly read only two out of twelve Ishihara color plates OD, and zero out of twelve OS. Goldmann visual field testing demonstrated a pericentral ring scotoma in each eye (). Funduscopy showed bilateral bull’s eye-appearing macular lesions () with no visible fundus flecks, vessel attenuation, optic nerve pallor or peripheral pigmentary changes. A scotopic and photopic full-field electroretinogram (ERG) of the right eye showed normal a- and b-wave amplitudes and implicit times. The patient reported no current or previous use of hydroxychloroquine or any other medication or toxic exposure that could be potentially responsible for the bull’s eye-appearing lesions in her maculae. The patient reported no family history of a retinal dystrophy, and examination of both parents and a brother () showed essentially normal maculae; only a few fine drusen near the fovea were observed in the examinations of both parents.
FIGURE 1. Pedigree of the family and segregation of the MMACHC mutations with the disease phenotype. The open circle and squares represent the unaffected female and male family members, respectively; the closed circle represents the affected female patient.
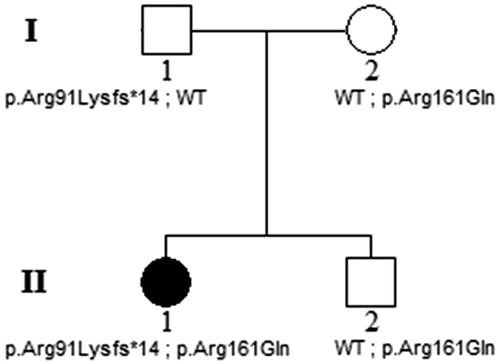
FIGURE 2. Goldmann visual fields of the left (A) and right (B) eyes from the initial visit in 2006, showing ring scotomas consistent with the bull’s eye-appearing macular lesions. Shaded areas indicate the target was not seen in that area. Fundus photos from 2006 (C, right eye and D, left eye) and 2013 (E and F) show some extension of RPE atrophy, apparent at the peripheral boundaries, as well as in the foveal region, in both eyes over 7 years. Near-infrared autofluorescence (G and H) from the 2013 follow-up visit shows hyper-autofluorescence at the fovea, surrounded by hypo-autofluorescence (bull’s eye), further surrounded by a ring of hyper-autofluorescence.
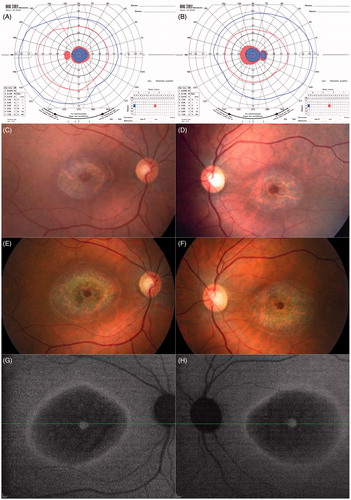
By the most recent visit in 2013 at age 35, the patient was still working at her job as a production planner, although her BCVA had dropped to 10/100−2 OD and 10/80 OS. Fundus photos (), and near-infrared autofluorescence () demonstrated bull’s eye macular dystrophy with relative preservation of the retinal pigment epithelium (RPE) at the foveolar region. Spectral-domain optical coherence tomography also showed preservation of the RPE at the fovea in the presence of marked foveal thinning (). However, the patient’s eccentric viewing and level of visual acuity evidenced a notable degree of compromised foveal function. A repeat full field ERG () of the right eye at that visit still showed amplitudes and implicit times that were within normal limits under both scotopic rod isolated and photopic cone isolated test conditions, whereas the scotopic rod and cone combined response b-wave was mildly reduced and delayed compared to normal.
FIGURE 3. Spectral domain optical coherence tomography line scans (right eye top and left eye bottom, corresponding to the location of the horizontal lines on the near-infrared autofluorescence images in , respectively) show intact RPE at the foveola in the presence of marked foveal thinning, surrounded by atrophy of the outer retinal layers and retinal pigment epithelium, with relatively more normal retinal structure at the margins of the line scans in both eyes.
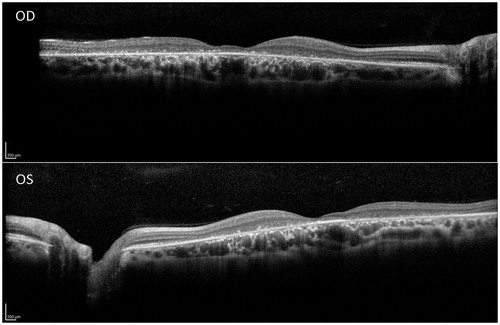
FIGURE 4. Full-field electroretinogram (Espion E3, Diagnosys LLC, Littleton, MA) of the patient’s right eye at age 35 during a follow-up visit. Boxes represent ranges of 13 visually-normal subjects. The cone isolated amplitudes and implicit times (top), and the rod isolated single flash response (bottom left) are within the normal range. The combined rod and cone single flash response (bottom right) is mildly reduced and delayed compared to normal.
Genetic Analysis
Since the bull’s eye maculopathy phenotype is frequent in patients with diseases caused by mutations in the ABCA4 gene,Citation8,Citation9 the patient was first screened for mutations in ABCA4 by direct sequencing with negative results. Consequently, all members of the family were subjected to whole exome sequencing (WES) at the Baylor College of Medicine Human Genome Sequencing Center as part of the Baylor-Hopkins Center for Mendelian Genomics. Exome capture was performed on the NimbleGen array with minor revisions, and sequencing was performed on the Illumina platform. Sequence analysis was performed using the HGSC Mercury analysis pipeline. Samples achieved an average of 90% of the targeted exome bases covered to a depth of 20X or greater. Possible disease-associated variants were determined by filtering based on minor allele frequency, predicted pathogenicity and by segregation analysis in all family members.
RESULTS
Screening of the ABCA4 gene on an array revealed no disease-associated variants. WES analysis uncovered two compound heterozygous mutations, c.G482A:p.Arg161Gln and c.270_271insA:p.Arg91Lysfs*14, in the MMACHC gene in the proband, which segregated with the disease in the family (). Sanger sequencing confirmed compound heterozygous mutant alleles in the proband and heterozygous alleles in carrier parents again documenting trans mutations in the patient. Both of these mutations have been frequently described as causes of cblC (http://omim.org/entry/609831).
Consequently, based upon the molecular diagnosis established by genetic studies, the patient was referred for biochemical laboratory testing, which showed a urine methylmalonic acid/creatinine level of 510 mmol/mol creatinine (reference range 0–3.60) and a urine homocysteine level of 51 mg/day (reference range 0–32). The patient was then referred for disease management by a pediatric genetics specialist. Physical examination at that visit was normal; specifically, no focal neurological abnormalities were found. She was subsequently prescribed intramuscular hydroxycobalamin (vitamin B12) injections, as well as carnitine supplementation due to low carnitine levels.
DISCUSSION
To our knowledge, no cases with ocular findings as the only presenting sign in an adult patient with methylmalonic aciduria and homocystinuria type C have been described before. In addition, maculopathy has not been described in late-onset cblC disease.Citation10 It is possible that subtle, sub-clinical bull’s eye macular changes have been missed in previously reported cases of late-onset cblC disease if sensitive tests such as SD-OCT, autofluorescence imaging or multifocal ERG were not performed.
The late-onset form of cblC is usually discovered due to neurological complaints and cognitive decline. The history of iron deficiency anemia and urinary tract infections in our case has been described in other cases of late-onset cblC disease,Citation11 although these are not unusual findings in an otherwise healthy adult female. Phenotypes associated with the two disease-causing mutations in the MMACHC gene, c.G482A:p.Arg161Gln and c.270_271insA:p.Arg91Lysfs*14, have been previously characterized. The c.270_271insA:p.Arg91Lysfs*14 variant is the most common MMACHC mutation (>40% of all disease-associated alleles), and is associated with a more severe, early-onset phenotype especially when found in homozygosity.Citation2,Citation12 The c.G482A:p.Arg161Gln mutation is more often associated with the milder, late-onset phenotype.Citation2,Citation11–13
Interestingly, the exact same combination of mutations has been described before in at least six cases with teenage or adult onset of cblC.Citation11–14,Citation15 All cases, however, presented with severe systemic symptoms, often including muscular abnormalities in lower extremities, loss of bowel and/or bladder function, progressive encephalopathy, neurological anomalies, and thromboembolic complications. No degenerative retinal changes were described in any of these patients.Citation13,Citation14 Patients who were compound heterozygous for the c.G482A:p.Arg161Gln and c.270_271insA:p.Arg91Lysfs*14 variants had variable ethnic origin, including Italian, German, Hispanic, and Chinese descent.
Perhaps the most relevant study with regards to the patient described in this report is the description of a patient with late-onset cblC presented by Tsai and colleagues,Citation11 since that patient matched the case described here by age, gender, and Hispanic ethnicity. However, in that case a 36-year-old woman had a constellation of systemic features including joint hypermobility, arthritis, chronic anemia, urogenital fistula, and spinal cord infarct. She also presented with emotional difficulties beginning in her teens and was diagnosed with depression and psychosis requiring hospitalization.Citation11 Bilateral cataracts were the only ocular findings described. Conversely, the patient described here, although matched by age, ethnicity, and gender, did not report any systemic abnormalities, other than iron deficiency anemia and urinary tract infections, but instead presented with an ocular finding (bull’s eye maculopathy) as a primary finding.
More macular dystrophy patients with mutations in the MMACHC gene will need to be identified before cblC becomes a serious consideration for a disease underlying a non-syndromic macular dystrophy. However, patients with bull’s eye-appearing macular lesions may be questioned about systemic symptoms, possibly prompting testing for methylmalonic acid and homocysteine levels. Even with overt neurological symptoms, the diagnosis of late-onset cblC disease can be easily missed or delayed,Citation7,Citation16 but urine or plasma markers of the disease can be easily detected with simple and inexpensive clinical biochemical laboratory tests.
The implementation of whole exome and whole genome sequencing has allowed identification of causes of metabolic diseases which previously have remained obscure.Citation17 These include new genes, including those for methylmalonic aciduria,Citation18 and many cases of phenotypic expansion of known loci; i.e. where the clinical phenotype has not directed the screening of underlying genetic causes.Citation19 Importantly, as shown here, clinical phenotype can be very divergent from those described, and genetic testing can unequivocally determine the cause of a disease. Screening of the MMACHC gene is an accessible option for confirmation of cblC disease. An early diagnosis in patients with cblC disease has added importance because often systemic symptoms can not only improve,Citation7,Citation16 but also may be prevented, delayed, or ameliorated with hydroxycobalamin treatment.Citation1
In conclusion, a patient with bull’s eye macular lesions and no neurological, psychiatric or cognitive symptoms was determined to have cblC, a potentially debilitating disorder if left untreated. Due to the precise molecular diagnosis, achieved through genetic studies and confirmed by follow-up biochemical laboratory tests, she was treated for her condition, thereby possibly improving her systemic prognosis.
Declaration of interest
The authors report no conflicts of interest. The authors alone are responsible for the content and writing of the paper.
This study is supported in part by NIH grants EY021163, EY019861, HG006542 and EY019007 (Core Support for Vision Research), by unrestricted funds from Research to Prevent Blindness (New York, NY) to the Department of Ophthalmology, Columbia University, and by the Pangere Family Corporation, The Chicago Lighthouse for People Who Are Blind or Visually Impaired (Chicago, IL).
References
- Carrillo-Carrasco N, Chandler RJ, Venditti CP. Combined methylmalonic acidemia and homocystinuria, cblC type. I. Clinical presentations, diagnosis and management. J Inherit Metab Dis 2012;35:91–102
- Lerner-Ellis JP, Tirone JC, Pawelek PD, et al. Identification of the gene responsible for methylmalonic aciduria and homocystinuria, cblC type. Nat Genet 2006;38:93–100
- Gizicki R, Robert MC, Gomez-Lopez L, et al. Long-term visual outcome of methylmalonic aciduria and homocystinuria, cobalamin C type. Ophthalmology 2014;121:381–386
- Thauvin-Robinet C, Roze E, Couvreur G, et al. The adolescent and adult form of cobalamin C disease: clinical and molecular spectrum. J Neurol Neurosurg Psychiatry 2008;79:725–728
- Gerth C, Morel CF, Feigenbaum A, et al. Ocular phenotype in patients with methylmalonic aciduria and homocystinuria, cobalamin C type. J AAPOS 2008;12:591–596
- Van Hove JL, Van Damme-Lombaerts R, Grunewald S, et al. Cobalamin disorder Cbl-C presenting with late-onset thrombotic microangiopathy. Am J Med Genet A 2002;111:195–201
- Ben-Omran TI, Wong H, Blaser S, et al. Late-onset cobalamin-C disorder: a challenging diagnosis. Am J Med Genet A 2007;143A:979–984
- Allikmets R, Singh N, Sun H, et al. A photoreceptor cell-specific ATP-binding transporter gene (ABCR) is mutated in recessive Stargardt macular dystrophy. Nat Genet 1997;15:236–246
- Michaelides M, Chen LL, Brantley MA Jr., et al. ABCA4 mutations and discordant ABCA4 alleles in patients and siblings with bull’s-eye maculopathy. Br J Ophthalmol 2007;91:1650–1655
- Carrillo-Carrasco N, Venditti CP. Combined methylmalonic acidemia and homocystinuria, cblC type. II. Complications, pathophysiology, and outcomes. J Inherit Metab Dis 2012;35:103–114
- Tsai AC, Morel CF, Scharer G, et al. Late-onset combined homocystinuria and methylmalonic aciduria (cblC) and neuropsychiatric disturbance. Am J Med Genet A 2007;143A:2430–2434
- Lerner-Ellis JP, Anastasio N, Liu J, et al. Spectrum of mutations in MMACHC, allelic expression, and evidence for genotype-phenotype correlations. Hum Mutat 2009;30:1072–1081
- Morel CF, Lerner-Ellis JP, Rosenblatt DS. Combined methylmalonic aciduria and homocystinuria (cblC): phenotype-genotype correlations and ethnic-specific observations. Mol Genet Metab 2006;88:315–321
- Bodamer OA, Rosenblatt DS, Appel SH, et al. Adult-onset combined methylmalonic aciduria and homocystinuria (cblC). Neurology 2001;56:1113
- Nogueira C, Aiello C, Cerone R, et al. Spectrum of MMACHC mutations in Italian and Portuguese patients with combined methylmalonic aciduria and homocystinuria, cblC type. Mol Genet Metab 2008;93:475–480
- Wang X, Sun W, Yang Y, et al. A clinical and gene analysis of late-onset combined methylmalonic aciduria and homocystinuria, cblC type, in China. J Neurol Sci 2012;318:155–159
- Yang Y, Muzny DM, Reid JG, et al. Clinical whole-exome sequencing for the diagnosis of mendelian disorders. N Engl J Med 2013;369:1502–1511
- Sloan JL, Johnston JJ, Manoli I, et al. Exome sequencing identifies ACSF3 as a cause of combined malonic and methylmalonic aciduria. Nat Genet 2011;43:883–886
- Bainbridge MN, Wiszniewski W, Murdock DR, et al. Whole-genome sequencing for optimized patient management. Sci Transl Med 2011;3:87re83