Abstract
Context: Chamaecyparis obtusa Sieb. & Zucc., Endlicher (Cupressaceae) forest bathing or aromatherapy has been shown in various studies to have biological functions such as anticancer, antiallergies, antiinflammatory, and antioxidant activity. However, no reports exist on the pharmacological or biological activities of the essential oil of C. obtusa (EOCO) or its effects on central nervous system.
Objective: The aggregation and formation of β-amyloid peptides (Aβ) into fibrils are central events in the pathogenesis of Alzheimer’s disease (AD), and overproduction and aggregation of Aβ into oligomers have been known to trigger neurotoxicity. In this study, we investigated the effects of inhaled EOCO on cognitive function and neuronal apoptosis in rats intrahippocampally injected with Aβ.
Materials and methods: To model AD, 4 μg of aggregated Aβ was injected into the hippocampus. To test the effects of EOCO, behavioral performance in the Morris water maze was tested 4 days after injection. After behavioral testing, brain sections were prepared for TTC staining and TUNEL assay.
Results: Inhaled EOCO protected spatial learning and memory from the impairments induced by Aβ1–40 injection. In addition, the behavioral deficits accompanying Aβ1–40-induced AD were attenuated by inhalation of EOCO. Furthermore, acetylcholinesterase (AChE) activity and neuronal apoptosis were significantly inhibited in rats treated with Aβ1–40 and EOCO compared to rats treated only with Aβ1–40.
Discussion and conclusion: EOCO suppressed both AD-related neuronal cell apoptosis and AD-related dysfunction of the memory system. Thus, the results of this study support EOCO as a candidate drug for the treatment of AD.
Introduction
Essential oils have been used for thousands of years in the treatment of various diseases, such as depression, pain, and cancer (CitationGraeff, 1993; CitationAloisi et al., 2002; CitationKomiya et al., 2006). Clinical trials have demonstrated that essential oils can improve sleep (CitationGraham et al., 2003), act as anticonvulsants (CitationYamada et al., 1994), and have other useful functions when inhaled. The essential oil of Chamaecyparis obtusa Sieb. & Zucc., Endlicher (Cupressaceae) is a volatile compound with natural antibiotic properties that protect against harmful insects, animals, and microorganisms. Inhalation with the essential oil of C. obtusa (EOCO) is known as C. obtusa forest bathing or C. obtusa aromatherapy. Chemical and pharmacological studies have shown that terpenoids, the major components of EOCO, are the active components that exert antigastropathic, antiinflammatory, and antioxidant activity. These activities of terpedoids were verified by inhibiting information of toxic lipid peroxide, recovering antioxidant enzyme such as superoxide dismutase (SOD) activity in other plants in vivo (CitationDurham et al., 1994; CitationChoi et al., 2003; CitationNam et al., 2006). Furthermore, it has been reported that the physiological effects of phytoncides contribute to the improvement of symptoms of various disorders, including accelerated aging, allergies, multiple sclerosis, and cancer (CitationNose et al., 2000; CitationOikawa et al., 2005). Recently, it was shown that the addition of phytoncides potently decreases stress hormone levels and increases natural killer (NK) cell activity and neurite outgrowth (CitationLi et al., 2006, Citation2009). These results raise the possibility that EOCO may be therapeutically useful in blocking the primary cause of neuronal death and memory impairment associated with neurodegenerative diseases. However, no reports exist on the pharmacological or biological activities of EOCO or its effects on learning and memory in rat models of amnesia and/or dementia.
Alzheimer’s disease (AD) is an age-related neurodegenerative disorder that is recognized as the most common form of dementia. It is clinically associated with cognitive impairment, loss of language and motor skills as well as changes in behavior (CitationHuang & Jiang, 2009). Furthermore, AD is well established to be associated with selective lesions of neuronal circuits in the neocortex, hippocampus, and basal forebrain cholinergic system. In this progressive dementia disorder, the accumulation of large amyloid plaques, mostly consisting of Aβ1–40 and Aβ1–42, has been found in the brain at autopsy. The extent of Aβ accumulation correlates with the progression of cognitive deficits and memory impairment, which prompted the suggestion that Aβ may play a significant role in memory degeneration in AD patients (CitationSchwarting et al., 1991; CitationKoudinov et al., 2009). The enzyme choline acetyltransferase (ChAT) is necessary for acetylcholine synthesis, and ChAT levels are significantly reduced in patients with AD (CitationPákáski et al., 2008). Moreover, addition of Aβ has been shown to potently suppress acetylcholine (ACh) release induced by high potassium in the hippocampus and cortex (CitationWatanabe et al., 2009). Therefore, selective suppression of cholinergic transmission by Aβ in the hippocampus and cortex could be part of the mechanism through which amyloid peptides elicit their neurodegenerative effect.
The current study is the first report of the effects of inhaled EOCO on Aβ-induced memory impairment in rats. We also examined whether inhalation of EOCO affects Aβ-induced cholinergic dysfunction and neuronal apoptosis. Our findings may provide a basis for preventative treatment of AD with EOCO.
Materials and methods
Plant materials
C. obtuse, fresh leaves were collected from the Wood-land, Jangheung, Jeollanamdo, South Korea in July 2010 and authenticated by Dr. Kim at Jeollanamdo Institute of Natural Resources Research (JINR), Jangheung, Jeollanamdo, South Korea, where an experimental sample had been deposited.
Reagents
Aβ1–40 was obtained from GL Biochem (Shanghai, China). 2,3,5-Triphenyl-terazoliumchloride, ACh, hydroxylamine, and dithionitrobenzoic acid were purchased from Sigma Chemical (St. Louis, MO). Donepezil was obtained from Eisai (Tokyo, Japan). All other reagents were of analytical grade.
Gas chromatography and mass spectrometry analysis
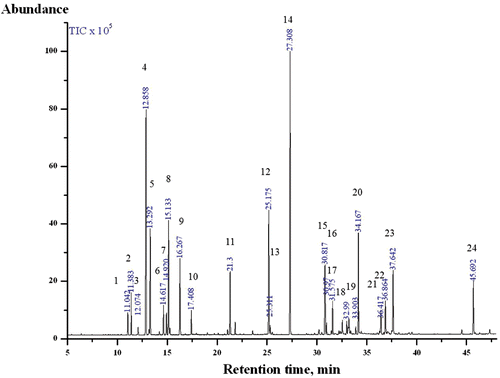
EOCO was produced by the water-vapor distillation method. The essential oil was analyzed by gas chromatography (GC)/mass spectrometry (MS). The GC (GC-2010, Shimadzu Corporation, Japan) was equipped with a DB-5MS column (30 m × 0.25 mm i.d., 0.25 μm film thickness, Agilent Technology, USA) and a split injection port (100:1). The injector and detector temperatures were maintained at 250°C and 280°C, respectively. Helium was used as the carrier gas at a flow rate of 1.65 mL/min. The initial oven temperature was maintained at 50°C for 5 min. The temperature was then raised to 100°C at a rate of 10°C/min, then to 280°C at a rate of 5°C/min and finally held at that temperature for 5 min. The electron ionization mode was used for MS (GCMS-QP 2010 Plus, Shimadzu Corporation, Japan), and the conditions were as follows: electron energy, 70 eV; scan range, 28–550 amu; and source temperature, 200°C. The chemical structure of each constituent was identified by comparing mass data with Wiley 8N Library data. The EOCO used in this study was standardized systemically. shows the chromatogram of EOCO.
Figure 1. Chromatogram of EOCO. 1) α-thujene; 2) α-pinene; 3) camphene; 4) β-phellandrene; 5) β-myrcene; 6) α-terpinene; 7) ρ-cymene; 8) limonene; 9) γ-terpinene; 10) α-terpinolene; 11) α-terpineol; 12) bornyl ester; 13) 2-bornyl aetate; 14) α-terpinyl acetate; 15) thujopsene; 16) (+)-δ-cadinene; 17) (+)-epi-bicyclosesquiphellandrene; 18) (+)-ρ-himachalene; 19) (+)-α-longipinene; 20) elemol; 21) α-cedrol; 22) γ-eudesmol; 23) β-eudesmol; 24) beyerene. (Compounds occupying more than 0.4% peak area are shown.)
Animals
Male Sprague-Dawley rats (weight range: 220–240 g) were purchased from the Central Lab Animal Inc. (Seoul, Korea) and housed seven per cage in a light-controlled room (lights on from 8:00 AM to 8:00 PM) at a temperature of 22 ± 2°C and humidity of 50 ± 5% with food and water available ad libitum. All experimental procedures were conducted in accordance with guidelines relevant to the care of experimental animals, as approved by the Jeollanamdo Institute for Natural Resources.
Inhalation and groups
Inhalation of EOCO was performed using a vaporization system cage (VSC), which allowed quantitation of the volume of EOCO inhaled to the rats. The VSC was made of acrylate board (50 cm × 60 cm × 50 cm) and was equipped with an electric fan that injected oxygen and vaporized and distributed EOCO. Rats were randomly assigned into one of four groups: the vehicle-injection plus saline-administered group (control group), the Aβ-injection plus saline administered group (Aβ1–40 group), the Aβ-injection plus donepezil-administered group (Donepezil group), and Aβ-injection plus EOCO-inhalation group (EOCO group). Donepezil (1.5 mg/kg bw/day) was administered intragastrically once daily for 7 days after injection of Aβ into the hippocampus. EOCO (1 mL/cage) was inhaled in the VSC for two consecutive hours for 30 days before injection of Aβ into the hippocampus and 7 days after injection.
Aβ1–40 preparation
Aβ1–40 was initially dissolved in a small volume of DMSO, and diluted to a final stock concentration of 1 μg/μL in artificial cerebrospinal fluid (ACSF: 145 mM NaCl, 2.7 mM KCl, 1.2 mM CaCl2, 1.0 mM MgCl2) and aggregated at 37°C for 5 days prior to use (CitationEkinci et al., 1999; CitationKim et al., 2011).
Aβ1–40-induced AD rat model
The rats were anesthetized with isoflurane (3%, i.a.), and placed in a stereotaxic instrument (Harvard, USA). An incision was made on the scalp of each rat, and the skull was adjusted to position bregma and lambda on the same horizontal plane. Small burr holes were drilled, and Aβ1–40 (4 μg) or vehicle was injected into the hippocampi (−3.0 mm, 2.2 mm, −2.8 mm) at a rate of 1 μL/min with a 5 μL Hamilton syringe. The hole was then covered with dental acrylic cement and the scalp was closed with sutures. After surgery, each rat was injected with penicillin in the hindquarter muscle (100,000 U) and individually housed after the operation. All behavioral tests were started at day 2 after surgery in unanesthetized freely moving rats.
Morris water maze test
The water maze test was performed according to standard methods with some modification (CitationMorris, 1984) in a circular pool (diameter: 180 cm, height: 75 cm) filled to a depth of 25 ± 1 cm. The transparent platform (24 cm) was submerged approximately 1 cm below the surface of the water, and nonfat milk was added to the water to make it opaque. The first day of the experiment was a swim training day during which rats swam for 60 s in the absence of the platform. On each day following the first training day, the rats were given four sessions, with an intertrial interval of 5 min. The starting point changed for each session, but the location of the platform was fixed during the entire test period. The time from being placed in the pool to reaching the platform was measured. On the day after the last training session, each rat was subjected to a probe trial (90 s) in which no platform was present. The time spent in the target quadrant (the quadrant in which the platform had previously been located) was taken as a measure of spatial memory retention. All data were collected and analyzed using Smart video tracking system (Panlab, USA).
Passive avoidance test
The rats were tested for memory retention deficits using a passive avoidance apparatus (Iwoo Scientific Co, Seoul, Korea). The apparatus consisted of a two-compartment dark/light shuttle box with a guillotine door separating the compartments. The dark compartment had a stainless steel shock grid floor. During the acquisition trial, each rat was placed in the light chamber. After a 60 s habituation period, the guillotine door was opened, and the latency for the animal to enter the dark chamber was recorded, and termed initial latency. Rats with an initial latency of greater than 60 s were excluded from further experiments. Immediately after the rat had entered the dark chamber, the guillotine door was closed and an electric foot shock (75 V, 0.2 mA, 50 Hz) was delivered through the floor grid with a stimulator for 3 s. Five seconds later, the rat was removed from the dark chamber and returned to its home cage. Eight hours later, this test was repeated and the latency to enter the dark chamber was recorded. Latency in this test was termed retention latency.
Vertical pole test
The vertical pole test was performed as previously described (CitationOgawa et al., 1985) with minor modifications. Rats were tested for measuring grip strength and sensorimotor performance on a vertical pole (2 cm in diameter, 60 cm high). Animals were placed on the center of the pole, which was fixed in vertical position. Animals with deficits in grip strength and sensorimotor performance fall off the pole. Rats were habituated to the task in two trials per day for 2 days. On the testing day (third day), three measures were taken over three trials per rat.
Rotarod test
Performance on a rotarod task (five-lane accelerating rotarod; Jeung Do Bio & Plant, Seoul, Korea) was used to measure motor balance and coordination. Rats were placed on a horizontal rotating rod (diameter, 8 cm; rotation speed, 20 rpm) and were left on the rod for 5 min or until they fell off. Falling off the rod activated a switch that automatically stopped a timer. Five rats separated by large disks were tested simultaneously. On the testing day, each rat was submitted to three trials with an intertrial interval of 10 min.
ACh content and AchE activity assays
ACh content was determined according to the method of CitationVincent et al. (1958). Aliquots (20 μL) of brain homogenate and 50 μL of 1% hydroxylamine were added to a 96-well multiplate, incubated for 15 min at 25°C, and then 250 μL FeCl3 (in 0.1 N HCl, pH 1.2 ± 0.2) was added. Absorbance was read at 540 nm and calibrated with a blank.
AchE activity was determined by the colorimetric method described by CitationEllman et al. (1961). DTNB (0.01 mM, 20 μL) and 10 μL of brain homogenate (Crude enzyme) in 0.1 mM sodium phosphate buffer (pH 8.0) were added to a 96-well multiplate and incubated for 5 min at 25°C. The reaction was then initiated by adding 10 μL of 0.1 M acetylthiocholine. The hydrolysis of acetylthiocholine was monitored by the formation of yellow 2-nitro-5-sulfidobenzene-carboxylate anion as the result of the reaction of DTNB with thiocholine (released by enzymatic hydrolysis of acetylthiocholine) for 10 min, at a wavelength of 412 nm.
TTC staining
At the end of behavioral evaluation, rats were sacrificed for 2,3,5-triphenyltetrazolium chloride (TTC) staining. Brains were quickly removed and placed in ice-cold saline solution then sectioned at 2 mm intervals using rat brain matrix. Slices were submerged in 2% TTC for 5 min at 37°C in the dark then removed and placed in 4% paraformaldehyde, pH 7.4 in 0.1 M phosphate buffer.
TUNEL staining
Rats were anesthetized and transcardially perfusion fixed with 4% formaldehyde in 0.1 M sodium phosphate buffer (pH 7.4). Brains were removed quickly and postfixed with the same fixation solution overnight at 4°C. Postfixed brains were embedded in paraffin, and 5 μm sections were obtained using a rotary microtome and placed on pretreated slides. For detection of in situ DNA fragmentation, TUNEL staining was performed using the Cell Death Detection kit (Roche Diagnostics GmbH, Germany) according to the manufacturer’s instructions with minor modifications. Fixed tissues were placed in a plastic jar containing 0.1 M citrate buffer (pH 6.0). Slides were exposed to 750 W microwave irradiation for 1 min and cooled rapidly by immediately adding double distilled water. Next, the slides were immersed in Tris-HCl (0.1 M, pH 7.5, containing 3% BSA and 20% normal bovine serum) for 30 min at 25°C, then rinsed three times with PBS. TUNEL reaction mixture was then added to the sections, and the slides were incubated for 60 min at 37°C in a humidified atmosphere in the dark. TUNEL-positive cells in the CA1 pyramidal cell layer were then counted under a fluorescence microscope by investigators blinded to the treatment (magnification in 40×, and scale bar = 20 um). Three slices of every rat brain were used for counting.
Data analysis
The results are expressed as mean ± S.D. and S.E. The data were statistically evaluated using Student’s t-test or one-way analysis of variance (ANOVA) followed by Duncan’s Multiple Range test to compare significant differences between the groups at p < 0.05.
Results
Qualitative analysis of EOCO
The EOCO was composed of 45 main compounds with significant differences in the contributions of major monoterpenes (67.97%) and sesquiterpenes (25.97%; ). The main monoterpene constituents were α-terpinyl acetate (16.82%), β-phellandrene (13.11%), β-myrcene (5.68%), limonene (6.49%), bornyl acetate (7.48%), γ-terpinene (4.28%), and α-terpineol (4.33%). The main sesquiterpene and diterpene constituents were elemol (6.22%), thujopsene (4.50%), β-eudesmol (4.13%), and beyerene (3.35%). According to CitationYang et al. (2007), the chemical composition of essential oil obtained from the leaves of C. obtuse was mainly α-terpinyl acetate (13.71%), sabinene (10.97%), isobornyl acetate (8.85%), and limonene (6.89%). Only systemically standardized samples were used to evaluate the following effects.
Table 1. Chemical composition of the essential oil extracted from Chamaecyparis obtusa.
Effects of EOCO on Aβ1–40-induced impairment of memory acquisition and retention
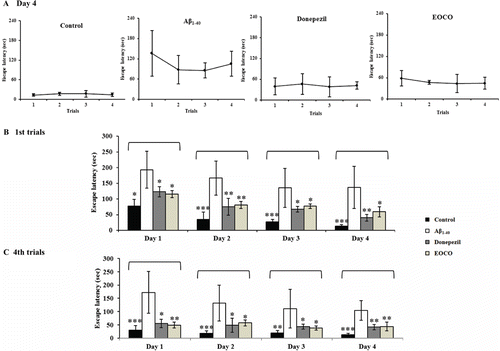
The effects of EOCO on spatial memory were investigated using the Morris water maze test (). shows the escape latencies for each group in trials 1–4 on day 4. The control group rapidly learned the location of the platform, as demonstrated by short escape latencies. The escape latencies decreased from the first trial to fourth trial and the first day to the fourth day due to normal learning. However, despite learning for 4 days, escape latencies of the Aβ1–40 group did not decrease (104.91 ± 37.04 sec) compared with controls (13.48 ± 5.08 s). In contrast, the escape latencies of the donepezil group (41.75 ± 10.40 s) and the EOCO group (44.22 ± 16.50 s) did decrease over the 4 days of testing.
Figure 2. Effects of EOCO on Aβ1–40-induced impairments of memory acquisition and retention in the Morris water maze test. Before receiving intrahippocampally injection of either ACSF or Aβ1–40 (4 μL), the rats inhaled EOCO (1 mL/cage) in a VSC for 30 days. Donepezil (1 mg/kg b.w./day) served as a positive control. The rats were given four trials each day for 4 consecutive days. (A) The effects of EOCO on the memory of rats impaired by injection of Aβ1–40 in the fourth day of the Morris water maze test. (B) The effects of EOCO on the memory of rats impaired by injection of Aβ1–40 in the first trial and fourth trials of the Morris water maze test over 4 days. The values shown are the mean latency ± S.D (n = 7); *p < 0.05, **p < 0.01, ***p < 0.001, significantly different from the Aβ1–40-treated group. Statistical significance was tested with the unpaired Student’s t-test.
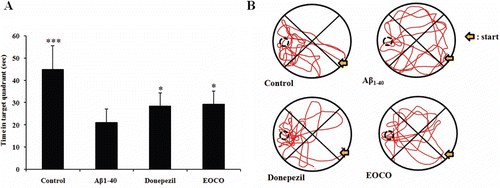
The results of the spatial probe trials are showed in . The time spent in the target quadrant (where the platform was previously placed) was significantly decreased in the Aβ1–40 group (21.06 ± 6.15 s) compared with the control group (44.90 ± 10.73 s, ***p < 0.001). In contrast, the time spent in the target quadrant by the donepezil group (28.55 ± 5.90 s, *p < 0.05) and the EOCO group (29.34 ± 5.85 s, *p < 0.05) increased remarkably. The effects of treatment group on the retention latencies, as revealed in the passive avoidance test, are shown in . In the Aβ1–40 group, retention latencies significantly decreased (114.32 ± 21.28 s) compared with the control group (268.69 ± 10.97 s, ***p < 0.001). However, the retention latency reduction induced by Aβ1–40 was alleviated by donepezil (200.34 ± 23.01 s, *p < 0.05) and EOCO treatments (192 ± 24.40 s, *p < 0.05).
Table 2. Effects of EOCO on Aβ1–40-induced impairment of memory acquisition and memory retention in the passive avoidance test.
Figure 3. Effects of EOCO on Aβ1–40-induced impairments of memory acquisition and retention in probe trials. (A) Effects of EOCO on probe trial sessions of the Morris water maze test. The time in the figure represents the cumulative time in the target quadrant of the pool in the 90 s probe trial. (B) Swimming pattern on probe trial sessions of the Morris water maze test. The values shown are the mean latency ± S.D (n = 7); *p < 0.05, **p < 0.01, ***p < 0.001, significantly different from Aβ1–40-treated group. Statistical significance was tested with the unpaired Student’s t-test.
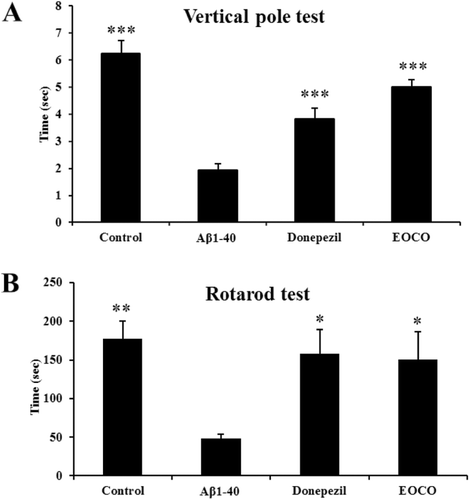
Effects of EOCO on Aβ1–40-induced behavioral deficits
The effects of EOCO on the behavioral deficits induced by Aβ were determined using the vertical pole and rotarod tests. Grip strength and sensorimotor performance measured by the vertical pole test were found to be significantly decreased in the Aβ1–40 group (1.93 ± 0.23 s) compared to the control group (6.25 ± 0.45 s, ***p < 0.001). Specifically, EOCO treatment significantly improved the time spent (5.01 ± 0.26 s, ***p < 0.001) on the vertical pole compared to the Aβ1–40 group (1.93 ± 0.23 s, ***p < 0.001, ). In the rotarod test, the Aβ1–40 group showed a significant reduction in time spent on the rotarod (48.08 ± 5.74 s, **p < 0.01) compared to the control group (177.17 ± 23.27 s, *p < 0.05), and EOCO treatment (150.01 ± 36.46 s, *p < 0.05) was found to be effective for partial recovery of balance and coordination compared to the Aβ1–40 group. No significant differences were observed in the EOCO group compared to the donepezil group ().
Figure 4. Effects of EOCO on Aβ1–40-induced behavioral deficits. (A) Effects of EOCO on Aβ1–40-induced sensorimotor deficits in the vertical pole test. (B) Effects of EOCO on Aβ1–40-induced motor coordination and balance deficits in the rotarod test. The values shown are the mean latency ± S.E. (n = 7); *p < 0.05, **p < 0.01, ***p < 0.001, significantly different from Aβ1–40-treated group. Statistical significance was tested with the unpaired Student’s t-test.
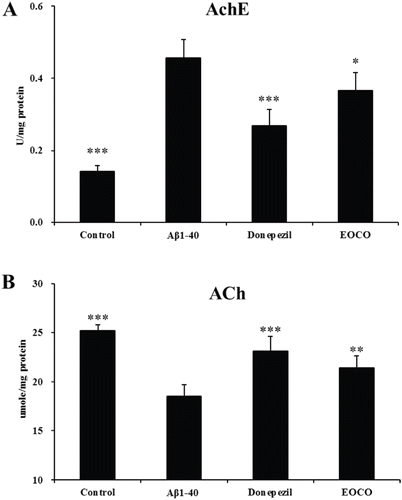
Effects of EOCO on AchE activity and ACh content in Aβ1–40-injected rats
AD is accompanied by synaptic dysfunction in the cholinergic system. The increased AchE activity and a significant loss of ACh correlate with cognitive impairments. We measured AchE activities using Ellman’s method with some modification. We found that AchE activity was significantly elevated (0.46 ± 0.05 U/mg protein, ***p < 0.001) and ACh content was decreased markedly (18.54 ± 1.17 umole/mg protein, ***p < 0.001, , ) in the Aβ1–40 group. However, after treatment with donepezil or EOCO, AchE activity was significantly decreased and ACh content was significantly increased compared with the Aβ1–40 group. In addition, the EOCO group had less of a reduction in AchE activity than the donepezil group.
Figure 5. Effects of EOCO on AchE activity (A) and ACh content (B) in Aβ1–40-injured rats. Results are expressed as the means ± S.D. (n = 7); *p < 0.05, **p < 0.01, ***p < 0.001, significantly different from only Aβ1–40-treated group. Statistical significance was tested with the unpaired Student’s t-test.
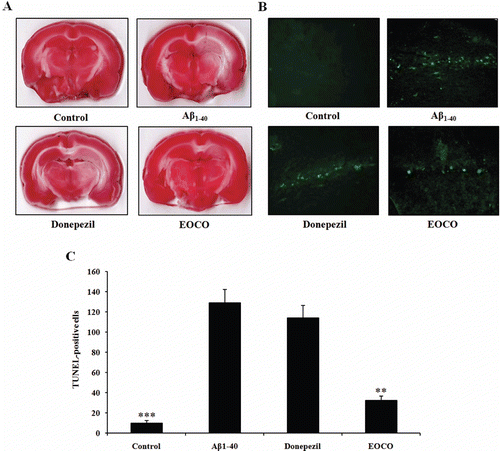
Effects of EOCO on Aβ1–40-induced hippocampal degeneration
Following behavioral evaluations, rats were sacrificed for TTC staining. shows representative TTC staining for each group. Normal brain tissue appeared uniformly red in the control group, while degenerating regions appeared white in the Aβ1–40 group. In addition, among animals treated with Aβ1–40, the volume of degenerating regions was significantly less in EOCO group than in Aβ1–40 group. However, no significant difference was observed in the volume of degenerating regions between donepezil group and Aβ1–40 group. Brain sections were also subjected to TUNEL staining. As shown in , no immunofluorescence was observed in hippocampal neurons of the control group. However, in both Aβ1–40 (129.01 ± 22.65 cells) and donepezil group (114.33 ± 20.98 cells), most of the hippocampal neurons were positive for TUNEL staining. Finally, a small number of TUNEL-positive cells were observed in EOCO group (42.33 ± 7.37 cells, **p < 0.01).
Figure 6. Effects of EOCO on Aβ1–40-induced neuronal degeneration and DNA strand breakage. (A) Effects of EOCO on Aβ1–40-induced neuronal degeneration assessed by TTC staining. Representative brain sections (2 mm thick) from EOCO-treated rats stained with 2% TTC showing neuronal degeneration. Red colored regions in the TTC-stained sections indicate nondegenerated regions and pale colored regions indicate degeneration. (B) Effects of EOCO on Aβ1–40-induced neuronal apoptosis assessed by TUNEL assay. Detection of hippocampal apoptotic cells was carried out using the TUNEL method. TUNEL-positive pyramidal neurons undergoing apoptosis were observed in the hippocampus (fluorescence). (C) Representative quantification of the number of TUNEL-positive cells. Results are expressed as the means ± S.E. (n = 7); *p < 0.05, **p < 0.01,***p < 0.001, significantly different from Aβ1–40-treated group. Statistical significance was tested with the unpaired Student’s t-test.
Discussion
The findings from this study clearly show that inhaled EOCO suppresses the impairments in spatial working memory induced by intrahippocampal injection of Aβ1–40. Using the Aβ hippocampal injection model, we further demonstrated that hippocampal neuronal cell apoptosis was induced by injection of Aβ1–40, and EOCO treatment antagonized Aβ-mediated neuronal apoptosis in vivo.
We compared learning and memory using the Morris water maze in non-Aβ1–40-injected rats and found no significant improvement in performance over 4 days between an EOCO inhalation and a control group. However, on the first trial on the first day of the Morris water maze test and probe test, escape latencies for the EOCO inhalation group were significantly decreased compared to escape latencies of the nontreated control group (data not shown). These results confirm the potential of EOCO as a neuroprotective agent against Aβ1–40-induced impairments of learning and memory acquisition.
Aβ1–40 injection into the hippocampus resulted in memory loss with AD-like features in rats. At the onset of AD pathogenesis, the fibrillogenesis of Aβ and the formation of insoluble amyloid plaques cause synaptic failure and clinical memory loss along with other cognitive deficiencies (CitationKoudinov et al., 2009). However, the newest versions of the amyloid theory posit that the major culprit is soluble Aβ oligomers rather than Aβ fibrils. It is known that soluble Aβ oligomers can disrupt synaptic memory mechanisms at extremely low concentrations via stress-activated kinases and oxidative/nitrosative stress mediators (CitationRowan et al., 2007). For these reasons, we used mixed-Aβ1–40 with soluble oligomeric and fibrillar Aβ1–40 in this study.
In the Morris water maze test, the EOCO-treated group exhibited significantly shorter escape latencies on the first trials of each day than the Aβ1–40-treated group for all 4 days; this finding suggests that EOCO improves long-term memory impaired by Aβ. Furthermore, short-term memory improvements were revealed by the significant differences in escape latencies between the first and second trial on day 1 in the EOCO-treated group. The EOCO-treated group also showed significant recovery of spatial memory retention abilities in the probe trial. Similar results were obtained in the passive avoidance test; EOCO treatment increased the retention latencies compared to the group that only received Aβ1–40 treatment. The results of these two behavioral tests show that EOCO protects spatial learning and memory function from Aβ1–40-induced impairments.
The behavioral effects of Aβ reflect the degree of neuronal dysfunction (Schwating et al., 1991). AD is accompanied by behavioral deficits. Similarly, after Aβ1–40 injection, several behavioral deficits became apparent, including deficits of balance and coordination and sensorimotor performance. The rotarod test is widely used to assess motor coordination, and this task has provided a rich source of information about the qualitative aspects of walking (CitationWhishaw et al., 2008). Compared to the control group, motor coordination was decreased only in the Aβ1–40-treated group, indicating EOCO provided significant protection against Aβ-induced motor impairments. On the vertical pole test, the Aβ1–40-treated group fell off the pole after a shorter latency than the control group. However, treatment with EOCO significantly increased the time spent on the vertical pole compared to the Aβ1–40-treated group. These results confirm that the behavioral deficits of the Aβ1–40-induced rat model of AD are attenuated by inhalation of EOCO.
Autopsy of AD patients has demonstrated a specific deficiency in ACh content and an excess of AchE (CitationDavies et al., 1976). Cholinergic transmission is terminated by ACh hydrolysis via AchE (CitationBallard et al., 2005); thus, AchE activity and ACh content are inversely related. In AD, AchE is usually decreased, not increased, as cholinergic neurons degenerate. However, increases in AchE have been observed in AD, although presumably the increase preceded neuronal degeneration (CitationKar et al., 2004). In this study, EOCO significantly inhibited AchE activity and increased ACh levels in the brain compared to the group treated only with Aβ1–40. Although we did not measure the levels of essential oil components in the blood of our animals, our findings demonstrate that inhalation of the fragrance apparently affected the nervous system, suggesting that some active components affected the Aβ signaling pathways in the CNS. These results are interesting and warrant further investigation.
The results from the present study provide possible mechanisms of EOCO in protecting neuronal cell death, especially Aβ-mediated death. First, EOCO’s action on the modulation of oxidative stress will diminish excessive activation of Aβ-metal ion complexes which were excessively released from injury to the CNS: according to this theory, Aβ and copper/zinc ions form an Aβ-metal ion complex, which produces reactive oxygen species (ROS), such as O2−, H2O2 and OH (CitationCurtain et al., 2001; CitationBarnham et al., 2004; CitationValko et al., 2005). Second, EOCO’s antagonistic effects of Ca2+ channels and N-methyl-d-aspartate receptors (NMDAr) will potentiate its neuroprotective action by regulating Ca2+ homeostasis: this theory posits that the collapse of Ca2+ homeostasis directly or indirectly induces neuronal apoptosis (Issacs et al., 2006; CitationKelly & Ferreira 2006; CitationArispe et al., 2007; CitationYoshiike et al., 2007). There are several widely known hypotheses regarding how Ca2+ homeostasis is lost, and they involve Ca2+ influx through voltage dependent Ca2+ channels (VDCCs), NMDAr and calcium channels formed directly by Aβ. Third, EOCO’s action on the hyperphosphorylation of tau protein: this theory posits that the loss of Ca2+ homeostasis induces hyperphosphorylation of tau, which causes tau to lose the capability to bind microtubules and subsequently induces neuronal apoptosis (CitationYamamoto et al., 2002; CitationIqbal et al., 2005; CitationMattson, 2007).
In all likelihood, the causes of neuronal loss are not independent events. Whatever the mechanisms of neuronal loss are, TTC staining and the TUNEL assay demonstrated that the volume of degenerated regions and the number of TUNEL-positive cells in the EOCO-treated group decreased compared to Aβ1–40-treated group. However, there was no change in these measures for the donepezil-treated group compared to the Aβ1–40-treated group. Donepezil has been reported as a promising palliative therapeutic agent for AD on the basis of its superior AchE inhibition and memory enhancement potency (CitationPang et al., 1996; CitationWang et al., 1999; CitationLiu et al., 2000). Although it has been shown that donepezil reverses memory deficits mainly via the inhibition on AchE, its exact multiple neuroprotective mechanisms remain elusive. Several studies have shown that donepezil did not show any protective effect against apoptosis by Aβ, NMDA or staurosporine (CitationZhang et al., 2002; CitationLi et al., 2005; CitationFu et al., 2006). Therefore, EOCO’s role of anti-apoptosis might be more beneficial than those that only inhibit AchE to treat AD and in particular to prevent the pathogenesis of AD and other related neurodegenerative disorders. We are currently investigating the effects of EOCO on NMDAr- and neuronal Ca2+ channels-mediated change of intracellular Ca2+ level and neuronal cell death, which are more relevant conditions of the delayed form of Aβ injury.
Although further experiments will be needed to uncover the detailed mechanisms of EOCO’s action on Aβ-induced memory impairment in rats, our data clearly demonstrate that inhaled EOCO suppresses impairment of spatial working memory and neuronal cell apoptosis induced by intrahippocampal injection of Aβ1–40. Therefore, for the first time, our findings provide in vivo evidence that EOCO offers protection against AD-related neuronal cell apoptosis and memory dysfunction and may be a useful therapeutic choice for the treatment of neurodegenerative disorders.
Conclusions
In the present study, we investigated the neuroprotective effects of EOCO against Aβ1–40-induced neuronal toxicity in rats. We found that EOCO ameliorated the deficits in spatial learning and memory function induced by Aβ1–40. In addition, we confirmed that the behavioral deficits associated with AD-like symptoms are attenuated by inhalation of EOCO. Furthermore, EOCO significantly inhibited AchE activity and neuronal apoptosis compared to the Aβ1–40-treated group. Taken together, the results of this study suggest that EOCO has neuroprotective effects against Aβ1–40-induced neuronal toxicity. Therefore, EOCO could be a useful neuroprotectant against Aβ1–40-induced neuronal damage.
Declaration of interest
This research was supported by Basic Science Research Program through the National Research Foundation of Korea (NRF) funded by the Ministry of Education, Science and Technology (2009-0076837) to S. Kim, and Jeollanamdo Institute for Natural Resources (JINR) Core-Competence Program grants to S. Kim.
References
- Aloisi AM, Ceccarelli I, Masi F, Scaramuzzino A. (2002). Effects of the essential oil from citrus lemon in male and female rats exposed to a persistent painful stimulation. Behav Brain Res, 136, 127–135.
- Arispe N, Diaz JC, Simakova O. (2007). Abeta ion channels. Prospects for treating Alzheimer’s disease with Abeta channel blockers. Biochim Biophys Acta, 1768, 1952–1965.
- Ballard CG, Greig NH, Guillozet-Bongaarts AL, Enz A, Darvesh S. (2005). Cholinesterases: Roles in the brain during health and disease. Curr Alzheimer Res, 2, 307–318.
- Barnham KJ, Masters CL, Bush AI. (2004). Neurodegenerative diseases and oxidative stress. Nat Rev Drug Discov, 3, 205–214.
- Choi J, Lee KT, Ha J, Yun SY, Ko CD, Jung HJ, Park HJ. (2003). Antinociceptive and antiinflammatory effects of Niga-ichigoside F1 and 23-hydroxytormentic acid obtained from Rubus coreanus. Biol Pharm Bull, 26, 1436–1441.
- Curtain CC, Ali F, Volitakis I, Cherny RA, Norton RS, Beyreuther K, Barrow CJ, Masters CL, Bush AI, Barnham KJ. (2001). Alzheimer’s disease amyloid-beta binds copper and zinc to generate an allosterically ordered membrane-penetrating structure containing superoxide dismutase-like subunits. J Biol Chem, 276, 20466–20473.
- Davies P, Maloney AJ. (1976). Selective loss of central cholinergic neurons in Alzheimer’s disease. Lancet, 2, 1403.
- Durham DG, Liu X, Richards RM. (1994). A triterpene from Rubus pinfaensis. Phytochemistry, 36, 1469–1472.
- Ekinci FJ, Malik KU, Shea TB. (1999). Activation of the L voltage-sensitive calcium channel by mitogen-activated protein (MAP) kinase following exposure of neuronal cells to beta-amyloid. MAP kinase mediates beta-amyloid-induced neurodegeneration. J Biol Chem, 274, 30322–30327.
- Ellman GL, Courtney KD, Andres V Jr, Feather-Stone RM. (1961). A new and rapid colorimetric determination of acetylcholinesterase activity. Biochem Pharmacol, 7, 88–95.
- Fu H, Li W, Lao Y, Luo J, Lee NT, Kan KK, Tsang HW, Tsim KW, Pang Y, Li Z, Chang DC, Li M, Han Y. (2006). bis(7)-Tacrine attenuates beta amyloid-induced neuronal apoptosis by regulating L-type calcium channels. J Neurochem, 98, 1400–1410.
- Graeff FG, Silveira MC, Nogueira RL, Audi EA, Oliveira RM. (1993). Role of the amygdala and periaqueductal gray in anxiety and panic. Behav Brain Res, 58, 123–131.
- Graham PH, Browne L, Cox H, Graham J. (2003). Inhalation aromatherapy during radiotherapy: Results of a placebo-controlled double-blind randomized trial. J Clin Oncol, 21, 2372–2376.
- Huang HC, Jiang ZF. (2009). Accumulated amyloid-beta peptide and hyperphosphorylated tau protein: Relationship and links in Alzheimer’s disease. J Alzheimers Dis, 16, 15–27.
- Iqbal K, Grundke-Iqbal I. (2005). Metabolic/signal transduction hypothesis of Alzheimer’s disease and other tauopathies. Acta Neuropathol, 109, 25–31.
- Isaacs AM, Senn DB, Yuan M, Shine JP, Yankner BA. (2006). Acceleration of amyloid beta-peptide aggregation by physiological concentrations of calcium. J Biol Chem, 281, 27916–27923.
- Kar S, Slowikowski SP, Westaway D, Mount HT. (2004). Interactions between beta-amyloid and central cholinergic neurons: implications for Alzheimer’s disease. J Psychiatry Neurosci, 29, 427–441.
- Kelly BL, Ferreira A. (2006). beta-Amyloid-induced dynamin 1 degradation is mediated by N-methyl-d-aspartate receptors in hippocampal neurons. J Biol Chem, 281, 28079–28089.
- Kim S, Rhim H. (2011). Effects of amyloid-β peptides on voltage-gated L-type Ca(V)1.2 and Ca(V)1.3 Ca(2+) channels. Mol Cells, 32, 289–294.
- Komiya M, Takeuchi T, Harada E. (2006). Lemon oil vapor causes an anti-stress effect via modulating the 5-HT and DA activities in mice. Behav Brain Res, 172, 240–249.
- Koudinov A, Kezlya E, Koudinova N, Berezov T. (2009). Amyloid-beta, tau protein, and oxidative changes as a physiological compensatory mechanism to maintain CNS plasticity under Alzheimer’s disease and other neurodegenerative conditions. J Alzheimers Dis, 18, 381–400.
- Li Q, Kobayashi M, Wakayama Y, Inagaki H, Katsumata M, Hirata Y, Hirata K, Shimizu T, Kawada T, Park BJ, Ohira T, Kagawa T, Miyazaki Y. (2009). Effect of phytoncide from trees on human natural killer cell function. Int J Immunopathol Pharmacol, 22, 951–959.
- Li Q, Nakadai A, Matsushima H, Miyazaki Y, Krensky AM, Kawada T, Morimoto K. (2006). Phytoncides (wood essential oils) induce human natural killer cell activity. Immunopharmacol Immunotoxicol, 28, 319–333.
- Li W, Pi R, Chan HH, Fu H, Lee NT, Tsang HW, Pu Y, Chang DC, Li C, Luo J, Xiong K, Li Z, Xue H, Carlier PR, Pang Y, Tsim KW, Li M, Han Y. (2005). Novel dimeric acetylcholinesterase inhibitor bis7-tacrine, but not donepezil, prevents glutamate-induced neuronal apoptosis by blocking N-methyl-d-aspartate receptors. J Biol Chem, 280, 18179–18188.
- Liu J, Ho W, Lee NT, Carlier PR, Pang Y, Han Y. (2000). Bis(7)-tacrine, a novel acetylcholinesterase inhibitor, reverses AF64A-induced deficits in navigational memory in rats. Neurosci Lett, 282, 165–168.
- Mattson MP. (2007). Calcium and neurodegeneration. Aging Cell, 6, 337–350.
- Morris R. (1984). Developments of a water-maze procedure for studying spatial learning in the rat. J Neurosci Methods, 11, 47–60.
- Nam JH, Jung HJ, Choi J, Lee KT, Park HJ. (2006). The anti-gastropathic and anti-rheumatic effect of niga-ichigoside F1 and 23-hydroxytormentic acid isolated from the unripe fruits of Rubus coreanus in a rat model. Biol Pharm Bull, 29, 967–970.
- Nose K. (2000). Role of reactive oxygen species in the regulation of physiological functions. Biol Pharm Bull, 23, 897–903.
- Ogawa N, Hirose Y, Ohara S, Ono T, Watanabe Y. (1985). A simple quantitative bradykinesia test in MPTP-treated mice. Res Commun Chem Pathol Pharmacol, 50, 435–441.
- Oikawa S. (2005). Sequence-specific DNA damage by reactive oxygen species: Implications for carcinogenesis and aging. Environ Health Prev Med, 10, 65–71.
- Pákáski M, Kálmán J. (2008). Interactions between the amyloid and cholinergic mechanisms in Alzheimer’s disease. Neurochem Int, 53, 103–111.
- Pang YP, Quiram P, Jelacic T, Hong F, Brimijoin S. (1996). Highly potent, selective, and low cost bis-tetrahydroaminacrine inhibitors of acetylcholinesterase. Steps toward novel drugs for treating Alzheimer’s disease. J Biol Chem, 271, 23646–23649.
- Rowan MJ, Klyubin I, Wang Q, Hu NW, Anwyl R. (2007). Synaptic memory mechanisms: Alzheimer’s disease amyloid beta-peptide-induced dysfunction. Biochem Soc Trans, 35, 1219–1223.
- Schwarting RK, Bonatz AE, Carey RJ, Huston JP. (1991). Relationships between indices of behavioral asymmetries and neurochemical changes following mesencephalic 6-hydroxydopamine injections. Brain Res, 554, 46–55.
- Valko M, Morris H, Cronin MT. (2005). Metals, toxicity and oxidative stress. Curr Med Chem, 12, 1161–1208.
- Vincent D, Segonzac G, Vincent MC. (1958). [Colorimetric determination of acetylcholine by the Hestrin hydroxylamine reaction and its application in pharmacy]. Ann Pharm Fr, 16, 179–185.
- Wang H, Carlier PR, Ho WL, Wu DC, Lee NT, Li CP, Pang YP, Han YF. (1999). Effects of bis(7)-tacrine, a novel anti-Alzheimer’s agent, on rat brain AChE. Neuroreport, 10, 789–793.
- Watanabe T, Yamagata N, Takasaki K, Sano K, Hayakawa K, Katsurabayashi S, Egashira N, Mishima K, Iwasaki K, Fujiwara M. (2009). Decreased acetylcholine release is correlated to memory impairment in the Tg2576 transgenic mouse model of Alzheimer’s disease. Brain Res, 1249, 222–228.
- Whishaw IQ, Li K, Whishw PA, Gorny B, Metz GA. (2008). Use of rotarod as a method for the qualitative analysis of walking in rat. J Vis Exp, 22, 1030.
- Yamada K, Mimaki Y, Sashida Y. (1994). Anticonvulsive effects of inhaling lavender oil vapour. Biol Pharm Bull, 17, 359–360.
- Yamamoto H, Yamauchi E, Taniguchi H, Ono T, Miyamoto E. (2002). Phosphorylation of microtubule-associated protein tau by Ca2+/calmodulin-dependent protein kinase II in its tubulin binding sites. Arch Biochem Biophys, 408, 255–262.
- Yang JK, Choi MS, Seo WT, Rinker DL, Han SW, Cheong GW. (2007). Chemical composition and antimicrobial activity of Chamaecyparis obtusa leaf essential oil. Fitoterapia, 78, 149–152.
- Yoshiike Y, Akagi T, Takashima A. (2007). Surface structure of amyloid-beta fibrils contributes to cytotoxicity. Biochemistry, 46, 9805–9812.
- Zhang BF, Peng FF, Zhang JZ, Wu DC. (2002). [Protective effects of tacrine and donepezil against staurosporine-induced apoptotic death]. Yao Xue Xue Bao, 37, 98–102.