
Abstract
Context: Obesity is associated with a number of diseases with metabolic abnormalities such as type 2 diabetes (T2D).
Objective: We investigate the effects of tectorigenin on 3T3-L1 preadipocyte differentiation and adipocytokines secretion.
Materials and methods: The effects of tectorigenin on adipocyte differentiation were studied using Oil Red O staining. Effects of tectorigenin on adipogenesis-related genes expression and adipocytokines secretion were measured by the real-time quantitative RT-PCR and ELISA method, respectively. Reporter gene assays were performed to determine the PPARγ and NF-κB transactivation. We also used [3H]-2-deoxy-d-glucose to study the glucose uptake, and the IKK/NF-κB signaling pathway was assessed by western blot analysis. HFD/STZ rats were used to evaluate the therapeutic efficacies of tectorigenin.
Results: Tectorigenin 10, 25, 50, and 75 μM inhibited 3T3-L1 adipogenesis and related genes transcription. TNF-α-induced changes of IL-6, MCP-1, as well as adiponectin in 3T3-L1 were markedly reversed by tectorigenin at 75 μM. Further investigation using reporter gene revealed that tectorigenin was a partial PPARγ agonist with an IC50 value of 13.3 μM. Tectorigenin improved basal and insulin-stimulated glucose uptake in mature 3T3-L1 adipocytes. Moreover, tectorigenin antagonized TNF-α-induced NF-κB transactivation and p65 nuclear translocation. Although tectorigenin (50 and 100 mg/kg) displayed the ability to promote insulin sensitivity and improve glucose metabolism in HFD/STZ rats, it did not cause significant side effects such as body weight gain, fluid retention, or cardiac hypertrophy.
Discussion and conclusion: These results suggest that tectorigenin may ameliorate hyperglycemia by blocking preadipocyte differentiation and adipocytokines secretion in which PPARγ and NF-κB signaling pathways were involved.
Introduction
The incidence of obesity is increasing dramatically in the world, and with it, comes important pathological consequences such as type 2 diabetes mellitus and cardiovascular disease (Han et al., Citation2013). It has been suggested that adipose tissue plays an important role in controlling whole-body glucose homeostasis in both normal and disease states. Thus, discovery of new strategies that regulate adipocyte function might ultimately lead to therapeutic modalities that can prevent insulin resistance and type 2 diabetes mellitus (Guilherme et al., Citation2008).
The nuclear receptor PPARγ, which is an important transcription factor in the development and function of the adipose tissue, participates in biological pathways of adipocyte differentiation, insulin sensitivity, and inflammation. During the differentiation, PPARγ is activated and then promotes the expression of downstream adipose-specific genes involved in adipose phenotype and glucose metabolism (Hong et al., Citation2011). Furthermore, activated PPARγ antagonizes the function of pro-inflammatory transcription factors such as NF-κB, thereby decreasing the expression of pro-inflammatory cytokines and diminishing the inflamed state in the adipose tissue (Tontonoz & Spiegelman, Citation2008).
Due to the critical role of PPARγ in fat cell development, great efforts have been made to identify the transactivated ligands of PPARγ to treat metabolic disorders (Rosen et al., Citation1999). There are several ligands with different affinities that have been identified, including natural fatty derivatives, 15-deoxy-Δ12, 14-prostaglandin J2, and antidiabetic thiazolidinediones (TZD) (van Bilsen & van Nieuwenhoven, Citation2010). As full PPARγ agonists, the TZD members, rosiglitazone and pioglitazone, exert insulin-sensitizing actions largely through pleiotropic effects in adipose tissue. However, the clinical use of TZD is often associated with undesirable side effects including peripheral edema, congestive heart failure, and weight gain. In particular, European Medicines Agency (EMA) has concluded that the risks to patients’ hearts outweigh the benefits of treatment with rosiglitazone, and recommended the withdrawal of the diabetes drug from the market. In contrast, ligands with partial PPARγ agonism, such as halofenate and INT131, have been reported to display significant insulin-sensitizing activity but minimal adipogenesis (Allen et al., Citation2006; Motani et al., Citation2009). Therefore, new efforts should be focused on searching for selective PPARγ modulators with better therapeutic profile.
In recent years, tectorigenin has attracted considerable interests due to its anti-proliferative, anti-inflammatory, and antioxidant activities via scavenging radicals and inhibiting aldose reductase and β-glucuronidase (Fang et al., Citation2008; Lee et al., Citation2003; Moon et al., Citation2006; Vigne et al., Citation1990). It is also reported that intraperitoneal administration of tectorigenin to streptozotocin-induced rats significantly reduced the blood glucose, total cholesterol, LDL- and VLDL-cholesterol (Lee et al., Citation2000). However, to our knowledge, the effect of tectorigenin on adipocyte differentiation has not been reported to date. Here, we reported that tectorigenin inhibits fat cell differentiation and adipocytokines secretion. Furthermore, we identified tectorigenin as a partial agonist of PPARγ and provided new insight into the role of tectorigenin in improvement of glucose uptake and antagonism of NF-κB activation. In vivo investigation showed that tectorigenin could promote insulin sensitivity and improve glucose metabolism, but evoke less side effects. Thus, this study indicates a potential therapeutic role for tectorigenin in the treatment of diabetes, obesity, and metabolic syndrome.
Materials and methods
Materials and reagents
Tectorigenin is supplied by the Division of Chinese Materia Medica and Natural Products, National Institute for the Control of Pharmaceutical and Biological Products (NICPBP), China. Isobutylmethylxanthine (IBMX), GW9662, rosiglitazone, dexamethasone, and insulin were obtained from Sigma-Aldrich (St. Louis, MO). TRIzol reagent was purchased from Invitrogen (Carlsbad, CA). Antibodies to p-IKK, IKBα, IKKα, p65, and PARP were purchased from Santa Cruz Biotechnology (Santa Cruz, CA). Antibodies to insulin receptor substrate 1 (IRS1) and Akt1 were purchased from Upstate Biotechnology (Lake Placid, NY). Antibodies to Akt1 phosphorylated at a position equivalent to threonine 308 (Akt-pT308) were purchased from Cell Signaling Technology (Beverly, MA). Mouse preadipocyte 3T3-L1 was from American Type Culture Collection (ATCC, Manassas, VA). Mouse PPARγ plasmid (pCMX-mPPARγ) was kindly provided by Dr. Ronald M. Evans of the Salk Institute for Biological Studies (La Jolla, CA).
Cell culture and adipogenic differentiation
3T3-L1 cells were grown and maintained in DMEM supplemented with 10% fetal bovine serum (HyClone, Logan, UT). For adipocyte differentiation, 3T3-L1 cells were grown in 24-well plates to full confluence for 2 d and then induced by differentiation medium (DM) containing 10 μg/ml insulin, 1 μM dexamethasone, and 0.5 mM IBMX. After 4 d induction, the medium was refreshed with DMEM plus 10% fetal bovine serum for differentiation at 37 °C and 10% CO2 atmosphere. After 7 d of differentiation, cells were harvested to evaluate lipid accumulation by Oil Red O staining or the triglyceride (TG) assay.
Oil Red O staining and triglyceride assay
After 7 d of differentiation, the cells were washed gently with PBS and fixed with 4% paraformaldehyde (pH 7.4) for 30 min. Lipid droplets were stained with 0.5% Oil Red O in 60% isopropanol for 15 min. The Oil Red O was removed and the plates were rinsed with water and dried at 37 °C. Then, isopropanol was used to extract the dye from the cells and the absorbance was measured at 510 nm.
For triglyceride assay, the cells in 24-well plates were washed gently with PBS and lysed with 0.1% NP-40. Triglyceride content was determined using the triglyceride GPO-Trinder kit (St. Louis, MO). The absorbance was measured at 540 nm, and triglyceride was normalized to protein concentration determined by the BCA method.
Glycerol-3-phosphate dehydrogenase (GPDH) activity determination
The GPDH assay was performed using a GPDH activity assay kit (Takara Bio Inc., Shiga, Japan). Differentiated 3T3-L1 adipocytes were washed twice with PBS and harvested into enzyme extraction buffer. Cells were sonicated and GPDH activity was determined according to the instruction of the manufacturer.
Total RNA isolation, cDNA preparation, and quantitative real-time PCR
Total RNA from 3T3-L1 cells was extracted with the TRIzol reagent (Invitrogen, Carlsbad, CA) and reverse transcribed (RT) using Superscript II First-strand Synthesis SuperMix (Invitrogen, Carlsbad, CA). Primers and real-time PCR conditions were described previously (Hassan et al., Citation2007; Takahashi et al., Citation2009). For RT-PCR, SYBR® Premix Ex Taq™ (Takara Bio Inc., Dalian, China) was used. Final PCR products were subjected to graded temperature-dependent dissociation to verify that only one product was amplified. Reactions with no RT sample and no template were included as negative controls. Relative quantitative evaluation of target gene levels was performed by the comparative CT (cycle threshold) method (Livak & Schmittgen, Citation2001) and performed in triplicates.
Western blot analysis
3T3-L1 cells were lysed in a lysis buffer containing 50 mM Tris-HCl (pH 7.5), 2 mM ethylenediaminetetraacetic acid (EDTA), 150 mM NaCl, 0.5% deoxycholate, 0.1% sodium dodecyl sulfate (SDS), 1 mM NaF, 1 mM Na3VO4, 1 mM phenylmethylsulfonyl fluoride (PMSF), 1 mM dithiothreitol (DTT), 1 μg/mL leupeptin, 1 μg/mL aprotinin, and 1% Triton. Nuclear and cytoplasmic proteins of 3T3-L1 cells were extracted using a nuclear and cytoplasmic protein extracting kit (Pierce, Rockford, IL) according to the standard protocol of the manufacturer. The protein concentration was determined using a BCA kit (Pierce, Rockford, IL). Equal amounts of protein from each sample were subjected to SDS-PAGE and blotted on the PVDF membrane, which was incubated for 2 h at room temperature with blocking buffer (5% non-fat milk, 0.1% Tween 20, in TBS, pH 7.6) and then probed with primary antibodies overnight at 4 °C. After incubation with the appropriate secondary antibodies, the immunoreactive band was detected by an ECL Western blotting detection system (GE Healthcare, Pittsburgh, PA) and subsequently photographed by a LAS-3000 luminescent image system (Fujifilm, Tokyo, Japan).
Cytokines ELISA
The concentrations of IL-6, MCP-1, and adiponectin in conditioned culture medium from 3T3-L1 adipocytes were measured by ELISA kits obtained from R&D Systems (Minneapolis, MN) according to the method recommended by the manufacturer. Each concentration was determined from the standard curve and calculated as per mg of the total cell protein.
[3H]-2-Deoxy-d-glucose uptake assay
After 7 d of induction, fully differentiated 3T3-L1 adipocytes were treated with test compounds for 24 h. At day 8 of differentiation, the cells were rinsed with serum-free DMEM and incubated for 2 h in serum-free DMEM. The cells were incubated with or without 100 nM insulin in KRPH buffer at 37 °C for 30 min. Then, the buffer was replaced with 10 μCi/mL of [3H]-2-deoxy-d-glucose in KRPH buffer supplemented with 100 μM 2-deoxy-d-glucose and incubated at 37 °C for 10 min. The cells were rinsed with ice-cold PBS and lysed with 0.2 N NaOH. Lysate (200 μL) was added to a scintillation vial and 2 ml of scintillation fluid was added. The radioactivities of the mixed vials were counted. The rest of the lysate was used to determine the protein concentration using the BCA protein assay method. The glucose uptake values are expressed as “pmol radioactive 2-deoxyglucose taken up per minute and per mg protein” (Takahashi et al., Citation2009).
Cell transfection and reporter gene assays
For transient transfection, 3T3-L1 cells were seeded in a six-well plate with 10% FBS/DMEM. Mouse PPARγ vector (pCMX-mPPARγ), together with the PPRE-Luc reporter and β-galactosidase plasmids, was co-transfected into 3T3-L1 cells using Lipofectamine™ (Invitrogen, Carlsbad, CA). After 24 h transfection, the medium was changed to DMEM containing 10% FBS, and the cells were cultured for a further 24 h. Luciferase activity was determined by using the luciferase assay system (Promega, Madison, WI) according to the instructions of the manufacturer, and then normalized to β-galactosidase activity.
For NF-κB transactivation assay, pNF-κB-Luc reporter (Stratagene, San Diego, CA) and pSV-β-galactosidase plasmids were co-transfected into 3T3-L1 cells according to the instructions of the manufacturer. Luciferase activity and β-galactosidase activity were determined as described above.
Type II diabetic rats and treatments
Animal experimentation was conducted in accordance with the regulations approved by the Animal Care and Use Committee, Fudan University. The type II diabetic rat model system was developed as previously described (Wu et al., Citation2005). Eight-week-old male Sprague–Dawley rats (Shanghai SLAC Laboratory Animals Co., Shanghai, China) were housed (four per cage) at 22.5 ± 0.7 °C in a 12:12 h light:dark cycle. Animals were fed a HFD (41% fat, 41% carbohydrate, and 18% protein) or a SCD (12% fat, 60% carbohydrate, and 28% protein) and watered ad libitum. After 2 weeks on HFD, rats were anesthetized and injected into the tail vein with STZ (35 mg/kg body weight in 0.1 M citrate-buffered saline, pH4.5). The development of diabetes was confirmed by determining blood glucose concentrations 72 h after STZ administration.
The control and diabetic groups (n = 8) were then treated once daily for 2 weeks by oral gavage with vehicle (0.25% methylcellulose, 10 ml/kg), rosiglitazone (20 mg/kg), or tectorigenin (50 and 100 mg/kg). At the end of experiments, individual body weights, heart weights, and extracellular fluid volume were recorded, and glucose concentrations were determined under fasting and non-fasting conditions.
Evaluation of insulin sensitization
Peripheral insulin resistance was assessed with an insulin tolerance test (ITT) described previously (Wu et al., Citation2005). Briefly, fasted rats were administered with a neutral insulin solution (2 U/kg) by slow intravenous injection through a tail vein. Blood samples were collected at 0, 10, 20, 30, and 60 min following administration of insulin. Blood glucose concentrations were immediately measured with a blood glucose monitor. KITT represents the percent decline in blood glucose concentration per minute and is calculated according to the formula: KITT = (0.693/t1/2) × 100.
Statistical analysis
Results are expressed as mean ± SEM. Statistical analysis was performed using one-way analysis of variance (ANOVA). The intergroup comparisons (post hoc analysis) among the data with equal variances were made by the LSD method, while Tamhane’s T2 method was used for the data with unequal variances. Results were considered significant at p < 0.05.
Results
Tectorigenin-inhibited 3T3-L1 preadipocyte differentiation
In a screen effort to identify novel natural compounds capable of modulating adipogenesis, we found that tectorigenin could potently inhibit 3T3-L1 preadipocyte differentiation. As seen in , 3T3-L1 was differentiated to mature adipocyte at day 8 by the differentiation medium (DM) containing insulin, IBMX, and dexamethasone. Accumulation of intracellular fat drops, as well as cell morphological changes, suggested the occurrence of adipogenesis in 3T3-L1. Treatment with tectorigenin blocked 3T3-L1 differentiation, as demonstrated by using Oil Red O staining (). At a concentration of 50 μM, tectorigenin significantly antagonized adipocyte differentiation.
Figure 1. Effects of tectorigenin on 3T3-L1 differentiation. (A) Tectorigenin inhibited 3T3-L1 cells adipocyte differentiation induced by DM. The cells were stained with Oil Red O on day 8. GM, growth medium; DM, differentiation medium; Tec, tectorigenin. The concentrations of tectorigenin were 25, 50, and 75 μM. After 7 d of differentiation, the cells were harvested, and (B) triglyceride contents or (C) GPDH activity was determined as described in Materials and methods. Data (mean ± SEM) are representative of three independent experiments. #p < 0.01 versus GM; *p < 0.05, **p < 0.01, and ***p < 0.001 versus control (DM induction).
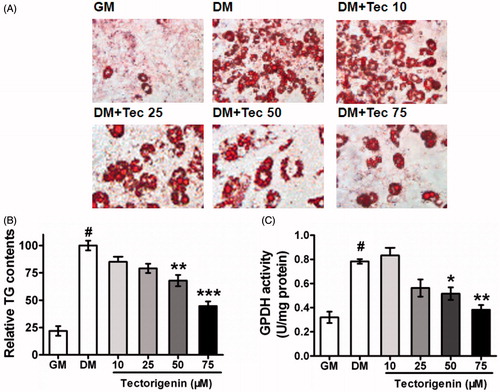
depicted the degree of differentiation by determining the intracellular TG content in 3T3-L1 using the triglyceride GPO-Trinder assay. Tectorigenin at 25 μM, 50 μM, and 75 μM significantly down-regulated intracellular TG levels by 20.9%, 32.1%, and 55.3%, respectively, compared with the control group treated with differentiation medium only. Consistent with the intracellular TG accumulation, GPDH activity was also markedly attenuated in cells treated with tectorigenin ().
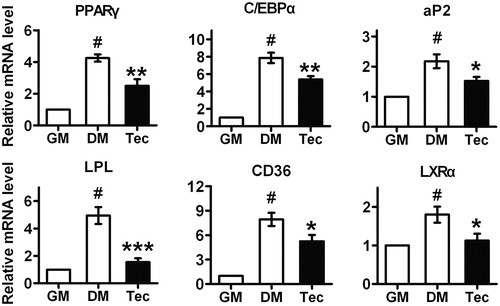
Tectorigenin suppressed the expression of adipogenesis-related genes in 3T3-L1
It has been suggested that PPARγ and C/EBPα are activated during DM induction of 3T3-L1 adipocyte differentiation and PPARγ activity is essential to adipogenesis both in vivo and in vitro (Rosen & Spiegelman, Citation2006). Therefore, we investigated the effect of tectorigenin on the adipogenesis-related transcription factors’ expression by using quantitative real-time PCR. The results indicated that exposure of 3T3-L1 to tectorigenin for 8 d significantly inhibited the transcription of PPARγ and C/EBPα (). Since the transcription of PPARγ was blocked by tectorigenin, we hypothesized that expression of PPARγ target genes may also be inhibited. Indeed, under tectorigenin treatment, the mRNA levels of aP2, LPL, CD36, and LXRα were markedly attenuated during adipocyte differentiation (). These results provided detailed evidence for tectorigenin-mediated anti-adipogenesis.
Figure 2. Effects of tectorigenin on adipogenesis-related genes expression during 3T3-L1 differentiation. At 7 d, after inducing differentiation, total RNA was isolated and mRNA level of the indicated genes were measured by real-time quantitative RT-PCR. GM, growth medium; DM, differentiation medium; Tec, tectorigenin 50 μM. Data (mean ± SEM) are representative of three independent experiments. #p < 0.01 versus GM; *p < 0.05, **p < 0.01, and ***p < 0.001 versus DM induction.
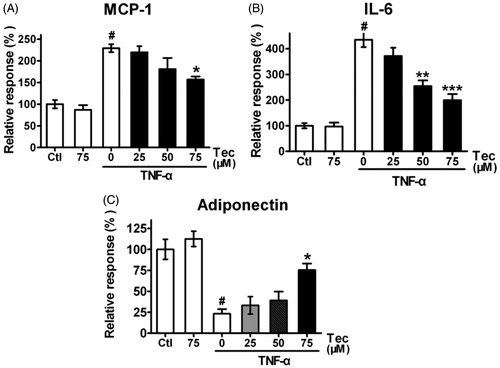
Tectorigenin modulated TNF-α-induced changes of IL-6, MCP-1, and adiponectin secretion
An emerging feature of obesity and type 2 diabetes is their linkage with chronic inflammation that begins in white adipose tissue and eventually becomes systemic (Chuang & McIntosh, Citation2011). Thus, we investigated the impact of tectorigenin on the production of the key pro-inflammatory cytokines, IL-6 and MCP-1, by 3T3-L1 cells. Tectorigenin alone displayed no effect on the secretion of both IL-6 and MCP-1, as presented in . However, tectorigenin inhibited TNF-α-induced IL-6 () and MCP-1 () production, implying the potent anti-inflammatory activities. Moreover, tectorigenin not only promoted the adiponectin secretion but also potently reversed the decrease in adiponectin protein level induced by TNF-α at concentrations from 25 to 75 μM ().
Figure 3. Effects of tectorigenin on adipocytokines expression in 3T3-L1 cells treated with TNF-α. 3T3-L1 adipocytes were pre-incubated with 0–75 μM Tec (tectorigenin) for 6 h and exposed to TNF-α (5 ng/mL) for 16 h. The secreted proteins (A) IL-6, (B) MCP-1, and (C) adiponectin in the conditioned medium were measured using ELISA kits. Data (mean ± SEM) are representative of four independent experiments. #p < 0.01 versus control; *p < 0.05, **p < 0.01, and ***p < 0.001 versus TNF-α (5 ng/mL).
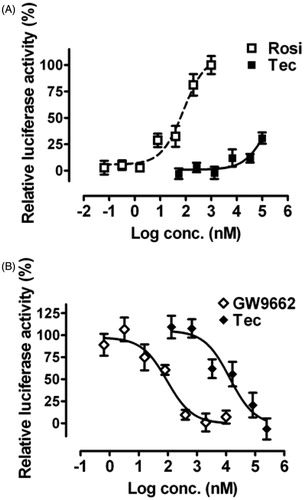
Tectorigenin modulated PPARγ signaling pathway
Thereafter, a mouse PPARγ-PPRE luciferase reporter gene assay was performed to characterize the effects of tectorigenin on PPARγ signaling in HEK293 cells. In the agonist mode, tectorigenin was able to enhance PPARγ transcription from a luciferase reporter gene (pPPRE-luc) under the control of a promoter containing 4 × PPRE, showing a maximum response about 31% compared with the response to rosiglitazone (). Meanwhile, tectorigenin antagonized transcription of reporter gene driven by 0.5 μM rosiglitazone in a dose-dependent manner (IC50 = 13.3 μM; ), thereby pointing to a mechanism of direct interaction between tectorigenin and PPARγ signaling.
Figure 4. Effects of tectorigenin on PPARγ transactivation. Reporter gene assays were performed in HEK293 cells cotransfected with mPPARγ and PPRE-luc plasmids. (A) Tectorigenin moderately activated PPRE-mediated transcription in HEK293 cells. Rosiglitazone (Rosi) was used as a positive control. Data (mean ± SEM) are percentage of 1 μM rosiglitazone (n = 3). (B) Tectorigenin antagonized rosiglitazone (0.5 μM)-mediated PPARγ transactivation (n = 3). GW9662 was used as a positive control.
Tectorigenin increased basal and insulin-stimulated glucose uptake in differentiated 3T3-L1 adipocyte
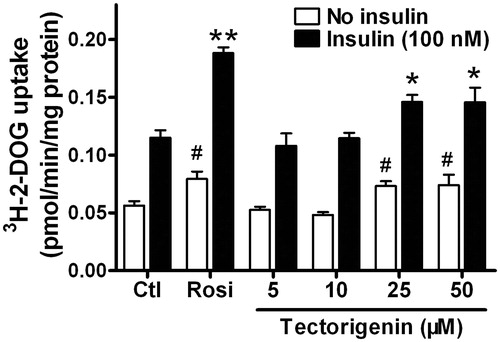
Having established that tectorigenin is a partial PPARγ agonist, we further examined the effects of tectorigenin and rosiglitazone on the glucose uptake in the presence or absence of insulin (100 nM) in differentiated 3T3-L1 adipocytes. As shown in , both basal and insulin-stimulated glucose uptakes were enhanced by tectorigenin in mature 3T3-L1 adipocytes. These results indicated that tectorigenin, similar to rosiglitazone, could ameliorate insulin resistance.
Figure 5. Tectorigenin promoted basal and insulin-stimulated glucose uptake in 3T3-L1 adipocytes. Differentiated 3T3-L1 adipocytes were treated with the indicated concentrations of tectorigenin. After 24 h incubation, 2-DOG uptake in adipocytes was measured in the absence or presence of 100 nM insulin for 30 min. Data (mean ± SEM) are representative of three independent experiments. Rosi, rosiglitazone (1 μM). #p < 0.05 versus the control group without insulin; *p < 0.05 and **p < 0.01 versus the control group with 100 nM insulin.
Tectorigenin inhibited IKK/NF-κB transactivation and p65 nuclear translocation
IKK/NF-κB activation is critical in the development of insulin resistance and metabolic dysfunction. Furthermore, NF-κB is a proinflammatory master switch in response to TNF-α to up-regulate some important inflammation factors, such as IL-6 and MCP-1 (Jain et al., Citation1999; Zhu et al., Citation2008). Inactivated NF-κB stays in cytosol in a complex of the inhibitory subunit IκBα. IκBα undergoes proteasome-dependent degradation immediately by IKK phosphorylation.
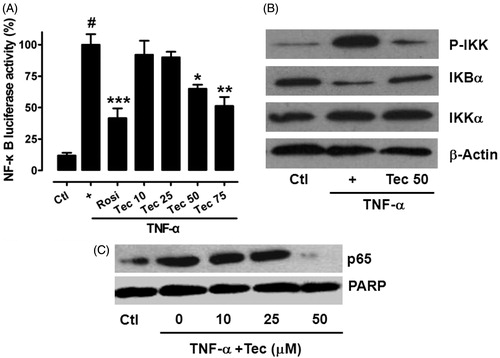
To obtain further evidence that tectorigenin may affect NF-κB pathway, a pNF-κB-Luc under the control of a promoter containing five copies of NF-κB response elements was employed in 3T3-L1 cells. As shown in , tectorigenin significantly inhibited NF-κB transactivation induced by TNF-α. Moreover, tectorigenin (50 μM) markedly attenuated TNF-α-induced IKK activation and IκBα degradation (), as well as the nuclear translocation of p65, a predominant member of NF-κB family (), in 3T3-L1 cells.
Figure 6. Tectorigenin inhibited IKK/NF-κB transactivation and p65 nuclear translocation. (A) Tectorigenin inhibited TNF-α-induced NF-κB transactivation in reporter gene assay. 3T3-L1 adipocytes transfected with pNF-κB-luc were treated with the indicated concentrations of tectorigenin with or without TNF-α (5 ng/mL) for 24 h, and then luciferase activity was determined. Data (mean ± SEM) are representative of three independent experiments. #p < 0.05 versus control; *p < 0.05, **p < 0.01, and ***p < 0.001 versus TNF-α (5 ng/mL). (B) 3T3-L1 adipocytes were treated with Tec at 50 μM for 24 h and then TNF-α at 5 ng/mL. Then cell lysates were resolved by SDS-PAGE and analyzed using antibodies against total and phosphorylated IKK and IκBα. (C) Tectorigenin inhibited translocation of p65 into nucleus as detected by Western blot.
Tectorigenin improved insulin sensitivity in the type 2 diabetic rats
Next, we investigated the in vivo effects of tectorigenin on insulin sensitivity using the type 2 diabetic rat model (HFD/STZ rats). Treatment of type 2 diabetic rats with tectorigenin resulted in an improvement in both fasting and fed glucose concentrations (). In vivo insulin sensitivity was evaluated using the glucose disappearance constant (KITT) during an ITT. In contrast to normal rats (control), the KITT in type 2 diabetic rats (model) decreased significantly, thereby indicating a significant decrease in insulin sensitivity. Administration of tectorigenin for 2 weeks significantly increased insulin sensitivity by 29% at 100 mg/kg dose in model rats (p < 0.01) ().
Figure 7. Effects of tectorigenin on insulin sensitivity in the type 2 diabetic rats. (A) Treatment with tectorigenin reduced the concentrations of blood glucose in both fasting and fed type 2 diabetic rats. Ctl, normal rats group; model, HFD/STZ rats group. #p < 0.05 versus control; *p < 0.05 and **p < 0.01 versus the model group. (B) Tectorigenin increased the glucose disappearance constant (KITT) during an ITT, which represents insulin sensitivity in vivo, in HFD/STZ rats. Ctl, normal rats group; model, HFD/STZ rats group. #p < 0.05 versus control; **p < 0.01 versus the model group. (C) Effects of tectorigenin on insulin-stimulated IRS1 and Akt phosphorylation and glut4 expression in adipose tissue ex vivo. Abdominal adipose tissues from HFD/STZ rats were minced and incubated with Rosi or tectorigenin for 4 h, exposed to 50 nM insulin for 20 min, and then lysed. Shown are western blots to detect the level of IRS1-pTy, IRS1, Akt-pT308, Akt, and glut4. Similar results were obtained from three independent experiments.
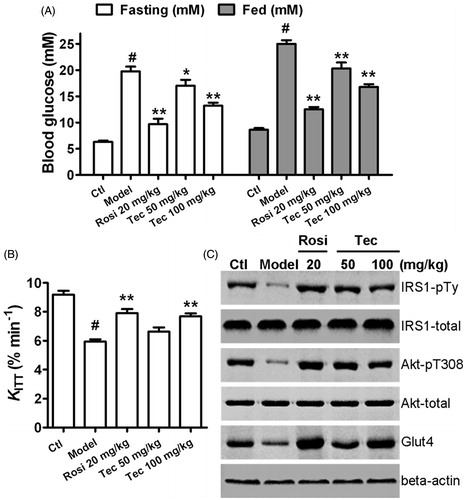
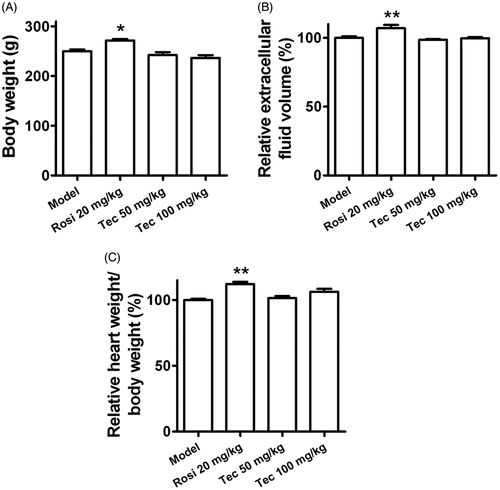
We also evaluated the effects of tectorigenin on insulin-stimulated IRS1 and Akt phosphorylation in adipose tissue ex vivo. As shown in , IRS-pTy and Akt-pT308 were significantly induced in the fat of the control rats and rosiglitazone-/tectorigenin-treated type 2 diabetic rats, but not in vehicle-treated type 2 diabetic rats (model). Furthermore, tectorigenin reversed the downregulation of glut4 expression in adipose tissue (). Taken together, these results suggest that insulin signaling is impaired in the adipose tissues of the type 2 diabetic rats and that such impairment could be improved by tectorigenin. Although tectorigenin displayed the ability to promote insulin sensitivity, it did not cause significant body weight gain, fluid retention, or cardiac hypertrophy ().
Figure 8. Effects of tectorigenin on body weight gain, fluid retention, or cardiac hypertrophy in the type 2 diabetic rats. (A) Rosi (20 mg/kg) increased the body weight of the type 2 diabetic rats, but tectorigenin (50 and 100 mg/kg) did not. Model, HFD/STZ rats group. *p < 0.05 versus the model group. (B) Tectorigenin did not increase the extracellular fluid volume as Rosi did in the type 2 diabetic rats. Model, HFD/STZ rats group. **p < 0.01 versus the model group. (C) Effects of tectorigenin on heart weight of the type 2 diabetic rats. Model, HFD/STZ rats group. **p < 0.01 versus the model group.
Discussion
Tectorigenin showed potent hypoglycemic and hypolipidemic effects in the streptozotocin-induced diabetic rats (Lee et al., Citation2000). However, the exact mechanism of the antidiabetic activity exemplified by tectorigenin remains unknown. In this study, we show for the first time that tectorigenin possesses inhibitory properties on 3T3-L1 differentiation and suppresses inflammatory responses in vitro. In addition, we also demonstrate that tectorigenin served as a PPARγ partial agonist in reporter gene assay and markedly improved basal and insulin-stimulated glucose uptake in mature 3T3-L1 cell system. Further experiments confirmed that tectorigenin could inhibit NF-κB signal pathway. Therapeutic effects of tectorigenin in HFD/STZ rats showed that it could improve insulin resistance and lower blood glucose, but evoke fewer side effects. Thus, our data provide potential molecular targets through which tectorigenin manifests it as a possible antidiabetic agent.
Activation of PPARγ by full agonist has been verified to cause adipocyte differentiation and increased adipose tissue mass, contributing to weight gain. Thus, a different approach to leveraging PPARγ antidiabetic therapeutic benefits would focus on minimizing side effects by limiting the spectrum of activation (Kintscher & Goebel, Citation2009). Based on this strategy, several PPARγ modulators including INT131, SPPARγM5, and halofenate were developed (Allen et al., Citation2006; Chang et al., Citation2008; Kintscher & Goebel, Citation2009). For example, INT131 shows selectivity among the full spectrum of PPARγ effects and has the desired, non-adipogenic profile (Kintscher & Goebel, Citation2009; Motani et al., Citation2009). The dissociation between antidiabetic effects and adverse effects exemplified by new generation of PPARγ modulators may represent a better therapeutic profile. In this study, identification of tectorigenin as a partial PPARγ agonist with potent hypoglycemic effects but minimal adipogenesis may contribute to the development of a novel class antidiabetic drug candidate.
It has been observed that inflammatory molecule MCP-1 upregulates in adipose tissues of obese and dysfunction, thus causing macrophage infiltration of the adipose tissue and inducing systemic insulin resistance (Xu et al., Citation2003). The level of adiponectin decreases in adipocytes and body circulation in insulin-resistant individuals (Hassan et al., Citation2007). Increasing evidence suggests that anti-inflammatory therapy improves insulin resistance because of the important pathogenic roles of cytokines in diabetes and obesity. Our results indicate that tectorigenin could downregulate TNF-α-induced expression of MCP-1 and IL-6. In addition, tectorigenin reversed TNF-α-induced changes of secretion of adiponectin, thus contributing to enhanced glucose uptake.
NF-κB is an ubiquitous transcription factor whose involvement in inflammatory disorders is well established in both in vitro and in vivo experimental systems (De Bosscher et al., Citation2003). NF-κB also plays a pivotal role in the TNF-α-induced upregulation of adipocytokines including IL-6 and MCP-1 in adipocytes (Jain et al., Citation1999; Zhu et al., Citation2008). Tectorigenin, as a partial PPARγ agonist, inhibited transcriptional activation of NF-κB. Furthermore, translocation of p65 to the nucleus, the key step to initiate the transcription of a plethora of inflammatory genes under the control of NF-κB, was blocked by tectorigenin. It is likely that the improved inflammatory response in adipocyte is a consequent of NF-κB antagonism by tectorigenin treatment.
Tectorigenin demonstrated insulin-sensitizing and hypoglycemic activities in the type 2 diabetic rat model. However, this partial PPARγ agonist did not promote body weight gain and fluid retention, or cause cardiac hypertrophy as the full agonist, rosiglitazone, did. The observation that tectorigenin potentiated insulin signaling pathway in adipose tissue ex vivo may have important implications for partial PPARγ agonists as long-term therapeutics. Since the major pharmacological activities of PPARγ modulators are thought to be mediated by alterations in gene expression, further investigation of tectorigenin will focus on its effects on gene expression profiles in vivo.
Conclusion
The present study provides new experimental evidence supporting the postulated property of tectorigenin as a functional PPARγ partial agonist capable of inhibiting 3T3-L1 differentiation and inflammatory responses. Furthermore, the anti-diabetic effects exerted by tectorigenin both in vitro and in vivo are not accompanied with adipogenesis. These findings presented here highlight the possible therapeutic use of tectorigenin in metabolic disorders.
Acknowledgements
We thank Dr. Ronald M. Evans for providing mouse PPARγ vector.
Declaration of interest
The authors report that they have no declarations of interest. This study was supported by grants from the National Natural Science Foundation of China (No. 81302853), Specialized Research Fund for the Doctoral Program of Higher Education of China (No. 20110071120071), Shanghai Bureau of Health (Nos. 2009Y009 and 2009Y069) and Shanghai Xuhui District Central Hospital (No. 2011XHCH01).
References
- Allen T, Zhang F, Moodie SA, et al. (2006). Halofenate is a selective peroxisome proliferator-activated receptor gamma modulator with antidiabetic activity. Diabetes 55:2523–33
- Chang CH, McNamara LA, Wu MS, et al. (2008). A novel selective peroxisome proliferator-activator receptor-gamma modulator-SPPARgammaM5 improves insulin sensitivity with diminished adverse cardiovascular effects. Eur J Pharmacol 584:192–201
- Chuang CC, McIntosh MK. (2011). Potential mechanisms by which polyphenol-rich grapes prevent obesity-mediated inflammation and metabolic diseases. Annu Rev Nutr 31:155–76
- De Bosscher K, Vanden Berghe W, Haegeman G. (2003). The interplay between the glucocorticoid receptor and nuclear factor-kappaB or activator protein-1: Molecular mechanisms for gene repression. Endocr Rev 24:488–522
- Fang R, Houghton PJ, Hylands PJ. (2008). Cytotoxic effects of compounds from Iris tectorum on human cancer cell lines. J Ethnopharmacol 118:257–63
- Guilherme A, Virbasius JV, Puri V, Czech MP. (2008). Adipocyte dysfunctions linking obesity to insulin resistance and type 2 diabetes. Nat Rev Mol Cell Biol 9:367–77
- Han Y, Jung HW, Bae HS, et al. (2013). The extract of Cinnamomum cassia twigs inhibits adipocyte differentiation via activation of the insulin signaling pathway in 3T3-L1 preadipocytes. Pharm Biol 51:961–7
- Hassan M, EI Yazidi C, Landrier JF, et al. (2007). Phloretin enhances adipocyte differentiation and adiponectin expression in 3T3-L1 cells. Biochem Biophys Res Commun 361:208–13
- Hong Y, Kim MY, Yoon M. (2011). The anti-angiogenic herbal extracts Ob-X from Morus alba, Melissa officinalis, and Artemisia capillaris suppresses adipogenesis in 3T3-L1 adipocytes. Pharm Biol 49:775–83
- Jain RG, Phelps KD, Pekala PH. (1999). Tumor necrosis factor-alpha initiated signal transduction in 3T3-L1 adipocytes. J Cell Physiol 179:58–66
- Kintscher U, Goebel M. (2009). INT-131, a PPARgamma agonist for the treatment of type 2 diabetes. Curr Opin Investig Drugs 10:381–7
- Lee HW, Choo MK, Bae EA, Kim DH. (2003). Beta-glucuronidase inhibitor tectorigenin isolated from the flower of Pueraria thunbergiana protects carbon tetrachloride-induced liver injury. Liver Int 23:221–6
- Lee KT, Sohn IC, Kim DH, et al. (2000). Hypoglycemic and hypolipidemic effects of tectorigenin and kaikasaponin III in the streptozotocin-induced diabetic rat and their antioxidant activity in vitro. Arch Pharm Res 23:461–6
- Livak KJ, Schmittgen TD. (2001). Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods 25:402–8
- Moon HI, Jung JC, Lee J. (2006). Aldose reductase inhibitory effect by tectorigenin derivatives from Viola hondoensis. Bioorg Med Chem 14:7592–4
- Motani A, Wang Z, Weiszmann J, et al. (2009). INT131: A selective modulator of PPAR gamma. J Mol Biol 386:1301–11
- Rosen ED, Sarraf P, Troy AE, et al. (1999). PPAR gamma is required for the differentiation of adipose tissue in vivo and in vitro. Mol Cell 4:611–17
- Rosen ED, Spiegelman BM. (2006). Adipocytes as regulators of energy balance and glucose homeostasis. Nature 444:847–53
- Takahashi N, Goto T, Taimatsu A, et al. (2009). Bixin regulates mRNA expression involved in adipogenesis and enhances insulin sensitivity in 3T3-L1 adipocytes through PPARgamma activation. Biochem Biophys Res Commun 390:1372–6
- Tontonoz P, Spiegelman BM. (2008). Fat and beyond: The diverse biology of PPARgamma. Annu Rev Biochem 77:289–312
- van Bilsen M, van Nieuwenhoven FA. (2010). PPARs as therapeutic targets in cardiovascular disease. Expert Opin Ther Targets 14:1029–45
- Vigne R, Gourdou I, Mazarin V, et al. (1990). Regulatory genes of visna virus. Dev Biol Stand 72:213–22
- Wu Y, Ouyang JP, Wu K, et al. (2005). Rosiglitazone ameliorates abnormal expression and activity of protein tyrosine phosphatase 1B in the skeletal muscle of fat-fed, streptozotocin-treated diabetic rats. Br J Pharmacol 146:234–43
- Xu H, Barnes GT, Yang Q, et al. (2003). Chronic inflammation in fat plays a crucial role in the development of obesity-related insulin resistance. J Clin Invest 112:1821–30
- Zhu J, Yong W, Wu X, et al. (2008). Anti-inflammatory effect of resveratrol on TNF-alpha-induced MCP-1 expression in adipocytes. Biochem Biophys Res Commun 369:471–7