Abstract
In the pathway of anticancer drug development, we designed and synthesized some 6H-indolo[2,3-b]quinoxaline derivatives (which act as DNA intercalators) by structural modification. The structure of the 6H-indolo[2,3-b]quinoxaline derivatives was confirmed by IR, NMR, Mass and elemental analysis. The compounds (IDQ-5, IDQ-10, IDQ-11, IDQ-13, and IDQ-14) exhibited significant in vitro activity against a human leukemia (HL-60) cell line. The QSAR derived for modeling the cytotoxic activity of 6H-indolo[2,3-b]quinoxaline derivatives suggests that candidate structures for increased cytotoxic potency should incorporate cyclic substituents or substituents with primary carbon atoms.
Introduction
Cancer is the second most common cause of death in the world, after heart disease. The World Health Organization (WHO) estimated that by 2020, 20 million new cases of cancer will be diagnosed each year, and 75% of these will occur in nations that between them have only 5% of resources. At present, it is estimated that ∼1 million new cancer cases per annum will be recorded. The effectiveness of cancer chemotherapy is mostly limited by the lack of selectivity of anticancer agents and the occurrence of intrinsic or acquired resistance, leading to significant side effects and sometimes failure of treatmentsCitation1–4.
Anticancer drug toxicities include nausea, vomiting, alopecia, bone marrow depression, and many more. In the course of anticancer drug development, it is necessary to develop extremely potent and less toxic compounds. The discovery of new compounds with antitumoral activity has become one of the goals of medicinal chemistryCitation6,Citation7. Drug design is an effort to develop drugs on a rational basis. The design of drugs reduces trial and error factors as well as the cost of screening large numbers of molecules. Analog design is most fruitful in the study of pharmacologically active molecules that are structurally specific. Their biological activity depends on the nature and details of their chemical structure.
A literature survey showed that over the past 10 years, the search for new cytotoxic agents has mainly followed classical approaches: structural modifications to conventional molecules, new natural products and modifications of them, potential synthetic compounds and chemical conjugates. Research in this area has not been without benefits, however, for it has produced much information on the synthesis and antitumoral properties of hundreds of compounds, which have been tested on diverse tumoral cell linesCitation8–12. In the present work, it was attempted to design and synthesize some compounds with the 6H-indolo[2,3-b]quinoxaline nucleus as polycyclic pharmacophore and evaluate their cytotoxic activity.
6H-Indolo-[2,3-b]quinoxaline is an analog of the cytotoxic agent ellipticine. The first 6H-indolo-[2,3-b]quinoxaline derivatives were prepared in 1895 by the condensation of 1,2-diaminobenzene with isatin; these compounds have the combined structural features of indoles and quinoxalinesCitation13. In recent years, 6H-indolo[2,3-b]quinoxaline derivatives have received much attention, due to their variety of pharmacological activities such as anticancer activity and antiviral activity against herpes simplex virus type 1 (HSV-1), cytomegalovirus (CMV), varicella-zoster virus (VZV), etc.Citation14–16. It has been reported that the mechanism of the antiviral and cytotoxic action of the derivatives involves intercalation into the DNA helix and, thus, disturbance of the processes that are vital for DNA replicationCitation14,Citation17. Hence, the indolo[2,3-b]quinoxaline nucleus was considered as a template for the design and development of DNA intercalating agents with cytotoxic activities in this study.
Chemistry
The 6H-indolo[2,3-b]quinoxaline derivatives were synthesized as illustrated in Scheme 1. Isatins, which served as key intermediates, were prepared from the appropriately substituted anilines using Sandmeyer methodologyCitation18,Citation19. Condensation of the synthesized isatins with the appropriate o-phenylenediamines in glacial acetic acid or hydrochloric acid and ethanol or formic acid at reflux temperature afforded 6H-indolo[2,3-b]quinoxaline derivatives (IDQ-1 to IDQ-4, IDQ-7, and IDQ-9) in good yields. The N-ethyl derivatives of 6H-indolo[2,3-b]quinoxalines (IDQ-6 and IDQ-8) were generated by ethyl iodide–potassium carbonate alkylation of compounds IDQ-1 and IDQ-7 at 85°C for 15 h. 6H-Indolo[2,3-b]quinoxaline-7-carboxamide (IDQ-5) was obtained from amide bond formation between an activated form of the corresponding acid (IDQ-2) and dimethyl amine. The synthesis of N-alkyl-6H-indolo[2,3-b]quinoxaline-9-amine (IDQ-10 to IDQ-14) was achieved by heating a reaction mixture containing 9-bromo-6H-indolo[2,3-b]quinoxaline (IDQ-7) and the appropriate secondary amine in dimethylformamide (DMF) at 120°C.
Scheme 1. Synthesis of 6H-indolo[2,3-b]quinoxaline derivatives. A: Conc. HCl, sodium sulfate, hydroxylamine HCl, reflux for 15 min; B: PPA/H2SO4, 56°C for 6 h/120°C for 6 h; C, D: o-phenylenediamine or diaminobenzoic acid, acetic acid/HCOOH, reflux for 1–7 h; E: CDI, dry DMF, stir at room temperature for 4 h, DCM, MgSO4; F: dry DMF, 120°C, 30 min–2 h; G: K2CO3, dry DMF, 85–100°C for 12–16 h.
![Scheme 1. Synthesis of 6H-indolo[2,3-b]quinoxaline derivatives. A: Conc. HCl, sodium sulfate, hydroxylamine HCl, reflux for 15 min; B: PPA/H2SO4, 56°C for 6 h/120°C for 6 h; C, D: o-phenylenediamine or diaminobenzoic acid, acetic acid/HCOOH, reflux for 1–7 h; E: CDI, dry DMF, stir at room temperature for 4 h, DCM, MgSO4; F: dry DMF, 120°C, 30 min–2 h; G: K2CO3, dry DMF, 85–100°C for 12–16 h.](/cms/asset/ee5e66ec-f8de-426f-a114-bb7c64b20c50/ienz_a_419247_f0001_b.gif)
Experimental
Materials used
Reagents, starting materials, and solvents were purchased from common commercial suppliers (CDH, S.D. Fine, Loba, E. Merck, Lancaster, etc.). The melting points were determined in open capillary tubes on a Jindal melting point apparatus and are reported uncorrected. H1 nuclear magnetic resonance (NMR) spectra were recorded on a Bruker Avance DRX-2000 (300 MHz, Fourier transform (FT) NMR) spectrometer in dimethylsulfoxide (DMSO) and CDCl3, with tetramethylsilane (TMS) as internal standard. Fast atom bombardment (FAB) mass spectra were recorded on a Jeol SX 102/DA-6000 mass spectrometer/data system using argon/xenon (6 kV, 10 mA) as the FAB gas. The accelerating voltage was 10 kV and the spectra were recorded at room temperature. m-Nitrobenzyl alcohol (NBA) was used as the matrix. Infrared (IR) absorption spectra were recorded on a Jasco FT/IR – 470 Plus machine, using KBr by diffuse reflectance method. Ultraviolet (UV) absorption studies were done on a Shimadzu Pharmaspec-UV 1700 spectrophotometer to determine λmax of the compounds. Elemental analysis (C, H, N) was done on a PerkinElmer CHN rapid analyzer. All the compounds gave satisfactory analysis within ±0.4% of the theoretical values. The purity and reaction progress were monitored by thin layer chromatography (TLC) using silica gel-G on a glass plate as the stationary phase and different polarities of solvents as the mobile phase. Visualization was accomplished with UV light and/or iodine vapor.
Synthesis of 6H-indolo[2,3-b]quinoxaline derivatives
The synthesis of the selected 6H-indolo[2,3-b]quinoxaline derivatives is outlined in .
Isatin
Isatin was synthesized as per the reported procedureCitation18,Citation19.
Isatin-7-carboxylic acid
Isatin-7-carboxylic acid was synthesized as per the reported procedureCitation18,Citation19.
Isatin-5-carboxylic acid
A solution of p-aminobenzoic acid (3.15 g), chloral hydrate (3.5 g), and hydroxylamine hydrochloride (2.86 g) in concentrated sulfuric acid (2.5 mL) and water (150 mL) was heated at 90°C for 30 min and then kept at 4°C for 16 h. The cream-colored isonitroso intermediate (29.8 g (64%)) was filtered off, washed with water, and dried. This compound (1.5 g) was added with stirring in portions over 30 min to concentrated sulfuric acid (7.5 g) maintained at 60–65°C. The mixture was then heated at 95°C for 1 h and poured onto ice (60 g). The resulting brown solid was filtered, dissolved in 1M NaOH solution, and filtered, and the filtrate was taken to pH 2 with concentrated HCl to give isatin-5-carboxylic acid. (65%, MP 274–275°C).
5-Bromo-isatin
This was synthesized as per the reported procedureCitation18,Citation19.
6H-Indolo[2,3-b]quinoxaline (IDQ-1)
A solution of isatin (10 mmol) and o-phenylenediamine (30 mmol) in acetic acid (10 mL) was refluxed for 15 min and the precipitate obtained on cooling for 10 h. A pale reddish-yellow color precipitate was obtained. The product was filtered and washed with water. The crude product was extracted with chloroform and the obtained product was recrystallized from acetic acid. TLC was performed with methanol:chloroform (3:7).
1H NMR (300 MHz, δ/ppm in DMSO-d6): 4.9 (br, s, 1H, NH), 6.55–6.57 (d, J = 5.5 Hz, 1H, Ar-H), 6.75–6.77 (d, J = 6.1 Hz, 1H, Ar-H), 7.07–7.10 (d, J = 9.2 Hz, 1H, Ar-H), 7.32–7.34 (d, J = 6.2 Hz, 1H, Ar-H), 7.60–7.62 (d, J = 6.2 Hz, 2H, Ar-H), 7.71–7.73 (d, J = 9.0 Hz, 1H, Ar-H), 7.95 (d, J = 5 Hz, 1H, Ar-H); IR (KBr/cm−1): 3452 (N-H, stretching), 3057 (C-H (Ar, stretching)), 1531 (C=N, stretching); FAB-MS m/z [M]+: 219; Elemental Analysis: Calcd. (Found) (%) for C14H9N3: C 76.70 (76.77), H 4.14 (4.26), N 19.17 (19.25).
6H-Indolo[2,3-b]quinoxaline-3-carboxylic acid (IDQ-2)
A solution of isatin (10 mmol) and diaminobenzoic acid (30 mmol) in acetic acid (10 mL) and ethanol (5 mL) was refluxed for 30 min and the precipitate obtained on cooling for 10 h. A reddish-yellow color precipitate was obtained. The product was filtered and washed with water. The crude product was extracted with chloroform and ethyl acetate and the obtained product was recrystallized from DMSO. TLC was monitored with acetic acid:methanol:chloroform (1:3:6).
1H NMR (300 MHz, δ/ppm in CDCl3): 6.25 (br, s, 1H, NH), 6.66–6.68 (d, J = 5.5 Hz, 1H, Ar-H), 6.79–6.81 (d, J = 6.1 Hz, 1H, ArH), 7.07–7.25 (m, 3H, Ar-H), 7.53–7.5 (d, J = 9.0 Hz, 1H, Ar-H), 7.72–7.73 (d, J = 9.2 Hz, 1H, ArH), 12.69 (br, s, 1H, COOH); IR (KBr/cm−1): 3061 (C-H (Ar, stretching)), 1771 (C=O carboxylic, stretching), 1514 (C=N, stretching), 1427 (C-O-H carboxylic, stretching); FAB-MS m/z [M]+: 263; Elemental Analysis: Calcd. (Found) (%) for C15H9N3O2: C 68.44 (68.47), H 3.45 (3.46), N 15.96 (15.95).
6H-Indolo[2,3-b]quinoxaline-7-carboxylic acid (IDQ-3)
A solution of isatin-7-carboxylic acid (10 mmol) and o-phenylenediamine (30 mmol) in acetic acid (10 mL) and DMF (1 mL) was refluxed for 30 min and a pink-yellow color precipitate was obtained on cooling for 10 h. The product was filtered and washed with water. The obtained product was recrystallized from DMSO. TLC was monitored with acetic acid:methanol:chloroform (1:3:6).
1H NMR (300 MHz, δ/ppm in CDCl3): 5.6 (br, s, 1H, NH), 7.40–7.42 (d, J = 5.4 Hz, 1H, Ar-H), 7.53–7.55 (d, J = 6.1 Hz, 1H, Ar-H), 7.69–7.71 (d, J = 5.5 Hz, 1H, Ar-H), 8.09–8.11 (d, J = 6.6 Hz, 1H, Ar-H), 8.32–8.34 (d, J = 6.0 Hz, 1H, Ar-H), 8.48–8.50 (d, J = 5.8 Hz, 1H, Ar-H), 9.06 (s, 1H, Ar-H), 12.68 (br, s, 1H, COOH); IR (KBr/cm−1): 3575 (N-H polynuclear, stretching), 3102 (C-H (Ar, stretching)), 1706 (C=O carboxylic, stretching), 1515 (C=N, stretching), 1464 (C-O-H carboxylic, stretching); FAB-MS m/z [M]+: 263; Elemental Analysis: Calcd. (Found) (%) for C15H9N3O2: C 68.44 (68.64), H 3.45 (3.25), N 15.96 (15.78).
6H-Indolo[2,3-b]quinoxaline-9-carboxylic acid (IDQ-4)
A solution of isatin-9-carboxylic acid (10 mmol) and o-phenylenediamine (30 mmol) in acetic acid (10 mL) and DMF (1 mL) was refluxed for 30 min and a pink-yellow color precipitate was obtained on cooling for 10 h. The product was filtered and washed with water. The obtained product was recrystallized from DMSO. TLC was monitored with acetic acid:methanol:chloroform (1:3:6).
1H NMR (300 MHz, δ/ppm in DMSO-d6): 5.7 (br, s, 1H, N-H), 7.17–7.19 (m, 3H, Ar-H), 7.38–7.40 (d, J = 6.0 Hz, 1H, Ar-H), 7.57–7.59 (d, J = 6.3 Hz, 1H, Ar-H), 7.93–7.94 (d, J = 9.0 Hz, 1H, Ar-H), 8.17–8.20 (d, J = 9.3 Hz, 1H, Ar-H), 8.25 (br, s, 1H, Ar-H); IR (KBr/cm−1): 3060 (C-H (Ar, stretching)), 1774 (C=O carboxylic, stretching), 1524 (C=N, stretching), 1422 (C-O-H carboxylic, stretching); FAB-MS m/z [M]+: 263; Elemental Analysis: Calcd. (Found) (%) for C15H9N3O2: C 68.44 (68.30), H 3.45 (3.40), N 15.96 (15.95).
6H-Indolo[2,3-b]quinoxaline-3-carboxylic acid dimethylamide (IDQ-5)
A mixture of IDQ-2 (10 mmol) and CDI (N,N-carbonyl diimidazole) (130 mg) was stirred with dry DMF (8 mL) at room temperature for 48 h. To this mixture, dimethylamine (10 mmol) was added and stirred at room temperature for a further 30 min. The volatile components were then removed under vacuum and the residue was dissolved in dichloromethane (20 mL) and washed with water (2 × 20 mL) and dried over magnesium sulfate. The solvent was removed under vacuum to provide the crude product which was recrystallized with DMSO. TLC was developed with methanol:chloroform (4:6).
1H NMR (300 MHz, δ/ppm in DMSO-d6): 2.89 (s, 6H, CON(CH3)2), 6.15 (br, S, 1H, NH), 7.00–7.20 (m, 3H, Ar-H), 7.36–7.38 (d, J = 6.2Hz, 1H, Ar-H), 7.58–7.60 (d, J = 6.0 Hz, 1H, Ar-H), 7.65–7.68 (d, J = 9.2 Hz, 1H, Ar-H), 7.93–7.96 (d, J = 9.0 Hz, 1H, Ar-H); IR (KBr/cm−1): 3095 (C-H (Ar, stretching)), 2803 (C-H, stretching), 1692 (C=O amide, stretching), 1515 (C=N, stretching). FAB-MS m/z [M]+: 290; Elemental Analysis: Calcd. (Found) (%) for C17H14N4O: C 70.33 (70.30), H 4.86 (4.80), N 19.30 (19.25).
6-Ethyl-indolo[2,3-b]quinoxaline (IDQ-6)
A mixture of 6H-indolo[2,3-b]quinoxaline (10 mmol), ethyl iodide (20 mmol), and potassium carbonate (22 mmol) in dry DMF (35 mL) was heated at 85°C for 12 h. After cooling, the precipitate formed was filtered, washed with water, and recrystallized in DMF. TLC was performed using methanol:chloroform (3:7)
1H NMR (300 MHz, δ/ppm in DMSO-d6): 1.24–1.28 (t, J = 6.9 Hz, 3H, CH3), 3.11–3.23 (m, 2H, CH2), 7.70–7.71 (m, 4H, Ar-H), 8.11–8.15 (dd, J = 1.5, 8.9 Hz, 2H, Ar-H), 8.24–8.27 (dd, J = 1.5, 8.6 Hz, 2H, Ar-H); IR (KBr/cm−1): 3102 (C-H (Ar, stretching)), 2904 (C-H, stretching), 1547–1490 (C=N, stretching); FAB-MS m/z [M]+: 247; Elemental Analysis: Calcd. (Found) (%) for C16H13N3: C 77.71 (77.78), H 5.30 (5.26), N 16.99 (16.97).
9-Bromo-6H-indolo[2,3-b]quinoxaline (IDQ-7)
A solution of 5-bromo-isatin (10 mmol) and o-phenylenediamine (30 mmol) in formic acid (10 mL) was refluxed for 15 min and a reddish-yellow color precipitate was obtained on cooling for 10 h. The product was filtered and washed with water. The crude product was dissolved in hot DMSO and filtered. TLC was monitored with methanol:chloroform (3:7).
1H NMR (300 MHz, δ/ppm in DMSO-d6): 5.7 (br, s, 1H, NH), 7.31 (s, 1H, Ar-H), 7.49–7.50 (d, J = 1.2 Hz, 1H, Ar-H), 7.59–7.60 (d, J = 1.2 Hz, 1H, Ar-H), 8.08–8.11 (dd, J = 1.2, 8.4 Hz, 2H, Ar-H), 8.24–8.28 (dd, J = 1.2, 8.4 Hz, 2H, Ar-H); IR (KBr/cm−1): 3122 (C-H (Ar, stretching)), 1545 (C=N, stretching), 950 (C-Br, bending); FAB-MS m/z [M]+: 297; Elemental Analysis: Calcd. (Found) (%) for C14H8N3Br: C 56.40 (56.28), H 2.70 (2.76), N 14.09 (13.97).
6-Ethyl-9-bromo-indolo[2,3-b]quinoxaline (IDQ-8)
A mixture of 9-bromo-indolo[2,3-b]quinoxaline (10 mmol), ethyl iodide (20 mmol), and potassium carbonate (22 mmol) in dry DMF (35 mL) was heated at 85°C for 15 h. After cooling, the precipitate was filtered, washed with water, and recrystallized in DMF. TLC was run on methanol:chloroform (3:7).
1H NMR (300 MHz, δ/ppm in DMSO-d6): 1.23–1.27 (t, J = 6.9 Hz, 3H, CH3), 3.24–3.43 (m, 2H, CH2), 7.29 (s, 1H, Ar-H), 7.51–7.53 (d, J = 1.6 Hz, 1H, Ar-H), 7.62–7.64 (d, J = 1.6 Hz, 1H, Ar-H), 8.11–8.15 (dd, J = 1.6, 8.9 Hz, 2H, Ar-H), 8.28–8.32 (dd, J = 1.6, 8.9 Hz, 2H, Ar-H); IR (KBr/cm−1): 3140 (C-H (Ar, stretching)), 2965 (C-H, stretching), 1567 (C=N, stretching), 990 (C-Br, bending); FAB-MS m/z [M]+: 326; Elemental Analysis: Calcd. (Found) (%) for C16H12N3Br: C 58.91 (58.78), H 3.71 (3.66), N 12.88 (12.97).
9-Bromo-[6H-indolo[2,3-b]quinoxaline]-3-carboxylic acid (IDQ-9)
A solution of 5-bromo-isatin (10 mmol) and diaminobenzoic acid (30 mmol) in acetic acid (10 mL) and ethanol (5 mL) was refluxed for 30 min and a greenish-yellow color precipitate was obtained on cooling for 10 h. The crude product was filtered and washed with water and recrystallized in DMSO. TLC was monitored with acetic acid:methanol:chloroform (1:3:6).
1H NMR (300 MHz, δ/ppm in DMSO-d6): 6.02 (br, s, 1H, NH), 7.59 (s, 1H, Ar-H), 7.81–7.82 (d, J = 1.6 Hz, 1H, Ar-H), 7.84–7.85 (d, J = 1.8 Hz, 1H, Ar-H), 8.05–8.06 (d, J = 1.8 Hz, 1H, Ar-H), 8.08–8.09 (d, J = 1.5 Hz, 1H, Ar-H), 8.25 (s, 1H, Ar-H), 10.25 (s, 1H, COOH); IR (KBr/cm−1): 3110 (C-H (Ar, stretching)), 1742 (C=O carboxylic, stretching), 1514 (C=N, stretching), 1462 (C-O-H carboxylic, stretching), 978 (C-Br, bending); FAB-MS m/z [M]+: 342; Elemental Analysis: Calcd. (Found) (%) for C15H8N3O2Br: C 52.66 (52.78), H 2.36 (2.36), N 12.28 (12.17).
N,N-Dimethyl-[6H-indolo[2,3-b]quinoxaline]-9-amine (IDQ-10)
A mixture of 9-bromo-6H-indolo[2,3-b]quinoxaline derivative (5.5 mmol) and dimethylamine (30 mmol) in dry DMF (5 mL) was heated at 120°C for 1 h. After cooling, the precipitate was filtered off and digested with hot ethyl acetate to give the product. The crude product was recrystallized with DMF/DMSO and TLC was monitored with methanol:chloroform (3:7).
1H NMR (300 MHz, δ/ppm in DMSO-d6): 2.51 (br, s, 6H, N(CH3)2), 5.90 (s, 1H, NH), 7.36 (s, 1H, Ar-H), 7.61–7.62 (d, J = 1.9 Hz, 1H, Ar-H), 7.72–7.73 (d, J = 1.5 Hz, 1H, Ar-H), 8.08–8.11 (dd, J = 1.2, 9.6 Hz, 2H, Ar-H), 8.24–8.27 (dd, J = 1.2, 9.6 Hz, 2H, Ar-H); IR (KBr/cm−1): 3120 (C-H (Ar, stretching)), 2890 (C-H, stretching), 1535 (C=N, stretching), 1050 (C-N, bending); FAB-MS m/z [M]+: 262; Elemental Analysis: Calcd. (Found) (%) for C16H14N4: C 73.26 (73.38), H 5.38 (5.16), N 21.36 (21.47).
N,N-Diethyl-[6H-indolo[2,3-b]quinoxaline]-9-amine (IDQ-11)
A mixture of 9-bromo-6H-indolo[2,3-b]quinoxaline derivative (5.5 mmol) and diethylamine (30 mmol) in dry DMF (5 mL) was heated at 120°C for 2–3 h. After cooling, the precipitate was filtered off and digested with hot ethyl acetate to give the product. The crude product was recrystallized with DMF/DMSO and TLC was monitored with methanol:chloroform (4:6).
1H NMR (300 MHz, δ/ppm in DMSO-d6): 1.2–1.26 (t, J = 6.9 Hz, 6H, (CH3)2), 3.20–3.34 (m, 4H, CH2), 5.98 (br, s, 1H, NH), 6.48–6.51 (d, J = 1.5 Hz, 1H, Ar-H), 8.06–8.10 (dd, J = 1.5, 8.6 Hz, 2H, Ar-H), 8.11–8.15 (d, 2H, Ar-H), 8.25–8.29 (dd, J = 1.5, 8.6 Hz, 2H, Ar-H); IR (KBr/cm−1): 3115 (C-H (Ar, stretching)), 2985 (C-H, stretching), 1535 (C=N, stretching); FAB-MS m/z [M + H]+: 290; Elemental Analysis: Calcd. (Found) (%) for C18H18N4: C 74.46 (74.78), H 6.25 (6.16), N 19.30 (19.28).
4-(6H-Indolo[2,3-b]quinoxalin-9-yl)morpholine (IDQ-12)
A mixture of 9-bromo-6H-indolo[2,3-b]quinoxaline derivative (5.5 mmol) and N-morpholine (30 mmol) in dry DMF (5 mL) was heated at 120°C for 30 min–2 h. After cooling, the precipitate was filtered off and digested with hot ethyl acetate to give the product. The crude product was recrystallized with DMF/DMSO and TLC was monitored with methanol:chloroform (3:7).
1H NMR (300 MHz, δ/ppm in DMSO-d6): 2.69–2.75 (m, 4H, CH2 morphonyl), 3.34–3.46 (m, 4H, CH2, morphonyl), 6.03 (br, s, 1H, NH), 7.33–7.36 (d, J = 7.8 Hz, 1H, Ar-H), 7.60 (s, 1H, Ar-H), 7.73–7.75 (d, J = 6.9 Hz, 1H Ar-H), 8.08–8.11 (dd, J = 1.2, 8.9 Hz, 2H, Ar-H), 8.25–8.28 (dd, J = 1.5, 8.9 Hz, 2H, Ar-H); IR (KBr/cm−1): 3150 (C-H (Ar, stretching)), 2885 (C-H, stretching), 1572 (C=N, stretching), 1475 (C-O, stretching); FAB-MS m/z [M]+: 304; Elemental Analysis: Calcd. (Found) (%) for C18H16N4O: C 71.04 (71.08), H 5.30 (5.26), N 18.41 (18.57).
9-(Piperidin-1-yl)-6H-indolo[2,3-b]quinoxaline (IDQ-13)
A mixture of 9-bromo-6H-indolo[2,3-b]quinoxaline derivative (5.5 mmol) and N-piperidine (30 mmol) in dry DMF (5 mL) was heated at 120°C for 5 h. After cooling, the precipitate was filtered off and digested with hot ethyl acetate to give the product. The crude product was recrystallized with DMF/DMSO and TLC was monitored with methanol:chloroform (3:7).
1H NMR (300 MHz, δ/ppm in DMSO-d6): 1.88–1.93 (m, 6H, CH2, piperidinyl), 2.50–2.52 (m, 4H, CH2, piperidinyl), 6.2 (br, s, 1H, NH), 7.59 (s, 1H, Ar-H), 7.78–7.79 (d, J = 1.2 Hz, 2H, Ar-H), 8.08–8.12 (dd, J = 1.2, 8.4 Hz, 2H, Ar-H), 8.25–8.28 (dd, J = 1.2, 8.4 Hz, 2H, Ar-H); IR (KBr/cm−1): 3122 (C-H (Ar, stretching)), 1520 (C=N, stretching), 1038 (C-N, bending); FAB-MS m/z [M]+: 302; Elemental Analysis: Calcd. (Found) (%) for C19H18N4: C 75.47 (75.78), H 6.00 (6.01), N 18.53 (18.57).
9-(4-Methylpiperizin-1-yl)-6H-indolo[2,3-b]quinoxaline (IDQ-14)
A mixture of 9-bromo-6H-indolo[2,3-b]quinoxaline derivative (5.5 mmol) and N-methyl piperazine (30 mmol) in dry DMF (5 mL) was heated at 120°C for 4–5 h. After cooling, the precipitate was filtered off and digested with hot ethyl acetate to give the product. The crude product was recrystallized with DMF/DMSO and TLC was monitored with methanol:chloroform (3:7).
1H NMR (300 MHz, δ/ppm in DMSO-d6): 2.46–2.52 (m, 4H, CH2), 2.73(s, 3H, CH3), 3.29–3.36 (m, 4H, CH2), 5.78 (br, s, 1H, NH), 7.30–7.31 (d, J = 1.5 Hz, 1H, Ar-H), 7.36 (s, 1H, Ar-H), 7.90–7.91 (d, J = 1.8 Hz, 1H, Ar-H), 8.08–8.11 (dd, J = 1.9, 9.0 Hz, 2H, Ar-H), 8.25–8.28 (dd, J = 1.9, 9.0 Hz, 2H, Ar-H); IR (KBr/cm−1): 3132 (C-H (Ar, stretching)), 2885 (C-H, stretching), 1572 (C=N, stretching), 1475 (C-O, stretching); FAB-MS m/z [M]+: 317; Elemental Analysis: Calcd. (Found) (%) for C19H19N5: C 71.90 (71.78), H 6.03 (6.06), N 22.07 (21.97).
In vitro cytotoxicity assay in human tumor cell lines
The cytotoxicity of the synthesized compounds was screened in the HL-60 cell line by adopting the well-established MTT colorimetric microculture assay methodCitation20,Citation21.
Increasing concentrations of each drug were placed with HL-60 tumor cell suspension (5 × 104 cells/well). Cell growth was determined 72 h later by adding 50 µL of 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) (2.5 mg/mL). This was reduced by mitochondrial dehydrogenase of viable cells in an insoluble blue formazan product during the 4 h contact period at 37°C. After the supernatant was removed, the formazan crystals were solubilized by adding DMSO (100 µL). These plates were read at 550 nm with an enzyme linked immunosorbent assay (ELISA) reader. At each dose level of the compounds tested, cell growth inhibition was expressed as a fractional decrease of 550 nm absorbance in the treated cultures with respect to control and reference compounds (cisplatin and 5-fluorouracil) ().
Table 1. Structural variations and cytotoxicity of 6H-indolo[2,3-b]quinoxaline derivatives.
In silico ADMET study
A Pentium 4 workstation and Pallas 6.1.1 software were used to calculate and predict the ADMET (absorption, distribution, metabolism, excretion, toxicity) properties of the moleculesCitation22,Citation23. ChemDraw ultra software was used to draw the structures of the compounds to be analyzed, which were saved as an MDL file. The sketched molecules were subjected to calculation of the following properties: drug-likeness, metabolism, toxicity etc.
Quantitative structure–activity relationship analysis
The quantitative structure–activity relationship (QSAR) study of the synthesized compounds was carried out employing MOE and Dragon softwareCitation24,Citation25 running on a Pentium 4 computer. The compounds were sketched in ChemDraw softwareCitation26. The lowest energy conformers and physicochemical descriptors were calculated from MOE and Dragon softwareCitation27 (). The correlation between the activity and the physicochemical descriptors was calculated using the partial least squares method.
Table 2. Molecular descriptors used for modeling cytotoxicity of 6H-indolo[2,3-b]quinoxaline derivatives.
The dataset comprises 14 6H-indolo[2,3-b]quinoxaline derivatives with cytotoxic activity against a human leukemia (HL-60) cell line. The cytotoxic activity of compounds in the series is reported as the IC50 value, where IC50 refers to the experimentally determined concentration required to inhibit 50% cell growth.
Statistical analysis was done by multiple linear regression using statistical software SYSTATCitation28. QSAR models for the series were constructed by stepwise variable selection, and best regressions were selected on the basis of the statistical quality. The quality of the regression models was judged from statistical parameters such as correlation coefficient R and squared correlation coefficient R2, Fisher ratio value, t-statistic, and standard error of the estimate (SEE). Guidelines for the acceptance of regressions were: correlation coefficient, R = 0.90 or higher (variance, R2 > 0.70); minimum intercorrelation between descriptors found in the same model (<0.6); Fisher ratio and t-statistic values indicating 95% level of significance.
The orthogonality of descriptors selected for formulating statistically significant correlations in multiple linear regression (MLR) calculations was established by generation of the correlation matrix and calculation of variance inflation factor (VIF) values. The VIF value was calculated from 1/1 – R2, where R2 is the multiple correlation coefficient of one descriptor’s effect regressed on the remaining molecular descriptors. VIF values larger than 5 indicate that the information of the descriptors may be hidden by the correlation of the descriptors.
Validation of the selected correlations was performed using the in-house program VALSTATCitation29. The reliability of the QSAR correlations was tested in a cross-validation method with determination of cross-validated R2 or Q2. Values of Q2 can be considered as proof of high predictive ability of the QSAR model. Further confirmation of the predictive power of the generated models was obtained by calculation of the standard deviation based on the predicted residual sum of squares (SPRESS) and standard deviation of error of prediction (SDEP) values.
Results and discussion
The series of 6H-indolo[2,3-b]quinoxaline derivatives was synthesized as per Scheme 1, and their physicochemical properties were calculated and are given in . The structures of the synthesized compounds were confirmed as follows. The FAB mass spectra of the synthesized compounds showed molecular weights which corresponded to molecular weights calculated from the respective molecular formulae. Further, the results of elemental analysis of the compounds showed that the experimental values coincided with the calculated values and errors lay within ±0.4% of the calculated values. H1 NMR spectra were used to determine the number and environment of hydrogen atoms.
Table 3. Physicochemical characteristic of 6H-indolo[2,3-b]quinoxaline derivatives.
Cytotoxic activity
The IC50 values of the synthesized 6H-indolo[2,3-b]quinoxaline derivatives were determined against the HL-60 cancer cell line employing the MTT assay in 96-well plates (). Cisplatin and 5-fluorouracil were used as reference compounds. Except for three compounds (IDQ-4, IDQ-7, and IDQ-9), all compounds were cytotoxic at micromolar concentrations. Compounds IDQ-5, IDQ-6, IDQ-10, IDQ-11, and IDQ-14 were found to be the most active in the present series. The nature of the substituent in ring A of 6H-indolo[2,3-b]quinoxaline appears to play a significant role in the cytotoxicity exhibited by these analogs. The presence of a free carboxylic group in the 6H-indolo[2,3-b]quinoxaline nucleus, with the exception of compound IDQ-2, irrespective of its position, decreases the cytotoxic potency to a major extent. However, the 6H-indolo[2,3-b]quinoxaline derivative with a carboxyl moiety on C-7 (IDQ-2) showed a marginal increase in cytotoxic potency. Conversion of the carboxylic group in IDQ-2 to the dimethyl carboxamido moiety (IDQ-5) caused a significant increase in cytotoxic activity. This marked increase in cytotoxic potency might be due to the compound’s structural similarity to acridine-4-carboxamide. Another important finding is that the N-ethyl derivatives of 6H-indolo[2,3-b]quinoxaline are more cytotoxic than their unsubstituted analogs. Bromo substitution at the C-9 atom of the 6H-indolo[2,3-b]quinoxaline decreases the cytotoxic activity, whereas replacing the bromo group with a secondary amino group improves the cytotoxicity; compound IDQ-10 with a dimethylamino group at the C-9 atom of 6H-indolo[2,3-b]quinoxaline was the most potent. Furthermore, increasing the carbon chain length or cyclicity of the alkyl group on the nitrogen atom reduces the cytotoxic potency.
In silico ADMET study
Clinical failure of most intercalator cytotoxic drugs is mainly due to their pharmacokinetic attributes rather than to pharmacodynamics. Hence, optimizing the pharmacokinetic properties during the stages of drug development is now widely accepted as being essential. With the abovementioned view, ADME attributes of the designed compounds were determined computationally prior to their synthesis. Moreover, solubility, pKa, and lipophilicity are integrally linked with chemical structureCitation30.
In order to validate the proposed compounds on the guidelines of the Lipinski “rule of 5” (molecular weight >500, logP >5, hydrogen bond donors (HBD) >5, and hydrogen bond acceptors (HBA) >10), an in silico ADMET study on the proposed compounds was carried out using Pallas 3.1.1.2 software. The relevance of the designed molecules with respect to the Lipinski rule of 5 is as followsCitation10,Citation31.
The molecular weight of the proposed compounds lies in the range 219.24–342.15, which is well within the limits prescribed by the Lipinski rule of 5. LogP is a crucial factor governing passive membrane partitioning and thereby influencing permeability. In light of this dependence, it has been suggested that absorption may be optimal within a logP range of 1–4. The logP value of most of the synthesized compounds lies between 1 and 4, with the exception of compound IDQ-8 (logP = 4.35). The optimum range of the calculated logP values of the proposed compounds shows that the compounds are most suitable for passive transcellular absorption across intestinal epithelia. Compound IDQ-8 possess logP >4, so it is highly likely that the compound can undergo renal reabsorption and will have a prolonged half-life (t1/2) value due to its high lipophilicity.
Atoms of both HBD type and HBA type are molecular determinants responsible for binding affinity and specificity, e.g. hydrogen bonds. Hydrogen bond acceptor and donor groups in the compound optimize the drug–receptor interaction. The number of hydrogen atoms, i.e. ≤10 hydrogen bond acceptors and ≤5 hydrogen bond donors in the proposed compounds, obeys the Lipinski rule of 5. Thus the proposed molecules may have good absorption or permeability properties through the biological membrane in addition to enhanced binding affinity and specificity toward the receptor.
The predicted drug-likeness index highlights whether or not the proposed compounds follow the Lipinski rule of 5. It is quite evident from the results recorded in that the Lipinski parameters calculated for the proposed compounds are in the ranges recommended by the Lipinski rule of 5, though adherence to the aforementioned rule is not an essential requirement for anticancer compounds.
Table 4. Lipinski parameters of the compounds.
The ionization state of a compound in vivo can be predicted by considering its pKa relative to pH in various body compartments. pKa is the primary determinant of the solubility of a compound. In vivo, this has a decisive bearing on oral absorption, following the premise that drugs must dissolve in the gastrointestinal (GI) tract to cross the intestinal membrane. The proposed compounds have optimum pKa (3.29–11.91), which suggests that they may be absorbed orally and undergo proper distribution between compartmentsCitation31,Citation32.
The formation of reactive metabolic intermediates is one of the causes of drug toxicity. Oxidation to electrophilic intermediates or reduction to nucleophilic radicals that can attach DNA or RNA and induce carcinogenicity are two major reactions by which toxicity is exerted. Although many leads are abandoned early on in the drug discovery stage due to a toxic metabolite, this does not always imply toxicity in a given drug candidate, since there are other factors that can make the metabolite toxic or non-toxic. The presence of a toxic metabolite, however, raises a red flag, which must be extensively examined in animal toxicity studiesCitation33.
Metabolites of the proposed compounds were predicted employing the MetaboloExpert module of Pallas software (). The 6H-indolo[2,3-b]quinoxaline derivatives IDQ-2 to IDQ-4 were also predicted to form conjugates of glutamine, glycine, and glucouronide types of secondary metabolites, whereas another carboxylic derivative IDQ-9 was predicted to form only o-glucouronide and glutamine conjugated products. None of the proposed compounds was predicted to form methylated or acetylated products, which are toxic in nature.
Figure 1. Position of the metabolites of 6H-indolo[2,3-b]quinoxaline derivatives (A), cisplatin (B), and 5-fluorouracil (C).
![Figure 1. Position of the metabolites of 6H-indolo[2,3-b]quinoxaline derivatives (A), cisplatin (B), and 5-fluorouracil (C).](/cms/asset/db384725-1cee-413b-a10f-d094d691553f/ienz_a_419247_f0002_b.gif)
The predicted toxicity studies indicated that the proposed compounds were free from neurotoxicity, immunotoxicity, and irritation. The 6H-indolo[2,3-b]quinoxaline compounds were devoid of significant teratogenicity. It is also noteworthy that the compounds predicted to form conjugated products are less likely to exhibit teratogenic toxicity (<17%). Furthermore, all the proposed compounds were predicted to have less (or no) teratogenic toxicity than the reference compound (5-fluorouracil (34%)).
Literature findings revealed that oxidation of aromatic rings can be prevented by substituting them with stronger electron withdrawing groups. With this rationale, the proposed compounds were designed to have electron withdrawing groups (NO2, COOH, etc.) in the nucleus, which prevents oxidation of the aromatic ring. Furthermore, these groups also have the ability to interact with the receptor (DNA) through hydrogen bonding, which might help to stabilize the intercalated drug–DNA complex.
QSAR studies
Multiple linear regression treatment of the molecular descriptors () calculated for 6H-indolo[2,3-b]quinoxaline derivatives and their corresponding activity parameters led to the development of several QSARs. The generated QSARs were thoroughly scrutinized for statistical validity and predictive potential as per the criteria described in the experimental section.
The best QSAR models derived for modeling the cytotoxic activity of 6H-indolo[2,3-b]quinoxaline derivatives are as follows.
Model 1
–logIC50 = [3.059 (± 0.633)] + [0.281 (± 0.074)] nCp + [0.235 (± 0.147)] nCIC
n = 14, R = 0.93, R2 = 0.87, std = 0.09, F = 37.4, Q2 = 0.79, SPRESS = 0.12, SDEP = 0.11
Model 2
–logIC50 = [6.658 (± 1.748)] + [–3.433 (± 2.408)] Mp + [2.783 (± 2.135)] RBF + [–0.203 (± 0.171)] nCOOHPh
n = 14, R = 0.91, R2 = 0.84, std = 0.11, F = 17.0, Q2 = 0.65, SPRESS = 0.172, SDEP = 0.146
In the QSAR models given above, n is the number of data points, R is the correlation coefficient, R2 is the squared correlation coefficient, and F represents the Fisher ratio between the variances of calculated and observed activities (). The figures given in parentheses with ± sign in the model are 95% confidence limits.
Table 5. Correlation matrix for descriptors in QSAR models of series I.
The statistical relevance of models 1 and 2 is indicated as follows. The correlation coefficient values, R, show that they account for more than 90% of the variance in biological activity. The standard error of estimate is very low, demonstrating the accuracy of fit. The Fisher ratio values obtained for the QSARs exceed the tabulated value (8.51 for model 1 and 7.23 for model 2) by a large margin, as desired for a meaningful correlation. The coefficients are all significantly above the 95% level as established by their corresponding t-test values, given in . Furthermore, the calculated correlation matrix () and VIF values () indicate that the descriptors are not interrelated.
Table 6. Variance inflation factor (VIF) and t-values of descriptors in the QSAR models derived for series I.
Table 7. Experimental and predicted activity values of 6H-indolo [2,3-b]quinoxaline derivatives.
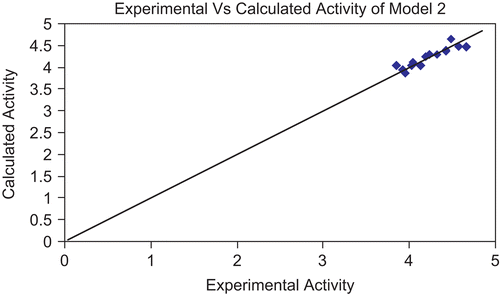
The above models (1 and 2) were validated using the leave-one-out (LOO) approach. The results of LOO cross-validations are given in . Plots of cross-validated activity values versus the experimental activity values of compounds in the training set using models 1 and 2 are shown in , respectively. Based on the cross-validated results, it is evident that the models exhibit good predictive capacity. Further confirmation of the predictive ability was obtained from the PRESS statistics of the QSARs, the uncertainty in the prediction (SPRESS) and standard error due to prediction (SDEP), which was less than 0.4.
Figure 2. Graph showing correlation between experimental activity and predicted activity of model 1.
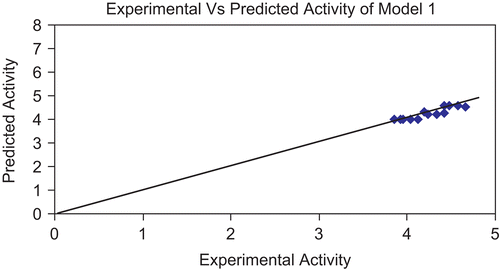
Figure 3. Graph showing correlation between experimental activity and calculated activity of model 1.
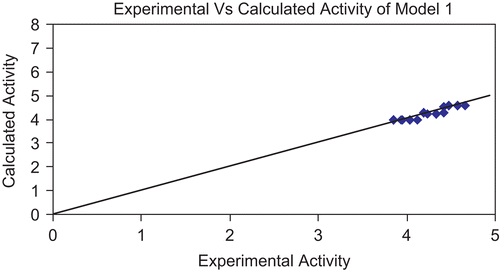
Figure 4. Graph showing correlation between experimental activity and predicted activity of model 2.
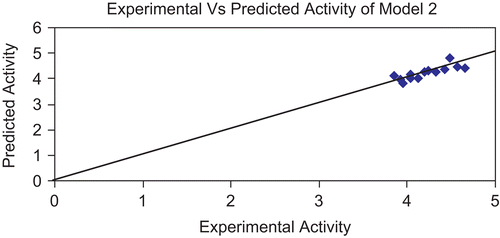
Figure 5. Graph showing correlation between experimental activity and calculated activity of model 2.
Model 1 was found to be the most important biparametric correlation for modeling the cytotoxic activity of 6H-indolo[2,3-b]quinoxaline derivatives. The molecular descriptors incorporated into the model in stepwise regression analysis are the functional group count descriptor nCp and constitutional descriptor nCIC. The descriptor nCp encodes information regarding the number of primary carbon atoms in the molecule; therefore, the positive coefficient associated with this term implies that an increase in the number of primary carbon atoms will cause a corresponding increase in the cytotoxic potency of 6H-indolo[2,3-b]quinoxaline derivatives. The second descriptor nCIC represents the number of rings in the molecule; thus, the positive coefficient of this descriptor in model 1 suggests that the good cytotoxic activity of 6H-indolo[2,3-b]quinoxaline derivatives can be increased by incorporating substituents with rings.
As no further biparametric correlations of statistical significance could be obtained for the dataset, the study was extended for triparametric correlations, as they are permitted for a dataset of 14 compounds in accordance with the lower limit of rule of thumbCitation34. Model 2 was found to be the most important triparametric correlation obtained in the stepwise multiple linear regression analysis. The model comprises the following descriptor terms: mean atomic polarizability (Mp), rotatable bond fraction (RBF), and functional group count descriptors (nCOOHPh). The descriptor Mp represents the polarizable components in the molecule. The negative slope of the descriptor in the model implies that the presence of polarizable substituents in the molecule is not conducive for cytotoxic activity. The second descriptor, RBF, in model 2 encodes information about the flexibility of the substituents in the molecule. The positive coefficient of the parameter indicates that an increase in flexibility of the molecules will cause a corresponding increase in their cytotoxic potency. The last descriptor in model 2 is nCOOHPh, which represents the aromatic carboxylic groups in the molecule. The negative sign associated with the coefficient of this descriptor suggests that the presence of carboxylic functional groups on the tetracyclic ring is not conducive for cytotoxic activity of the 6H-indolo[2,3-b]quinoxaline derivatives.
From the discussion above, it is quite evident that presence of polar substituents is highly disfavored for cytotoxic activity of 6H-indolo[2,3-b]quinoxaline derivatives, whereas the presence of hydrophobic moieties (as accounted for by the descriptors nCp and nCIC in model 1) appears to have a positive effect on the same. To be specific, the cytotoxic activity of the 6H-indolo[2,3-b]quinoxalines is profoundly influenced by the alkyl substituents in the R2 position and the tertiary amino moiety in the R4 position of the tertacyclic ring. However, the positive effect of molecular flexibility cannot be reasoned, but it is probably due to the fact that the dimethyl and diethylamino substituents at R4 exhibit better potency than the cyclic counterparts such as piperidino, morpholino, and piperazino groups. Finally, it may be concluded that the mechanism of cytotoxic activity exhibited by 6H-indolo[2,3-b]quinoxaline is nonspecific in nature, as indicated by the findings of the QSAR study. Though the DNA intercalating ability of 6H-indolo[2,3-b]quinoxalines is well defined in the literature, the cytotoxic activity of synthesized 6H-indolo[2,3-b]quinoxalines appears to be dependent on the ability to cross the cell membrane and establish an optimum intracellular concentration.
Conclusion
In the present study, the rationale for designing a series of novel cytotoxic agents bearing structural resemblance to anticancer acridines has been presented. In silico pharmacokinetic studies were also performed in order to validate the relevance of the proposed molecules on the basis of ADME and toxicity studies. Compounds with the desired pharmacokinetic profile were synthesized and structurally characterized. The novel 6H-indolo[2,3-b]quinoxaline derivatives exhibited significant activity in vitro against a human leukemia (HL-60) cell line. The data presented herein not only provide structural and mechanistic insight into newly synthesized compounds, but also provide new directions for the design of novel compounds, which will aid in the generation of more effective cytotoxic agents. In addition to this, QSAR studies were also performed on three series of compounds possessing cytotoxic activity in order to develop a set of guidelines for future design of potent analogs of the studied congeners that can be tailor-made to have enhanced anticancer activity. The results of the QSAR study presented here also lay the foundation for understanding the mechanism through which the molecules exert cytotoxic activity.
Acknowledgements
The authors are thankful to the Vice Chancellor, RGPV, Bhopal for providing laboratory facilities. Another of the authors (C.K.) would like to thank CSIR, New Delhi for providing a Senior Research Fellowship. The authors are also acknowledge CDRI, Lucknow and CNCI, Kolkata for their help in this work.
Declaration of interest: One of the authors (N.S.H.N.M.) would like to give his sincere thanks to AICTE, New Delhi for a grant from Career Award to Young Teachers.
References
- http://wwwpathology2.jhu.edu/bladder_cancer/glossary.cfm.
- Jackson BG. Mechanism-based target identification and drug discovery in cancer research. Science 2000;287:1969–73.
- American Cancer Society, Inc., Surveillance Research. Cancer Faiths and Figures, 2005. http://www.geocites.com.
- World Health Organization. Cancer. February 2006. www.who.int/cancer/en. Retrieved on 25-06-2007.
- Cancer Research UK. UK cancer incidence statistics by age. January 2007. www.cancerresearchuk.org. Retrieved on 25-06-2007.
- Luc P, Stefaan VD, Mei G, Ruoli B, Ernest H, Arnold V, et al. Synthesis and biological evaluation of dihydrobenzofuran lignans and related compounds as potential antitumor agents that inhibit tubulin polymerization. J Med Chem 1999;42:5475–81.
- Monish J, Chul-Hoon K. 1,2-Benzisoxazole phosphorodiamidates as novel anticancer prodrugs requiring bioreductive activation. J Med Chem 2003;46:5428–36.
- Congiu C, Cocco MT, Lilliu V, Onnis V. New potential anticancer agents based on the anthranilic acid scaffold. Synthesis and evaluation of biological activity. J Med Chem 2005;48:8245–52.
- http://jjco.oupjournals.org/cgi/content/full/32/suppl_1/S13.
- Oprea TI. Virtual screening in lead discovery: a viewpoint. Molecules 2002;7:51–62.
- Leslie WD, Antony JK, Graeme JF, Bruce CB, William AD. Positioning of the carboxamide side chain in 11-oxo-11H-indeno[1,2-b]quinolinecarboxamide anticancer agents: effects on cytotoxicity. Bioorg Med Chem 2001;9:445–52.
- Martin YC, Kutter E, Austel V. The Medicinal Chemist’s Approach, in Modern Drug Research: Path to Better and Safer Drugs. New York: Marcel Dekker, 1989:243–307.
- Katrin MD, Stefanie L, Martin H, Manuela H, Kristian W, Michael L, et al. Synthesis of 6H-indolo[2,3-b]quinoxaline-N-glycosides and their cytotoxic activity against human ceratinocytes (HaCaT). Org Biomol Chem 2008;6:4218–23.
- Wilhelmsson LM, Ngarita K, Jan B. Interactions of antiviral indolo[2,3-b]quinoxaline derivatives with DNA. J Med Chem 2008;51:7744–50.
- Harmenberg J, Wahren B, Bergman J, Akerfeldt S, Lundblad L. Antiherpesvirus activity and mechanism of action of indolo-(2,3-b)quinoxaline and analogs. Antimicrob Agents Chemother 1988;32:1720–4.
- Harmenberg J, Akesson JA, Graslund A, Malmfors T, Bergman J, Wahren B, et al. The mechanism of action of the anti-herpes virus compound 2,3-dimethyl-6(2-dimethylaminoethyl)-6H-indolo-(2,3-b)quinoxaline. AntiViral Res 1991;15:193–204.
- Paola BA, Brigitte B, William L, Christine B, France-Aimée A, Sylvain R, et al. DNA interaction and cytotoxicity of a new series of indolo[2,3-b]quinoxaline and pyridopyrazino[2,3-b]indole derivatives. Chem Biol Interact 2001;138:59–75.
- Silva JFM, Garden SJ, Pinto AC. The chemistry of isatins: a review from 1975 to 1999. J Braz Chem Soc 2001;12:273–324.
- Yen VQ, Buu-Hoi NP, Xuong ND. Fluorinated isatins and some of their heterocyclic derivatives. J Org Chem 1958;23:1858–61.
- Skehan P, Storeng R, Scudiero D, Monks A, Mcmahon J, Vistica D, et al. New colorimetric cytotoxicity assay for anticancer-drug screening. J Natl Cancer Inst 1990;82:1107–12.
- Mosmann T. Rapid colorimetric assay for cellular growth and survival: application to proliferation and cytotoxicity assays. J Immunol Methods 1983;65:55–63.
- Palas 3.1.1.2, ADME-Tox software. Sedona, AZ: CompuDrug International Inc., 2000.
- Moorthy NSHN, Rahul S, Hemendra PS, Gupta SD. Synthesis, biological evaluation and in silico metabolic prediction of flavanone derivatives. Chem Pharm Bull 2006;54:1384–90.
- MOE molecular modeling package. Montreal: Chemical Computing Group Inc., 2002.
- Dragon web version 3.0. Milano Chemometrics and QSAR Research Group, 2003. www.disat.uinmib.it.
- ChemOffice 2001. Bethesda, MD: Adept Scientific, CambridgeSoft Corp, UK, 2001.
- Lin A. QuaSAR-descriptors. J Chem Comput Group. http://www.chemcomp.com/Journal_of_CCG/Features/descr.htm.
- SYSTAT 10.2. Chicago, IL: Systat Software Inc., 2003.
- Gupta AK. VALSTAT. Indore, India: SGSITS, 2004.
- Beresford AP, Segall M, Tarbit MH. In silico prediction of ADME properties: are we making progress? Curr Opin Drug Discov Dev 2004;7:36–42.
- Hashida M. In silico prediction of pharmacokinetic properties. Yakugaku Zasshi 2005;125:853–61.
- McGee P. New in vitro, modeling tools may cut tox attrition.Drug Discov Dev 2005;8:55–60.
- White RE. High-throughput screening in drug metabolism and pharmacokinetic support of drug discovery. Annu Rev Pharmacol Toxicol 2000;40:133–57.
- Balaji S, Karthikeyan C, Hari Narayana Moorthy NS, Trivedi P. QSAR modeling of HIV-1 reverse transcriptase inhibition by benzoxazinones using a combination of P_VSA and pharmacophore feature descriptors. Bioorg Med Chem Lett 2004;14:6089–94.