Abstract
Ligands containing bulky aliphatic P1 residues exhibit a high affinity towards cytosolic leucine aminopeptidase, a bizinc protease of biomedical significance. According to this specificity, a series of phosphonic and phosphinic compounds have been put forward as novel putative inhibitors of the enzyme. These phosphonic and phosphinic compounds were derivatives of methionine and norleucine as both single amino acids and dipeptides. The designed inhibitors were synthesised and tested towards the peptidase isolated from porcine kidneys using an improved separation procedure affording superior homogeneity. Unexpectedly, organophosphorus derivatives of methionine and norleucine exhibited moderate activity with Ki values in the micromolar range.
Introduction
The leucine aminopeptidases [Citation1] are a group of exopeptidases that hydrolyse N-terminal amino acid residues from peptide and protein substrates with a much broader specificity than that indicated by their systematic nomenclature. They belong to the family of metallopeptidases, M1 and M17. Among the M17 members, the cytosolic leucine aminopeptidase (LAP, E.C. 3.4.11.1) [Citation2] has been the most extensively studied. Despite this, the biological significance of LAP, as well as many functional aspects of its mode of action, is still not fully understood. LAP is ubiquitous in microorganisms, plants and animals, is widespread in various tissues and organs and its role varies from organism to organism. The enzymes present in lower organisms are mostly responsible for proteolysis [Citation3], whereas those in plants are involved in the defence response system and wound healing [Citation4]. In mammals, the predominant function of LAP is considered to be its involvement in the maturation, activation, modulation and degradation of bioactive peptides [Citation1,Citation2,Citation5–7]. Recently, the leucine aminopeptidase has been proposed to be the key enzyme responsible for glutathione turnover in the liver, and thus influences the redox status of the cell [Citation8–10]. The processing of antigenic peptides by LAP has a minor role in epitope generation for presentation by the major histocompatibility complex class I molecules [Citation11,Citation12]. An altered action of the enzyme has been implicated in certain pathological states including HIV infection, inflammation, eye lens cataract and cancer [Citation13–16].
LAP can be considered a prototypical bimetallic aminopeptidase, since it contains two non-equivalent zinc ions that are both involved in substrate binding and catalysis. In the nineties, substantial efforts were dedicated to elucidating the mechanism of the catalytic reaction, these were mostly based on the crystal structures of the bovine lens enzyme, in both its free state and complexed with various inhibitors [Citation17–24]. This topic has promoted a number of theoretical studies on the environmental effects of zinc, the functional role of the active-site residues and the water channels in the homohexameric structure [Citation25–27]. The insight from these studies has lead to novel reflections on the action of a nucleophile in the active site and the reasons of the surprisingly broad specificity of the hydrolytic process. Due to the knowledge gathered and its biomedical importance, LAP is a good target for the rational design and development of novel inhibitors regulating its activity [Citation28]. Among de novo constructed targeted molecules, organophosphorus compounds, mostly aminophosphonates and phosphorus containing peptides, have probably contributed the most to this field [Citation29–35].
In this context, the phosphorus containing derivatives of norleucine (Nle) and methionine (Met) represent options not yet explored. Although analogous to the N-terminal residues in highly efficiently processed substrates [Citation9,Citation36,Citation37], the Nle and Met mimetics have not been considered as potential inhibitors of cytosolic leucine aminopeptidase. Here we report the validation of this hypothesis. The appropriate norleucine and methionine aminophosphonic and phosphinic analogues, as well as their phosphonate and phosphinate dipeptide derivatives were synthesised. In addition their affinity towards the enzyme was measured and discussed. Furthermore, an improved method for the purification of LAP from porcine kidneys has also been reported.
Materials and methods
General
The melting points were determined using a Boetius block and reported without correction. The IR spectra were recorded on a Bruker IFS 55 Equinox (Ettlingen, Germany). 1H and 13C NMR spectra were measured on a Bruker AVANCE-600 spectrometer (Karlsruhe, Germany) (1H at 600.1 MHz, 13C at 150.9 MHz), and 31P NMR spectra on a Bruker Avance 400 MHz spectrometer (31P {1H} at 162 MHz with 85 % H3PO4 as an external reference), in D2O or DMSO-d6 at 300 K. The 2D-correlation spectroscopy (2D-COSY) and 2D-H,C-heteronuclear single quantum coherence (2D-H,C HSQC) spectra were recorded and used for the structural assignment of the proton and carbon signals. High resolution mass spectra (HRMS) were obtained on a Fourier transform mass spectrometer (FTMS) LTQ-orbitrap XL (Thermo Fisher, Bremen, Germany) in the electrospray ionisation mode. Reverse phase HPLC (RP-HPLC) purification of the target compounds was performed as described previously by Pícha et al. [Citation38]. All chemicals were of analytical grade, obtained from commercial suppliers (Sigma-Aldrich and Fluka, Prague, Czech Republic and Poznań, Poland, Merck, Darmstadt, Germany, POCh, Gliwice, Poland) and were used without further purification. Biochemical materials were purchased from Pharmacia, (Uppsala, Sweden), Bio-Rad (Hercules, USA) and Millipore (Billerica, USA). Porcine kidney was obtained from a local slaughterhouse and stored at −20°C. A UV-Visible Cary 100 spectrophotometer (Varian, Australia) was used for the kinetic assays.
Synthesis
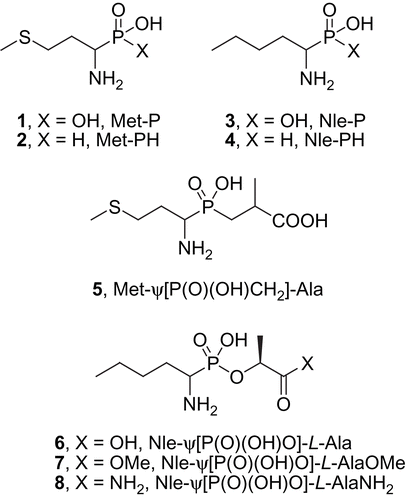
The phosphonic analogue of methionine (1, Met-P, for structures see ) was prepared from 3-(methylthio)propionaldehyde according to the method described previously by Stec and Kudzin [Citation39]. The phosphonic analogue of norleucine (3, Nle-P) was obtained from the respective aldehyde as described by Pícha et al. [Citation38,Citation40]. The syntheses of the phosphinic compounds Met-PH (2), Nle-PH (4) and Met-ψ[P(O)(OH)CH2]-Ala (5) were recently published elsewhere and we have only provided the detailed physico-chemical characteristics that weren′t reported in our previous communication here [Citation41]. The preparation of the phosphonate esters 6–8 was achieved by the coupling of monobenzyl 1-(N-benzyloxycarbonylamino)pentyl phosphonate with the respective derivatives of L-lactic acid and is described in detail elsewhere [Citation38,Citation40].
[1-(R,S)-amino-3-(methylsulphanyl)propyl]phosphonic acid (1)
Yield 36% (through two steps); mp 272–274°C (lit. mp 270–272°C [39]); IR (KBr): νmax (cm−1) 2966, 2910 br, 2293, 1583, 1182, 1030, 928, 558; 1H NMR (D2O + NaOD) δ 1.61 (m, J = 14, 10.7, 9.4, 7 and 4.6 Hz, 1H, C-CHaHb-C), 2.03 (m, J = 14, 9.7, 7.3, 7.3 and 3.2 Hz, 1H, C-CHaHb-C), 2.12 (s, 3H, CH3), 2.59 (ddd, J = 13, 9.4 and 7.3 Hz, 1H, S-CHaHb-C), 2.61 (ddd, J = 11.2, 10.7 and 3.2 Hz, 1H, N-CH-P), 2.75 (ddd, J = 13, 9.7 and 4.6 Hz, 1H, S-CHaHb-C); 13C NMR (D2O + NaOD) δ 16.74 (CH3), 33.91 (d, J(C,P) = 14.4 Hz, CH2), 33.93 (CH2), 52.01 (d, J(C,P) = 137.8 Hz, CH); 31P NMR (D2O + NaOD) δ 21.81; HRMS (ESI) calcd for C4H11NO3PS [M-H]− 184.0203; found: 184.0203.
[1-(R,S)-amino-3-(methylsulphanyl)propyl]phosphinic acid (2) [41]
Mp 232–233°C (lit. mp 231°C [42]); IR (KBr): νmax (cm−1) 2970, 2900 br, 1549, 1192, 1041, 966; 1H NMR (D2O) δ 1.99 (m, J = 14.9, 12.3, 8.6, 8 and 5.9 Hz, 1H, C-CHaHb-C), 2.12 (s, 3H, CH3), 2.2 (m, J = 14.9, 10.6, 8.3, 7.1 and 5.5 Hz, 1H, C-CHaHb-C), 2.69 (ddd, J = 13.7, 8 and 7.1 Hz, 1H, S-CHaHb-C), 2.76 (ddd, J = 13.7, 8.3 and 5.9 Hz, 1H, S-CHaHb-C), 3.32 (m, J = 10.8, 8.6, 5.5 and 1.4 Hz, 1H, N-CH-P), 7.03 (dd, J = 534.8 and 1.4 Hz, 1H, P-H); 13C NMR (D2O) δ 16.47 (CH3), 28.04 (CH2), 32.06 (d, J(C,P) = 9.8 Hz, CH2), 52 (d, J(C,P) = 91.6 Hz, CH); 31P NMR (D2O) δ 20.4; HRMS (ESI) calcd for C4H11NO2PS [M-H]− 168.0254; found: 168.0254.
[1-(R,S)-aminopentyl]phosphinic acid (4) [41]
Mp 235°C (lit. mp 230–232°C [43]); IR (KBr): νmax (cm−1) 3200–2500 br, 1546, 1192, 1057, 971; 1H NMR (D2O + NaOD) δ 0.9 (t, J = 7.3 Hz, 3H, CH3), 1.38 (m, 2H, CH2), 1.33 and 1.47 (m, 2H, CH2), 1.56 and 1.8 (m, 2H, CH2), 2.9 (m, 1H, N-CH-P), 6.87 (dd, J = 519 and 1.4 Hz, P-H); 13C NMR (D2O + NaOD) δ 13.68 (CH3), 22.37 (CH2), 27.64 (CH2), 28 (d, J(C,P) = 9.8 Hz, CH2), 51.09 (d, J(C,P) = 95.5 Hz, CH); 31P NMR (D2O + NaOD) δ 34.30; HRMS (ESI) calcd for C5H13NO2P [M-H]− 150.0689; found: 150.0690.
3-{[1-(R,S)-amino-3-(methylsulphanyl)propyl](hydroxy)phosphinyl}-2-(R,S)-methylpropanoic acid (5) (lyophilate) [41]
IR (KBr): νmax (cm−1) 2980 br, 2922, 1699, 1201, 1058; NMR (mixture of diastereoisomers in the ratio of ∼ 2:1; the signals were well resolved in the 13C NMR spectra, and the chemical shift of the major isomer has been reported first) 1H NMR (DMSO-d6) δ 1.19 (d, J = 7.1 Hz, 3H, C-CH3), 1.67 and 2.05 (m, 2H, P-CH2-C), 1.84 and 2.04 (m, 2H, C-CH2-C), 2.04 (s, 3H, S-CH3), 2.59 and 2.68 (m, 2H, S-CH2-C), 3.25 (m, 1H, N-CH-P), 8.13 (br, 2H, NH2); 13C NMR (DMSO-d6) δ 14.41 and 14.42 (S-CH3), 19.12 and 19.1 (d each, J(C,P) = 8.2 Hz, CH3), 27.65 and 27.78 (CH2), 29.73 and 29.72 (d each, J(C,P) = 8.8 Hz, S-CH2), 31.34 and 31.32 (d each, J(C,P) = 93.7 Hz, P-CH2), 33.54 (d each, J(C,P) = 3.9 Hz, CH), 48.19 and 48.08 (d each, J(C,P) = 92.6 Hz, N-CH-P), 176.9 and 176.85 (d each, J(C,P) = 9.6 Hz, COOH); 31P NMR (DMSO-d6) δ 36.07; HRMS (ESI) calcd for C8H17NO4PS [M-H]− 254.0621; found: 254.0622.
Enzyme purification and inhibitory activity measurements
The cytosolic leucine aminopeptidase from commercial sources has been found to be a non-homogeneous preparation which required further work up [Citation44]. Recently, the protein stopped being available for purchase. Therefore, the LAP used in our studies was isolated from porcine kidneys according to the standard literature procedure [Citation45–48], with the modification of a multi-step chromatographic purification (five consecutive steps). Consequently, the procedure consisted of: (1) extraction into 100 mM Tris-HCl buffer (pH 8, 50 mM NaCl, 10 mM 2-mercaptoethanol) and precipitation of the enzyme with acetone at −20°C; (2) classic fractionation with ammonium sulphate (40%-65% saturation); (3) gel filtration using a Sephadex G-25 column (100 mM Tris-HCl buffer, pH 8, 50 mM NaCl, 10 mM 2-mercaptoethanol); (4) ion-exchange chromatography on a DEAE-Sepharose column (100 mM Tris-HCl buffer, pH 8, NaCl gradient 0→0.25 M, 10 mM 2-mercaptoethanol); (5) gel filtration using a Sephacryl S-200 HR column (100 mM Tris-HCl buffer, pH 8, 100 mM NaCl, 10 mM 2-mercaptoethanol) and dialysis followed by (6) fractionation using a phenyl Sepharose 6 Fast Flow column (100 mM Tris-HCl buffer, pH 8, ammonium sulphate gradient 1.0→0 M, 10 mM 2-mercaptoethanol). After dialysis, final purification was carried out using Macro-Prep High Q (Bio-Rad, Hercules, USA) column chromatography (100 mM Tris-HCl buffer, pH 8, NaCl gradient 0.05→0.5 M, 10 mM 2-mercaptoethanol). The active fractions were pooled and concentrated by ultrafiltration using a YM-10 membrane. All procedures were performed at 4°C. The enzyme was stored as a suspension in an ammonium sulphate solution (3.5 M (NH4)2SO4, 100 mM Tris-HCl buffer, pH 8, 10 mM MgCl2).
In this manner, a homogeneous sample (1258-fold purification, ) of a superior purity to that of the commercial enzyme was obtained. Other noticeable advantages of the procedure reported here, was that the final preparations were considerably more stable and they did not change in activity on storage in the refrigerator at 4°C for several weeks. The protein had the correct molecular mass (apparent molecular weight 312 kD ± 27 kD for the homohexameric form) as determined by gel filtration on a Sephacryl S-200 HR column and its Michaelis constant for L-leucine p-nitroanilide was 0.72 ± 0.15 mM. The phosphonic analogue of homophenylalanine (hPhe-P, ) was used as a positive control in the inhibition of the enzyme activity measurements.
Figure 1. SDS-polyacrylamide gel electrophoresis after the enzyme purification (the molecular weights of the markers are indicated on the left). From the left: lane l: molecular weight markers, lane 2: the crude extract, lane 3: 40-65% ammonium sulphate precipitation, lane 4: Phenyl Sepharose fraction, lane 5: Macro-Prep High Q fraction. The gel was stained with Coomassie Brilliant R-250.
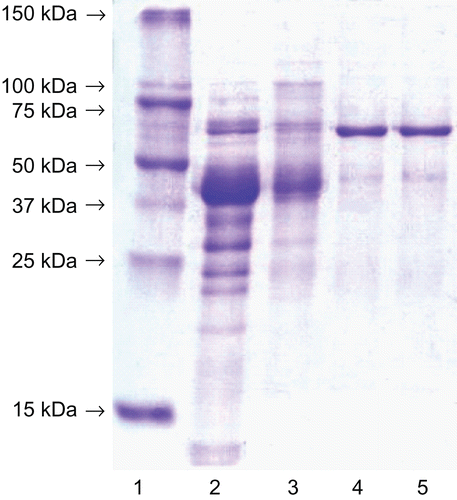
Figure 2. Selection of α-aminophosphonic acid inhibitors of LAP: analogues of natural (L-Val-P [Citation30] and L-Leu-P [Citation29]) and non-natural (hPhe-P and hTyr-P [Citation32]) amino acids, and their activity towards LAP.
![Figure 2. Selection of α-aminophosphonic acid inhibitors of LAP: analogues of natural (L-Val-P [Citation30] and L-Leu-P [Citation29]) and non-natural (hPhe-P and hTyr-P [Citation32]) amino acids, and their activity towards LAP.](/cms/asset/56b3868d-ff2d-40dc-ad43-7e83a3427939/ienz_a_482047_f0002_b.gif)
Figure 3. Newly designed and synthesised inhibitors of leucine aminopeptidase derivatives of methionine and norleucine.
Kinetic inhibition assays were performed as described earlier [Citation32–34]. LAP was assayed in 75 mM triethanolamine hydrochloride buffer (pH = 8.4, containing 5 mM MgCl2) at 25°C. The substrate (L-leucine-p-nitroanilide) was dissolved in DMSO and added to the assay buffer followed by the enzyme. The hydrolysis of the substrate was monitored by the change in absorbance measured at 405 nm. All the inhibitor solutions were prepared in the assay buffer. The assay solution, which contained 0.05 mL of the substrate solution (0.2, 0.4, 0.6, 0.8, 1 mM final concentrations), 0.25 mL of an inhibitor solution (concentration dependent on the inhibitor), the enzyme solution (20 μg/ml final concentration) and adjusted to a 1 mL final volume. The calculations were performed using three methods: Lineweaver-Burk, Dixon and semi-inhibition plots. The values of the inhibition constants presented are the averages of the three values. For compound 5, the Ki value was calculated from the dependence kapp versus inhibitor concentration. The kinetic constants for slow-binding were: k1 = 34389.43 M−1 × min−1, k2 = 0.13416 min−1.
Results and discussion
Leucine aminopeptidase is a hydrolase with broad band specificity that cleaves substrates bearing distinct N-terminal amino acid residues [Citation2,Citation7,Citation27], varying from neutral to both acidic and basic. However, those substrates containing N-termini of a hydrophobic character, such as leucine, methionine, isoleucine and valine, are processed noticeably faster. Proline remains the only naturally occurring amino acid that is not recognised by LAP when it is in the P1 position. A similar pattern of ligand affinity can be observed for the structurally related aminophosphonic acids, when applied as inhibitors of LAP. The analogues of natural hydrophobic aliphatic amino acids, such as valine and leucine, appeared to be the most efficient inhibitors with a Ki = 0.15 and 0.23 μM, respectively (for the R (L) enantiomers, Val-P [Citation30] and Leu-P [Citation29], ). Non-coded arylalkyl derivatives, exemplified by phosphonic homophenylalanine and homotyrosine, were bound preferentially to an even greater extent (Ki = 0.14 and 0.12 μM, respectively, for the racemic mixture, hPhe-P and hTyr-P [Citation32], ). The aforementioned structure was further modified to the phosphinic pseudodipeptides that appeared to be the most active organophosphorus inhibitors of LAP developed to date [Citation33,Citation35].
Surprisingly, there is no data in the literature on LAP inhibition by organophosphorus derivatives of methionine and norleucine although peptides with natural Met or Nle N-termini are among the most efficiently cleaved substrates. The kinetic parameters reported for the hydrolysis of Nle-X in the early works of Smith et al. [Citation36,Citation37] and recently for Met-X [Citation9] indicated they are processed slightly faster than the corresponding leucine derivatives. Therefore, to verify their potency towards leucine aminopeptidase we tested the phosphonic (1, Met-P, ) and phosphinic methionine analogues (2, Met-PH) as well as their aliphatic, non-sulphur norleucine counterparts (3, Nle-P and 4, Nle-PH). An additional reason for our interest in the latter compounds (3 and 4) was the promising affinity (Ki ≤ 1.0 μM) to LAP found for 1-amino-n-hexanephosphonic [Citation29] and 1-amino-n-octanephosphonic acid [Citation32], which are homologous to the Nle-P ligands of LAP. Both the methionine and norleucine compounds were further elongated to obtain the phosphinate (5, Met-ψ[P(O)(OH)CH2]-Ala) and phosphonate lactic acid esters (6–8, Nle-ψ[P(O)(OH)O]-Ala derivatives). This analysis evaluated the influence of the P1′ fragment introduced into the essentially designed structures and the C-terminus status on the inhibitory potency of the respective compounds.
The measured inhibition activity towards leucine aminopeptidase of the synthesised aminophosphonates and aminophosphinates is presented in . The tested compounds appeared to be moderate competitive inhibitors of LAP, with the phosphinic peptide 5 exhibiting slow-binding kinetics. The latter observation was not unexpected since other representatives of this group of pseudopeptides have been reported to display similar kinetics [Citation29,Citation30,Citation33]. Although the actual reasons of this remained elusive, a protein conformational change or slow displacement of an active-site water molecule has been suggested as the factors responsible [Citation29].
To our surprise, the studied ligands did not follow the general rules previously found for inhibition of LAP by phosphonic and phosphinic acids [Citation29,Citation30,Citation32,Citation49]. The analogues of methionine 1 (Ki = 22.6 μM, ) and norleucine 3 (Ki = 58 μM) did not exhibit comparable affinity to the corresponding valine or leucine derivatives, being approximately 20-fold (Met-P) and 50-fold (Nle-P) less potent than the racemic mixture of one of the most active aliphatic aminophosphonate compound Val-P (Ki = 1.2 μM [Citation29]). This indicates that the novel compounds are located unfavourably in the enzyme S1 binding pocket.
Table 1. The inhibitory activity of the phosphorus containing amino acid and dipeptide derivatives of methionine and norleucine towards the cytosolic leucine aminopeptidase.
Some particularly interesting observations were made for the α-aminophosphinic acids 2 and 4. Met-PH and Nle-PH were practically equipotent with their phosphonic counterparts (compare entries 1 versus 2 and 3 versus 4 in ). Previous reports suggested there should be significantly weaker binding by the zinc ions present in the active site, and thus show a large disparity in their Ki values (for example, for leucine analogues this disparity exceeded two orders of magnitude) [Citation49,Citation50]. Thus, the difference in inhibitory activity of compounds 1–4 depended only on the structure of their side chain substituents. The positive influence of the bulkier sulphur atom containing residue versus the aliphatic residue of the same length can easily be seen. This observation suggests that the presence of the norleucine side chain causes greater distortion in the optimal binding of these inhibitors.
The phosphonate esters Nle-ψ[P(O)(OH)O]-Ala (compounds 6–8) were found to be several times less active than the amino acid they originated from. In general, almost any modification, such as replacement or protection, that excluded one of the three oxygen atoms bound to phosphorus from interacting with the LAP active site, resulted in similar or even greater loss of inhibitory efficiency (for representative examples see also reference [Citation49]). Nevertheless, some interesting findings on the status of the C-termini of these compounds were observed. For instance, the methyl ester 7 was moderately tolerated by the enzyme, and surprisingly, was a slightly stronger inhibitor than the free acid 6. This seems to be a new observation since the non-protected carboxylate has been thought to be the best C-terminus and thus preferentially bound in the active site [Citation33]. The amide 8, which is the closest analogue to the substrate structure, was totally discriminated.
The phosphinic dipeptide analogue 5 appeared to be the only positive elongation of the fundamental amino acid structure. Although it was tested as a mixture of four diastereoisomers it appeared to be a low micromolar inhibitor of LAP (Ki = 3.64 μM, ). However, as seen before, this compound did not reach the level of inhibition found for the analogues bearing the favourable leucine side chain in the P1 position, for example Leu-ψ[P(O)(OH)CH2]-Gly (Ki = 0.33 μM) [Citation33].
Apparently, derivatives of methionine and norleucine cannot serve as privileged P1 residues for construction of efficient inhibitors of the leucine aminopeptidases. This is unexpected, and in contrast with the substrate preferences of the enzyme, as well as with the general trends observed earlier. The non optimal contacts with the S1 binding pocket of LAP seems to be a straightforward explanation for this fact. This, most likely, results in a slightly different binding mode and distortion of the complexation of the phosphorus fragment by the zinc ions, which is the most important feature for tight binding. Such an unpredictable mode of binding has already been reported for the phosphinate and phosphonamidate dipeptide mimetics, rationally designed as favorable inhibitors of LAP [Citation34,Citation51]. A recent hypothesis, based on a correlation of kinetic parameters obtained for a set of fluorogenic substrates with the activity of α-aminophosphonic inhibitors of aminopeptidase N (an M1 peptidase with a specificity related to LAP) [Citation52], can represent a complementary clarification. According to these data, it wasn′t the turnover velocity (expressed by the kcat/Km) but only the strength of substrate binding (described by the Km value) that predicted the most reliably structural features responsible for the inhibitory potency. Thus, the appropriate P1 residue incorporated into the α-aminophosphonate core would be indispensable for the tight binding of a ligand to an aminopeptidase.
Conclusion
Leucine aminopeptidase is probably the most comprehensively characterised bizinc target hydrolase with regards to the regulation of its activity by organophosphorus compounds. A broad collection of aminophosphonates and phosphopeptides mimicking the tetrahedral transition state of amide bond hydrolysis allowed for a systematic structure – activity relationship determination. Methionine and norleucine derivatives were exceptions and had not been studied, even though evidence for the preferential cleavage of the appropriate substrates by the enzyme has been observed. To fill this gap, a set of phosphonic and phosphinic analogues of these amino acids and their dipeptides were synthesised and their inhibitory activity towards LAP was evaluated. The enzyme was separated from porcine kidneys using an improved procedure. The studied compounds appeared to be moderate ligands of LAP, with affinity expressed by their Ki values in the micromolar range. An additional observation indicating the unusual behaviour of inhibitors in the active site was also seen. The linear four atom P1 residues, even those containing a sulphur atom in their structure, are most likely to be not bulky or extended enough to bind tightly in the S1 binding pocket and cause effective complexation of the zinc ions by the phosphoryl moiety at the same time.
Declaration of interest
The authors report no conflicts of interest. This project was supported by a grant from the Grant Agency of the Czech Republic (203/06/1405, to J.J.), by the Chemical Genetics Consortium of the Ministry of Education, Youth and Sports of the Czech Republic (LC060777, to J.J.), by Research Project Z4 055 0506 of the Academy of Sciences of the Czech Republic (to IOCB) and by the Polish Ministry of Science and Higher Education (N N302 159937, to A.M.).
References
- Matsui M, Fowler JH, Walling LL. Leucine aminopeptidases: diversity in structure and function. Biol Chem 2006;387:1535–1544.
- Sträter N, Lipscomb WN. Leucyl aminopeptidase (animal). In: Barrett AJ, Rawlings ND, Woessner JF eds. Handbook of Proteolytic Enzymes. London: Elsevier, 2004:896–901.
- Colloms SD. Leucyl aminopeptidase PepA. In: Barrett AJ, Rawlings ND, Woessner JF eds. Handbook of Proteolytic Enzymes. London: Elsevier, 2004:905–908.
- Walling LL. Leucyl aminopeptidase (plant). In: Barrett AJ, Rawlings ND, Woessner JF eds. Handbook of Proteolytic Enzymes. London: Elsevier, 2004:901–904.
- Taylor A. Aminopeptidases: towards a mechanism of action. Trends Biochem Sci 1993;18:167–171.
- Taylor A. Aminopeptidases: structure and function. FASEB J 1993;7:290–298.
- Lowther WT, Matthews BW. Metalloaminopeptidases: common functional themes in disparate structural surroundings. Chem Rev 2002;102:4581–4607.
- Jösch C, Klotz L-O, Sies H. Identification of cytosolic leucyl aminopeptidase (EC 3.4.11.1) as the major cysteinylglycine-hydrolysing activity in rat liver. Biol Chem 2003;384:213–218.
- Cappiello M, Lazzarotti A, Buono F, Scaloni A, D′Ambrosio C, Amodeo P, Méndez BL, Pelosi P, Del Corso A, Mura U. New role for leucyl aminopeptidase in glutathione turnover. Biochem J 2004;378:35–44.
- Cappiello M, Alterio V, Amodeo P, Del Corso A, Scaloni A, Pedone C, Moschini R, De Donatis GM, De Simone G, Mura U. Metal ion substitution in the catalytic site greatly affects the binding of sulfhydryl-containing compounds to leucyl aminopeptidase. Biochemistry 2006;45:3226–3234.
- York IA, Goldberg AL, Mo XY, Rock KL. Proteolysis and class I major histocompatibility complex antigen presentation. Immunol Rev 1999;172:49–66.
- Towne CF, York IA, Neijssen J, Karow ML, Murphy AJ, Valenzuela DM, Yancopoulos GD, Neefjes JJ, Rock KL. Leucine aminopeptidase is not essential for trimming peptides in the cytosol or generating epitopes for MHC class I antigen presentation. J Immun 2005;175:6605–6614.
- Pulido-Cejudo G, Conway B, Proulx P, Brown R, Izaguirre CA. Bestatin-mediated inhibition of leucine aminopeptidase may hinder HIV infection. Antiviral Res 1997;36:167–177.
- Beninga J, Rock KL, Goldberg AL. Interferon-gamma can stimulate post-proteasomal trimming of the N terminus of an antigenic peptide by inducing leucine aminopeptidase. J Biol Chem 1998;273:18734–18742.
- Taylor A, Daims M, Lee J, Surgenor T. Identification and quantification of leucine aminopeptidase in aged normal and cataractous human lenses and ability of bovine lens lap to cleave bovine crystallins. Curr Eye Res 1982;2:47–56.
- Umezawa H. Screening of small molecular microbial products modulating immune responses and bestatin. Recent Results Cancer Res 1980;75:115–125.
- Burley SK, David PR, Taylor A, Lipscomb WN. Molecular structure of leucine aminopeptidase at 2.7-A resolution. Proc Natl Acad Sci USA 1990;87:6878–6882.
- Burley SK, David PR, Sweet RM, Taylor A, Lipscomb WN. Structure determination and refinement of bovine lens leucine aminopeptidase and its complex with bestatin. J Mol Biol 1992;224:113–140.
- Kim H, Burley SK, Lipscomb WN. Re-refinement of the X-ray crystal structure of bovine lens leucine aminopeptidase complexed with bestatin. J Mol Biol 1993;230:722–724.
- Kim H. Lipscomb WN. X-ray crystallographic determination of the structure of bovine lens leucine aminopeptidase complexed with amastatin: formulation of a catalytic mechanism featuring a gem-diolate transition state. Biochemistry 1993:32:8465–8478.
- Kim H. Lipscomb WN. Structure and mechanism of bovine lens leucine aminopeptidase. Adv Enzymol Relat Areas Mol Biol 1994;68:153–213.
- Sträter N, Lipscomb WN. Transition state analogue L-leucine phosphonic acid bound to bovine lens leucine aminopeptidase: X-ray structure at 1.65-Å resolution in a new crystal form. Biochemistry 1995;34:9200–9210.
- Sträter N, Lipscomb WN. Two-metal ion mechanism of bovine lens leucine aminopeptidase: active site solvent structure and binding mode of L-leucinal, a gem-diolate transition state analog, by X-ray crystallography. Biochemistry 1995;34:14792–14800.
- Sträter N, Sun L, Kantrowitz ER, Lipscomb WN. A bicarbonate ion as a general base in the mechanism of peptide hydrolysis by dizinc leucine aminopeptidase. Proc Natl Acad Sci USA 1999;96:11151–11155.
- Schürer G, Horn AHC, Gedeck P, Clark T. The reaction mechanism of bovine lens leucine aminopeptidase. J Phys Chem B 2002;106:8815–8830.
- Erhardt S, Jaime E, Weston J. A water sluice is generated in the active site of bovine lens leucine aminopeptidase. J Am Chem Soc 2005;127:3654–3655.
- Jaime E, Kluge S, Weston J. On the origin of the broad-band selectivity of bovine lens leucine aminopeptidase. ARKIVOC 2007:77–95.
- Grembecka J, Kafarski P. Leucine aminopeptidase as a target for inhibitor design. Mini Rev Med Chem 2001;1:133–144.
- Giannousis PP, Bartlett PA. Phosphorus amino acid analogues as inhibitors of leucine aminopeptidase. J Med Chem 1987;30:1603–1609.
- Lejczak B, Kafarski P, Zygmunt J. Inhibition of aminopeptidases by aminophosphonates. Biochemistry 1989;28:3549–3555.
- Lejczak B, Popiel de Choszczak M, Kafarski P. Inhibition of aminopeptidases by phosphonic acid analogues of aspartic and glutamic acids. J Enzyme Inhib Med Chem 1993;7:97–103.
- Drąg M, Grembecka J, Pawełczak M, Kafarski P. α-Aminoalkylphosphonates as a tool in experimental optimisation of P1 side chain shape of potential inhibitors in S1 pocket of leucine and neutral aminopeptidases. Eur J Med Chem 2005;40:764–771.
- Grembecka J, Mucha A, Cierpicki T, Kafarski P. Structure-based design and synthesis of dipeptide analogues as new inhibitors of leucine aminopeptidase. J Med Chem 2003;46:2641–2655.
- Vassiliou S, Xeilari M, Yiotakis A, Grembecka J, Pawełczak M, Kafarski P, Mucha A. A synthetic method for diversification of the P1′ substituent in phosphinic dipeptides as a tool for exploration of the specificity of the S1′ binding pockets of leucine aminopeptidases. Bioorg Med Chem 2007;15:3187–3200.
- Mucha A, Lämmerhofer M, Lindner W, Pawełczak M, Kafarski P. Individual stereoisomers of phosphinic dipeptide inhibitor of leucine aminopeptidase. Bioorg Med Chem Lett 2008;18:1550–1554.
- Smith AL, Polglase J. The specificity of leucine aminopeptidase II. Optical and side chain specificity. J Biol Chem 1949;180:1209–1223.
- Smith AL, Spackman DH. Leucine aminopeptidase V. Activation, specificity, and mechanism of action. J Biol Chem 1955;212:271–299.
- Picha J, Budesinsky M, Hanclova I, Sanda M, Fiedler P, Vanek V, Jiracek J. Efficient synthesis of phosphonodepsipeptides derived from norleucine. Tetrahedron 2009;65:6090–6103.
- Stec W, Kudzin ZH. Phosphohomocysteine derivatives. Synthesis 1980:1032–1034.
- Picha J, Budesinsky M, Sanda M, Jiracek J. Synthesis of norleucine-derived phosphonopeptides. Tetrahedron Lett 2008;49:4366–4368.
- Liboska R, Picha J, Hanclova I, Budesinsky M, Sanda M, Jiracek J. Synthesis of methionine- and norleucine-derived phosphinopeptides. Tetrahedron Lett 2008;49:5629–5631.
- Baylis EK, Campbell CD, Dingwall JG.1-Aminoalkylphosphonous acids. Part 1. Isosteres of the protein amino acids. J Chem Soc Perkin Trans 1 1984:2845–2853.
- Dingwall JG, Baylis EK, Campbell CD. α-Amino-phosphonous acids for inhibiting bacteria and yeast. US Patent 4147780 1979.
- Van Wart HE, Lin SH. Metal binding stoichiometry and mechanism of metal ion modulation of the activity of porcine kidney leucine aminopeptidase. Biochemistry 1981;20:5682–5689.
- Ledeme N, Hennon G, Vincent-Fiquet O, Plaquet R. Purification and enzymatic properties of an L-leucine aminopeptidase from swine liver. Biochim Biophys Acta 1981;660:262–270.
- Kawata S, Imamura T, Ninomiya K, Makisumi S. Purification and characterization of an aminopeptidase from porcine liver. J Biochem 1982;92:1093–1101.
- Spackman DH, Smith EL, Brown DM. Leucine aminopeptidase IV. Isolation and properties of the enzyme from swine kidney. J Biol Chem 1955;212:255–269.
- Lin B-X Du, X-L, Zhou L-G Hara, K, Su W-J Cao, M-J. Purification and characterization of a leucine aminopeptidase from the skeletal muscle of common carp (Cyprinus carpio). Food Chemistry 2008:108;140–147.
- Grembecka J, Sokalski WA, Kafarski P. Computer-aided design and activity prediction of leucine aminopeptidase inhibitors. J Comput Aided Mol Des 2000;14:531–544.
- Grembecka J, Sokalski WA, Kafarski P. Quantum chemical analysis of the interactions of transition state analogs with leucine aminopeptidase. Int J Quant Chem 2001;84:302–310.
- Mucha A, Kunert A, Grembecka J, Pawełczak M, Kafarski P. A phosphonamidate containing aromatic N-terminal amino group as inhibitor of leucine aminopeptidase – design, synthesis and stability. Eur J Med Chem 2006;41:768–772.
- Drag M, Bogyo M, Ellman JA, Salvesen GS. Aminopeptidase fingerprints. An integrated approach for identification of good substrates and optimal inhibitors. J. Biol. Chem. 2010;285:3310–3318.