Abstract
In order to understand how rimantadine (RMT) inhibits the proton conductance in the influenza A M2 channel via the recently proposed “allosteric mechanism”, molecular dynamics simulations were applied to the M2-tetrameric protein with four RMTs bound outside the channel at the three protonation states: the 0H-closed, 1H-intermediate and 3H-open situations. In the 0H-closed state, a narrow channel with the RMT-Asp44-Trp41 H-bond network was formed, therefore the water penetration through the channel was completely blocked. The Trp41-Asp44 interaction was absent in the 1H-intermediate state, whilst the binding of RMT to Asp44 remained, which resulted in a weakened helix-helix packing, therefore the channel was partially prevented. In the 3H-open state it was found that the electrostatic repulsion from the three charged His37 residues allowed the Trp41 gate to open, permitting water to penetrate through the channel. This agreed well with the potential of the means force which is in the following order: 0H > 1H > 3H.
Introduction
The influenza A viruses, both the impending isolates such as H5N1 and the recent new strain (H1N1), are a vital and emergent problem of the global flu pandemic [1–2]. One of the important targets for disruption of the replication process is based on the fact that the integral M2 membrane protein pH gated channel is required in the early and late stages of infection [Citation3–5]. Amantadine (AMT) and rimantadine (RMT) are the first effective drugs licensed for influenza treatment and function as antivirals via inhibition of the function of the M2 proton transporter. Besides pore blocking [Citation6–7], which is a conventional mechanism, a new novel allosteric mechanism where four RMTs were found to bind outside the pore, was recently proposed based on an NMR study [8]. This highlights the need to understand how RMT controls the gating residue of the M2-channel. The present study aims to provide information at the molecular level for the development of more effective drugs.
The M2 protein of the influenza A virus functions as a proton selective channel that is activated at the low pH of the endosome after endocytosis of the virus. In the early stages of infection, the M2 proton transporter leads to acidification of the viral interior from the acidic endosomal compartment following cellular endocytosis and this is required for the unpacking of the viral genome via release of viral nucleoproteins prior to viral replication. Whilst in the later stages of infection, the M2 proton port is required to transport protons from the transgolgi membrane to the host cell cytoplasm to equalise the pH and so prevent premature conformational rearrangement of the newly synthesised haemagglutinin during transport to the host cell surface membrane [Citation5]. The M2 protein is structurally a homotetramer which is stabilised in part by the disulphide bridges between the N terminal domain cysteine residues near the membrane, with each 97 amino acid monomer being composed of a 24-residue N-terminal extracellular domain, an α-helix single transmembrane (TM) domain of 19 residues and a 54-residue cytoplasmic domain [9]. The TM helix of M2 (M2-TM) spans the hydrophobic region of the membrane and includes a few hydrophilic residues at either end. The M2-TM forms tetrameric bundles and binds adamantane-based drugs, acting as the full M2 protein does, both in the micelles [Citation10] and in the lipid bilayers [Citation11–14].
Although the model structures of M2-TM are experimentally [Citation12–17] and computationally available [Citation18–29], high resolution structures have only recently been determined [Citation7,Citation8]. X-ray diffraction analysis has been used to solve the M2-TM structures under neutral and low pH conditions. The crystal structure revealed that a single AMT molecule in the pore of the channel was surrounded by Val27, Ala30, Ser31 and Gly34 [Citation27,Citation28]. In contrast, Schnell and Chou reported an NMR structure for the peptide spanning residues 18–60 in detergent micelles at high pH [Citation8], where four RMT molecules were bound outside of the protein helix, facing the lipid bilayer and located in the membrane environment at the end of the helix towards the cytoplasmic face of the channel (). Drug binding includes interactions with Leu40-Arg45 residues and, in particular the hydrogen bond formation between the –NH2 group of RMT and the −COO− group of Asp44 appears to be important [Citation8]. This newly proposed channel inhibition model is known as the allosteric mechanism [Citation29]. The novel external drug binding was suggested to stabilise the closed state, thus making the channel more rigid and difficult to rearrange the four helices to allow the channel to open. Although the NMR structure of the M2-TM region seems to be very well solved, the drug-binding site around Asp44 is not well defined and RMT adopts too many possible conformations. Even within a single NMR structure, each of the four RMT molecules displays a noticeable variability in the hydrogen bond and hydrophobic interactions with the M2 binding residues [Citation30].
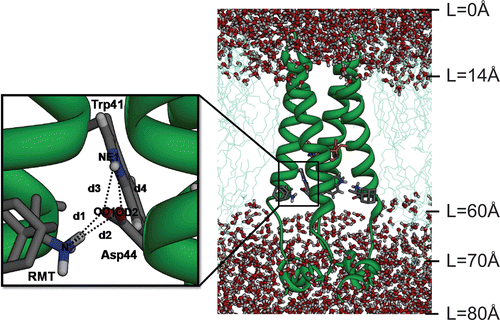
Figure 1. The initial structure of the tetrameric M2 protein complexed with four rimantadines (RMTs) bound outside the channel in the pre-equilibrated lipid bilayer-water pieces where L represents the distance along a channel axis starting from the extracellular site. The structure of the His37 tetrad has been coloured orange. Close view of the RMT-Asp44-Trp41 hydrogen bond network with definitions of the d1–d4 distances are also shown.
In the present study, molecular dynamics (MD) simulations were carried out for the tetrameric M2 protein channel complexed with four RMTs at the three protonation states of the His37 tetrad, corresponding to: the closed (0H), intermediate (1H) and open (3H) channel conformations. The main goal was to provide detailed information at the molecular level of how RMT inhibits the M2 proton conductance through the allosteric mechanism in terms of drug-target interactions, the conformation of the Trp41 channel gate, the water density and the potential of mean force (PMF) of water permeation along the channel axis.
Methodology
Preparation of the simulated systems
The NMR structures of the M2 tetrameric helix (residues 18–60) with the C-terminal base complexed with four RMTs, as taken from the Protein Data Bank [PDB entry code: 2RLF [Citation8], were used as the initial coordinates for the MD simulations. The simulations were carried out for the three different protonation states of the His37 residues, where all four His37 residues were considered neutral entities for the closed channel, one was positively charged for the intermediate channel and three of them were positively charged for the open channels, as a function of pH [Citation8,Citation12,Citation31,Citation32]. For simplicity, the simulations were denoted hereafter as 0H, 1H and 3H for the non-, mono- and triple-protonated states, respectively. In all the complexes, the protonation state of the ionisable amino acid residues were assigned in the following manner: side chain of Asp(24,44) and Glu(56) were considered to be negatively charged while that of Arg(45,53) was treated as positively charged. Therefore, the interhelical salt bridge between the Asp44 and Arg45 was well formed [Citation8]. Regarding drug interactions, the relatively high pKa values of 10.4 [Citation33] made the side chain of the RMT have a protonated form (R–NH3+) [Citation28] and consequently this –NH3+ group was in contact with the –COO− group of Asp44 at the allosteric binding region [Citation8]. Each system was separately built according to the designed protonation state of the His37 tetrad with the RMT inhibitor bound and then inserted into a pre-equilibrated lipid bilayer, initially made up of 77 molecules of 1-palmitoyl-2-oleoyl-sn-glycerol-3-phosphatidylcholine (POPC) lipid [Citation34] embedded in 3760 molecules of FLEXSPC water [Citation35]. The simulated systems were neutralised by counterions and the solvated box dimensions were set to 70 Å × 70 Å × 80 Å.
MD simulations
All calculations were performed using the GROMACS 3.2.1 package [Citation36] with the GROMACS force field [Citation37]. The inhibitor’s topology file was taken from our previous studies [28, 38]. The whole structure was optimised by the steepest descent minimisations. In the next stage, the system was equilibrated for 0.5 ns with position restraints on the protein atoms to improve the packing of the lipid bilayer around the protein, followed by a free MD simulation of 16 ns during which time the structural coordinates were saved every 0.5 ps.
For all the simulations, the periodic boundary condition with fixed pressure P, temperature T, and number of atoms N (NPT ensemble) was employed. The LINCS algorithm [Citation39] was applied to constrain the bond lengths and the angles involving hydrogen atoms and a 2 fs time step was used. The systems were coupled separately to a Berendsen temperature bath [Citation40] at 310 K using a coupling constant τT = 0.1 ps. The pressure (1 bar) was kept constant by semi-isotropic coupling of the system to a Berendsen pressure bath [40]. The long-range interactions were restricted to within the twin-range cutoffs of 1.2 nm for both van der Waals interactions and electrostatic interactions, computed using the Particle Mesh Ewald (PME) algorithm [41]. The analysis phase was from 8 ns to 16 ns, in which the convergences of energies, temperature, pressure and global root mean square displacement (RMSD) were used to verify the equilibrium of the system.
Free energy barrier for water permeation
The energy barrier of the water transport throughout the M2 channel was calculated with an analogous treatment according to Raschke and Levitt [43]. The PMF of the water permeation along the channel axis can be derived from the water density plot according to:
where R is the gas constant, T is the temperature in Kelvin and w(r) is the probability density of the water distribution in a spherical region within a radius of r. Here, the probability density is given as:
where ρr and ρtotal are the local and total water densities, respectively. In the water density calculation, r is extended to 80 Å to ensure that water molecules in the aqueous zone (bulk water) are taken into account. ρtotal represents the bulk water, which presumably has a total probability equal or near to 1. By employing this assumption, ρtotal has the maximum value of ρr .
Results and discussion
System stability
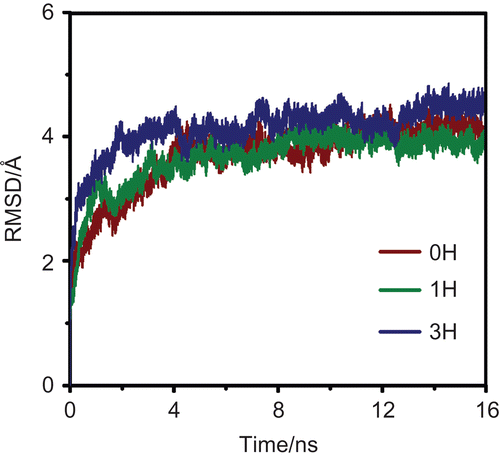
The RMSD for the M2-RMT complexes in the 0H, 1H and 3H states were calculated and used for monitoring the stability of the systems. The calculations were carried out relative to the initial structure (heavy atoms only) of the M2-RMT complexes and the results are shown in . The RMSD plots indicated that all systems reached equilibrium after the 8 ns simulations.
Figure 2. The root mean square displacements (RMSD) for the M2-RMT complexes in the 0H, 1H and 3H states.
Protein-protein and drug-protein interactions
The hydrogen bond network of RMT-Asp44-Trp41 () has been proposed to be a vital determinant in the inhibitory mechanism of RMT when bound outside of the M2 channel [Citation8]. To extract such information from the MD simulations, the percentage hydrogen bonding of the important central residue (Asp44) with the RMT inhibitor (RMT-Asp44), and the channel gating residue Trp41 (Asp44-Trp41) at the adjacent subunit, were evaluated based on the criteria of a proton donor-acceptor distance ≤ 3.5 Å and a donor-H-acceptor bond angle of ≥ 120° (). The RMT-Asp44-Trp41 hydrogen bond network was described in terms of the four distances, d1-d4 (), which represent the RMT-Asp44 (d1-d2) and Asp44-Trp41 (d3-d4) pair interactions (). The plots were separately determined for the four subunits (I–IV) of the 0H, 1H and 3H protonated states of the M2 protein.
Table 1. Percentage of hydrogen bonds in the three (0H, 1H and 3H) protonation states between (i) the ammonium group of RMT and M2 residues and (ii) the Asp44 and Trp41 gating residues.
Figure 3. The distributions of the d1–d4 distances, as defined in , for the three simulated systems: 0H, 1H and 3H, at the subunits I–IV of the M2 channel.
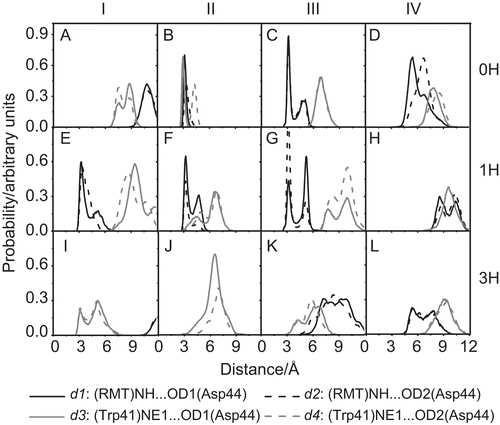
The complete RMT-Asp44-Trp41 hydrogen bond network was only present in the 0H-closed state, where the RMT-Asp44 and Asp44-Trp41 interactions were strongly and clearly detected. The sharp and narrow peaks for subunit II at the d1 and d2 distances of ~3.5 Å () indicated the two hydrogen bonds between the RMT and Asp44, with occupations of 31% and 14% (). At the same time, a strong and more secure hydrogen bond was formed between the Asp44 of subunit II and the Trp41 of subunit III at d3 of about 3.5 Å (), with an occupancy of 80% (). The presence of this hydrogen bond network among RMT, Asp44 and Trp41 indicated that close contact between the RMT ammonium group and the Asp44 carboxylate group could bring the bulky Trp41 indole ring into van der Waals contact to form a more stable Trp41 channel gate. In addition, the RMT-Asp44 interactions at subunit III (), indicated by the sharp peaks of the d1 and d2 at ~3.5 Å, also facilitate the formation of the closed conformation of the M2 channel.
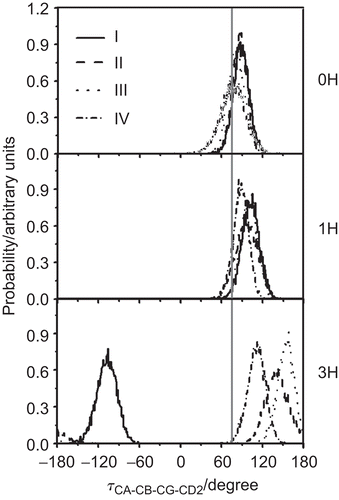
In the case of the intermediate channel conformation seen with the mono-protonated (1H) state, the shortest hydrogen bond distances were found for the RMT-Asp44 (d1-d2) interactions at the three subunits, I, II and III (), whilst the Asp44-Trp41 (d3-d4) distances were between ~7 and ~10 Å (). Therefore, only the rather weak interactions between the RMT and Asp44 were found (<20% occupation, ). The results indicated that Asp44 in the 1H state cannot play a role as the centre of the hydrogen bond network between the inhibitor and the gating residue Trp41. The situation is totally different for the higher protonated state, 3H, where almost no interaction amongst these three molecules was detected. This can be seen as the broad peak positions at longer distances of d1-d4 (), without any hydrogen bond occupancy (). This observation implies that the strong electrostatic repulsion among the three protonated His37 imidazole rings in the 3H state destabilises the helix–helix packing, leading to the conformational rearrangement to break interactions between Trp41 and Asp44, and so allow the Trp41 gate to flip open (more details in the “Conformation of the Trp41 gating residue” section) and reduces the ability of RMT to bind to Asp44.
Water density across the M2 channel
To determine the inhibition ability of RMT at the allosteric site of the M2 channel, the water density profile as a function of the distance (L) along the pore-axis of the channel, starting from the N-terminal site, was evaluated In addition, the distribution patterns of the His37 proton selectivity and the Trp41 gate opening residues were analysed in order to illustrate the movement of these critical residues ().
Figure 4. The water densities and the distribution patterns of the His37 selectivity and Trp41 gating residue positions in the free M2 protein and M2-RMTs complex.
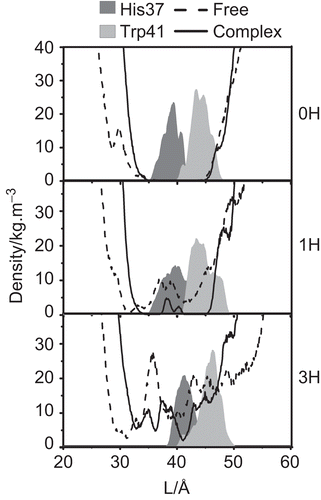
In the 0H-closed state of the M2 channel, a zero water density was observed in the channel pore at 35 Å < L < 45 Å for both the free and the RMT-bound forms. This indicated that water cannot penetrate through the channel when all four His37 residues are uncharged. With respect to the intermediate state (1H), although water entry was fully inhibited at ~30 Å in the free form (), a degree of water density was observed in the area of the His37 and Trp41 residues. Interestingly, in contrast a zero water density was found across this area in the M2-RMT complex. The results detected for both 0H and 1H states lead us to conclude that the four RMTs bind to the external pore side (lipid facing pocket) in the systems with neutral (pH = 7.5) or one charged (pH = 7) histidine residues and this not only helps to stabilise the M2-channel in its closed conformation but also blocks water penetration.
In contrast to the 0H and 1H protonation states discussed above, a zero water density in the RMT bound M2 channels was not observed in the 3H state (). Although the water density in the M2-RMT complex was significantly lower than that of the free channel, the four RMT drugs bound outside the pore did not inhibit the water transport. This observation is in good agreement with a previous electrophysiological study where the binding of RMT outside of the M2 channel was found to not be the primary site for pharmacological inhibition or proton transport [Citation30]. In contrast, water penetration was completely prevented in the 3H state when RMT was bound inside the M2 pore [Citation28]. A clear picture of water passing through the pH sensor His37 and proton gate Trp41 residues in the 3H state can be ascribed as follows. At pH ≤ 6, the His37 imidazole ring is protonated leading to destabilisation of the helix-helix packing due to the strong electrostatic repulsion. This conformational rearrangement leads to a breaking of the interaction between Trp41 and Asp44 residues (see and details discussed in the “Protein-protein and drug-protein interactions” section) and allows the gate to flip to an open form. Therefore, adding of RMT to the channel at pH ≤ 6, when the channel is in the open form, cannot facilitate the tetrad Asp44 to play a role as a locking residue for the M2 channel.
The above notion agrees very well with the protein-protein and drug-protein interactions, in terms of the RMT-Asp44-Trp41 hydrogen bond network, where the complete hydrogen bond network was only found in the 0H state, and only moderate RMT-Asp44 hydrogen bonds were detected in the 1H state, while those interactions were completely lost for the highest (3H) protonated state.
Conformation of the Trp41 gating residue
shows the CA-CB-CG-CD2 torsion angle of the indole ring of the gating residue Trp41 in the three different channels. In the 0H and 1H states, the orientations of the torsional angles of the Trp41 indole rings of the four M2 subunits are considerably similar. All rotations occur within the range of 30° to 120° with a maximum at 90°. The implication is that, at the low (0H and 1H) protonation states, the tryptophans mostly lie in a configuration that is almost perpendicular to the channel axis, i.e. the channel is closed by the four indole rings, explaining why water cannot pass through the 0H and 1H channels as discussed previously. In contrast, considerable variation in the torsion angles of the four tryptophan sidechains was found between the four subunits in the 3H state; namely at −110°, 110°, 130° and 170° for subunits I–IV, respectively. The dramatic rotation by ~180° in the Trp41 of M2 subunit-I and the moderate rotation in the Trp41 residues of the other three M2 subunits on changing from the 0H and 1H state to the 3H state indicates the transformation of the channel from the closed (0H and 1H) to the open (3H) conformation, supported by the water density detected along the pore ().
Figure 5. The rotational angle defined by CA, CB, CG and CD2 of the M2 Trp41 gate when M2 is complexed with the four RMTs outside the pore at different protonation states (0H, 1H and 3H). I–IV denote the four M2 subunits. Here, the vertical grey solid line (at 75°) represents the tortional angle of the closed conformation of the Trp41 indole ring obtained from NMR structure.
Affinity of water permeation
To investigate how difficult it may be for water molecules to move through the M2 channel in the free and RMT complexed forms of both four RMTs binding outside- (M2-RMTout) and a RMT binding inside- (M2-RMTin) mediate in which the data of M2-RMTin was taken from our previous study [Citation28], the PMFs of water permeation along the channel axis (L) starting from the N-terminal site were evaluated and compared in .
Figure 6. The potential of mean force (PMF) of the excess proton along the pore channel axis (L) starting from the N-terminal site for the three protonation states, 0H, 1H and 3H of the free M2 (dashed line) and RMT complexes where the four RMTs were located outside (black solid line, M2-RMTout) and inside the ion channel (grey solid line, M2-RMTin).
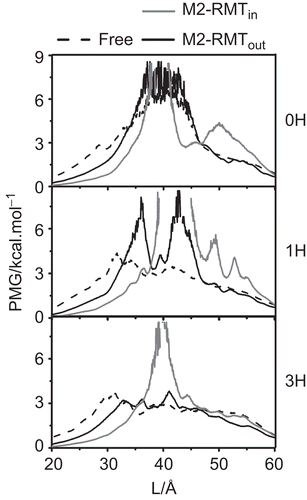
It is clearly seen that the energy barriers between the M2-RMTout complex and the free M2 protein for the 0H and 3H states are not notably different. However, this is not the case for the 1H state, where the maximum energy barrier of ~7.5 kcal·mol−1 for the complex at L ~36 Å and ~43 Å is significantly higher than that of the free form (~4 kcal·mol−1 at L ~32 Å). In addition, the energy barriers for these free and M2-RMTout complexed forms were found in the following order: 0H > 1H > 3H. In contrast, the PMFs for M2-RMTin observed at L~40, 42, 40 Å of 0H, 1H and 3H states, respectively, are significantly higher than those of the free and M2-RMTout complexes for all the protonation staes.
In the free form, the PMF barrier (taken from the maximum of each plot) of ~6 kcal mol−1 for the 0H state was significantly reduced to ~3-4 kcal·mol−1 in the 1H and 3H states. This supports the experimental evidence, where in the absence of RMT binding to the M2 channel water permeation was observed in the mono- (1H) and triple- (3H), but not the non- (0H) protonated states [Citation22,Citation28].
Regarding the M2 channel with RMTs bound outside, the 0H and 1H states display relatively high PMF energies of ~4-9 kcal·mol−1, suggesting that water permeation is energetically unfavourable in these systems. This is consistent with the zero water density plots of both the 0H and 1H states (). In contrast, the energy barrier for water transport is significantly lower at ~3 kcal·mol−1 in the 3H state of the M2-RMTout complex, consistent with the non-zero water density observed throughout the channel. Interestingly, the PMFs for M2-RMTin complexes in all the protonation states are significantly higher than ~7.5 kcal·mol−1 indicating no water transport was processed where a single RMT blocking inside the M2 channel. These results clearly demonstrated that the RMT binding inside the M2 pore has potentially inhibited the M2 machinery rather than the RMTs allosteric binding outside.
In summary, the information here leads us to conclude that the RMT-mediated inhibition via their binding to the outside of the pore in the closed and intermediate conformations at high pH conditions can prevent proton conductance by interrupting the formation of the water wire along the channel. In contrast, the transportation cannot be inhibited at a low pH condition, i.e. in the 3H-open conformation.
Conclusion
Based on the MD simulated results presented here, the inhibition efficiency of the four RMTs bound outside the M2 channel pore was considerably lower than that observed when bound inside the channel. The key parameter determining the drug outside binding efficiency is based on the hydrogen bond network centred on the Asp44 residue which interacts with RMT to further stabilise the gating channel by inter-subunit hydrogen bonding with the Trp41 gating residue. This network was eliminated when the positive charge was increased on the selective His37 residue, due to the decreased van der Waals contact of the Trp41 indole ring being affected by the strong electrostatic repulsion. The effect was manifested at pH ≤ 6 (triple protonation state on the His37 tetrad) where the RMT-Asp44-Trp41 hydrogen bond network was completely lost, leading to a rearrangement of the indole ring of the Trp41 gate to flip to an open conformation. The water permeation throughout the M2 channel that was consequently observed was ranked as 0H < 1H < 3H which is inversely consistent with the observed PMF. Taken together, the MD-based simulation results have clearly explained the role of RMT at the molecular level in the allosteric binding site of the M2 channel at high pH conditions but not at a low pH.
Acknowledgements
P.I. thanks the Post-Doctoral programme from the Commission on Higher Education. S.H. thanks the Centre of Excellence for Petroleum, Petrochemicals, and Advanced Materials, Chulalongkorn University. Research facilities, software packages and computing time were provided by the Computer Centre for Advanced Research and the Computational Chemistry Unit Cell, Faculty of Science, Chulalongkorn University. Molecular graphics images were produced using the UCSF Chimera package from the Resource for Biocomputing, Visualization, and Informatics at the University of California, San Francisco (supported by NIH P41 RR-01081).
Declaration of interest
This work was supported by The Thailand Research Fund.
References
- WHO, Global Alert and Response (GAR), Online at: http://www.who.int/csr/disease/avian_influenza/country/en/, accessed September 2009.
- WHO, Global Alert and Response (GAR), Online at: http://www.who.int/csr/don/2009_10_09/en/index.html, accessed October 2009.
- Helenius A. Unpacking the incoming influenza-virus. Cell 1992;69:577–578.
- Ciampor F, Bayley PM, Nermut MV, Hirst EM, Sugrue RJ, Hay AJ. Evidence that the amantadine-induced, M2-mediated conversion of influenza virus A hemagglutinin to the low pH conformation occurs in an acidic trans Golgi compartment. Virology 1992;188:14–24.
- Lamb RA, Holsinger LJ, Pinto LH. In: Wimmer E, ed. Receptor-mediated Virus Entry into Cells. New York: Cold Spring Harbor Laboratory Press, 1994:303–321.
- Hille B. Ionic Channels of Excitable Membranes. Sunderland, MA: Sinauer Associates, 2001.
- Stouffer AL, Acharya R, Salom D, Levine AS, Di Costanzo L, Soto CS, Tereshko V, Nanda V, Stayrook S, DeGrado WF. Structural basis for the function and inhibition of an influenza virus proton channel. Nature 2008;451:596–599.
- Schnell JR, Chou JJ. Structure and mechanism of the M2 proton channel of influenza A virus. Nature 2008;451:591–595.
- Wang C, Takeuchi K, Pinto LH, Lamb RA. Ion channel activity of influenza A virus M2 protein: Characterization of the amantadine block. J Virol 1993;67:5585–5594.
- Salom D, Hill BR, Lear JD, DeGrado WF. pH-dependent tetramerization and amantadine binding of the transmembrane helix of M2 from the influenza A virus. Biochemistry 2000;39:14160–14170.
- Nishimura K, Kim S, Zhang L, Cross TA. The closed state of a H+ channel helical bundle combining precise orientational and distance restraints from solid state NMR. Biochemistry 2002;41:13170–13177.
- Hu J, Fu R, Nishimura K, Zhang L, Zhou HX, Busath DD, Vijayvergiya V, Cross TA. Histidines, heart of the hydrogen ion channel from influenza A virus: Toward an understanding of conductance and proton selectivity. P Natl Acad Sci USA 2006;103:6865–6870.
- Cady SD, Hong M. Amantadine-induced conformational and dynamical changes of the influenza M2 transmembrane proton channel. P Natl Acad Sci USA 2008;105:1483–1488.
- Witter R, Nozirov F, Sternberg U, Cross TA, Ulrich AS, Fu RQ. Solid-state F-19 NMR spectroscopy reveals that Trp(41) participates in the gating mechanism of the M2 proton channel of influenza a virus. J Am Chem Soc 2008;130:918–924.
- Pinto LH, Dieckmann GR, Gandhi CS, Papworth CG, Braman J, Shaughnessy MA, Lear JD, Lamb RA, DeGrado WF. A functionally defined model for the M-2 proton channel of influenza A virus suggests a mechanism for its ion selectivity. P Natl Acad Sci USA 1997;94:11301–11306.
- Wang JF, Kim S, Kovacs F, Cross TA. Structure of the transmembrane region of the M2 protein H+ channel. Protein Sci 2001;10:2241–2250.
- Cady SD, Goodman C, Tatko CD, DeGrado WF, Hong M. Determining the orientation of uniaxially rotating membrane proteins using unoriented samples: A H-2, C-13, and N-15 solid-state NMR investigation of the dynamics and orientation of a transmembrane helical bundle. J Am Chem Soc 2007;129:5719–5729.
- Sansom MSP, Kerr ID, Smith GR, Son HS. The influenza A virus M2 channel: A molecular modeling and simulation study. Virology 1997;233:163–173.
- Zhong QF, Newns DM, Pattnaik P, Lear JD, Klein ML. Two possible conducting states of the influenza A virus M2 ion channel. Febs Lett 2000;473:195–198.
- Zhong QF, Husslein T, Moore PB, Newns DM, Pattnaik P, Klein ML. The M2 channel of influenza A virus: a molecular dynamics study. Febs Lett 1998;434:265–271.
- Ayton GS, Voth GA. Multiscale simulation of transmembrane proteins. J Struct Biol 2007;157:570–578.
- Kass I, Arkin IT. How pH opens a H+ channel: The gating mechanism of influenza a M2. Structure 2005;13:1789–1798.
- Chen HN, Wu YJ, Voth GA. Proton transport Behavior through the influenza a M2 channel: Insights from molecular simulation. Biophys J 2007;93:3470–3479.
- Wu YJ, Voth GA. A computational study of the closed and open states of the influenza A M2 proton channel. Biophys J 2005;89:2402–2411.
- Smondyrev AM, Voth GA. Molecular dynamics simulation of proton transport through the influenza A virus M2 channel. Biophys J 2002;83:1987–1996.
- Yi M, Cross TA, Zhou HX. A secondary gate as a mechanism for inhibition of the M2 proton channel by amantadine. J Phys Chem B 2008;112:7977–7979.
- Hay AJ, Wolstenholme AJ, Skehel JJ, Smith HH. The molecular basis of the specific anti-influenza action of amantadine. EMBO J 1985;4:3021–3024.
- Intharathep P, Laohpongspaisan C, Rungrotmongkol T, Loisruangsin A, Malaisree M, Decha P, Aruksakunwong O, Chuenpennit K, Kaiyawet N, Sompornpisut P, Pianwanit S, Hannongbu S. How amantadine and rimantadine inhibit proton transport in the M2 protein channel. J Mol Graph Model 2008;27:342–348.
- Huang RB, Du QS, Wang CH, Chou KC. An in-depth analysis of the biological functional studies based on the NMR M2 channel structure of influenza A virus. Biochim Bioph Res Commun 2008;377:1243–1247.
- Jing XH, Ma CL, Ohigashi Y, Oliveira FA, Jardetzky TS, Pinto LH, Lamb RA. Functional studies indicate amantadine binds to the pore of the influenza A virus M2 proton-selective ion channel. P Natl Acad Sci USA 2008;105:10967–10972.
- Cady S, Schmidt-Rohr K, Wang J, Soto C, DeGrado W, Hong M. Structure of the amantadine binding site of influenza M2 proton channels in lipid bilayers. Nature 2010;463:689–692.
- Ma C, Polishchuk A, Ohigashi Y, Stouffer A, Schön A, Magavern E, Jing X, Lear J, Freire E, Lamb R, DeGrado d, Pinto L. Identification of the functional core of the influenza A virus A/M2 proton-selective ion channel. Proc Natl Acad Sci U S A. 2009;106:12283–12288.
- Spector R. Transport of amantadine and rimantadine through the blood-brain barrier. J Pharm Exp Ther 1988;244:516–519.
- Forrest LR, Tieleman DP, Sansom MSP. Defining the transmembrane helix of M2 protein from influenza A by molecular dynamics simulations in a lipid bilayer. Biophys J 1999;76:1886–1896.
- Berendsen HJC, Postma JPM, van Gunsteren WF, Hermans J. Intermolecular Forces. In: Pullman B, ed. Dordrecht The Netherlands: D. Reidel Publishing Company, 1981. pp. 331–342.
- Lindahl E, Hess B, Spoel Dvd. GROMACS 3.0: a package for molecular simulation and trajectory analysis. J Mol Model 2001;7:306–317.
- Hermans J, Berendsen HJC, van Gunsteren WF, Postma JPM. A consistent empirical potential for water–protein interactions. Biopolymers 1984;23:1513–1518.
- Rungrotmongkol T, Intharathep P, Malaisree M, Nunthaboot N, Kaiyawet N, Sompornpisut P, Payungporn S, Poovorawan Y, Hannongbua S. Susceptibility of antiviral drugs against 2009 influenza A (H1N1) virus. Biochem Bioph Res Co 2009;385:390–394.
- Hess B, Bekker H, Berendsen HJC, Fraaije JGEM. LINCS: a linear constraint solver for molecular simulations. J Comp Chem 1997;18:1463–1472.
- Berendsen HJC, Postma JPM, van Gunsteren WF, DiNola A, Haak JR. Molecular dynamics with coupling to an external bath. J Chem Phys 1984;81:3684–3690.
- Darden T, York D, Pedersen L. Particle mesh Ewald: an N–log(N) method for Ewald sums in large systems. J Chem Phys 1993;98:10089–10092.
- Raschke TM, Levitt M. Detailed hydration maps of benzene and cyclohexane reveal distinct water structures. J Phys Chem B 2004;108:13492–13500.