Abstract
The 4-oxo-β-lactams (azetidine-2,4-diones) are potent acylating agents of the human leukocyte elastase (HLE), a neutrophil serine protease that plays a key role in several inflammatory diseases. A novel 4-oxo-β-lactam containing a N-(4-(phenylsulphonylmethyl)phenyl) group, 3, was designed as a potential mechanism-based inhibitor capable of undergoing elimination of phenylsulphinate upon Ser-195 acylation. Compound 3 was found to be a potent slow-tight binding inhibitor of HLE, presenting a remarkable second-order rate constant of 1.46 × 106 M−1s−1 and displaying selectivity over the proteinase 3 and cathepsin G. However, liberation of phenylsulphinate was not observed in the hydrolysis of 3 in both pH 7.4 phosphate buffer and human plasma. The Cmax values of 1207 μg/total blood, 179 μg/g spleen and 106 μg/g lung were determined by HPLC, following a single 30 mg/kg dose of 3 given intraperitoneally to NMRI mice, suggesting that the inhibitor distributes well into tissues. Although being a powerful selective inhibitor of HLE, 4-oxo-β-lactam 3 has a limited stability, being susceptible to off-target reactions (plasma and liver enzymes).
Introduction
Neutrophils are leukocytes and are the first line of defence against infection, they act by migrating from the blood into tissue to engulf pathogens in response to inflammatory stimuli. Neutrophils present a potent antimicrobial arsenal for the degradation of foreign material, including many degradative enzymes. In particular, human leukocyte elastase (HLE, EC 3.4.21.37) is a neutrophil serine protease released from neutrophils in response to stimuli and mediators [Citation1]. The unregulated activity of HLE, due to an imbalance between proteases and endogenous inhibitors (the predominant one being α1-antitrypsin), is implicated in the extracellular degradation of matrix proteins such as elastin and collagen and plays a key role in several inflammatory diseases, such as chronic obstructive pulmonary disease (COPD) and adult respiratory distress syndrome (ARDS) [Citation2–5]. In particular, CODP, which involves pulmonary emphysema and chronic bronchitis, is the fourth leading cause of death in the world [Citation6]. Furthermore, HLE also activates other proteases (e.g. metalloproteinases) and upregulates inflammation [Citation7,Citation8]. Thus, selective inhibitors of HLE have the potential to reestablish the protease/anti-protease balance, thereby providing a promising strategy for the treatment of COPD and other inflammatory disorders [Citation9–11]. While several low molecular weight synthetic inhibitors have been developed, toxicity and instability problems have disrupted their investigation [Citation10–12].
We have recently reported that the 4-oxo-β-lactams (azetidine-2,4-diones) are useful design scaffolds for serine protease inhibitors [Citation13]. The 4-oxo-β-lactams containing N-aryl or N-heteroarylthiomethyl groups were designed as potential mechanism-based inhibitors featuring a latent electrophilic quinone methide imine function, that upon further reaction with an active site nucleophilic residue (e.g. His-57), may ultimately lead to irreversible inactivation of the enzyme [Citation14]. Structure-activity relationship analysis showed that such 4-oxo-β-lactams afforded highly potent and selective inhibition of HLE, even at a very low inhibitor-to-enzyme ratio, as shown by the second-order rate constant for the inhibitor-enzyme association, kon, of 3.2 × 106 M−1s−1 for derivative 1 ().
Figure 1. Structures of elastase inhibitors 1-4.
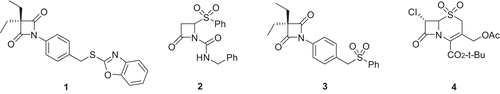
Despite the excellent inhibitory potency displayed by 1 and related derivatives, stability studies in both phosphate buffer and human plasma suggested that the departure of the thiol leaving group following 4-oxo-β-lactam ring-opening does not occur in the HLE inhibition pathway [Citation14]. Moreover, the highly hydrophobic nature of the 4-oxo-β-lactams (e.g. 1, log P = 4.48 [Citation15]) could well result in fast elimination from highly-perfused lungs and consequently a limited in vivo efficacy. We therefore turned our attention to the more polar sulphone counterparts. Aryl sulphonylmethyl moieties have been successfully incorporated into the design of mechanism-based HLE inhibitors, examples of which are the 1,2-benzisothiazol-3(2H)-one 1,1-dioxide [Citation16] and 1,2,5-thiadiazolidin-3-one 1,1-dioxide scaffolds [Citation17–21]. For example, Groutas et al. suggested that the latter inactivates HLE via a mechanism involving the departure of the corresponding sulphinic acid to form an electrophilic imine, while the less potent sulphide counterpart leads to acylation of the enzyme without loss of the leaving group [Citation18]. Moreover, we ourselves have unequivocally shown that β-lactam ring-opening of 2 () following nucleophilic attack at the lactam carbonyl carbon atom leads to the departure of phenylsulphinic acid [Citation22]. We thus envisaged that the 4-oxo-β-lactam sulphone derivative, 3, could lead to the expulsion of phenylsulphinate subsequent to acylation of Ser-195 and form an electrophilic quinone methide imine function in a mechanism similar to that depicted in . Here we wish to report the synthesis of 3 and its in vitro inhibitory activity against human neutrophil serine proteases HLE, proteinase 3 and cathepsin G, and the cysteine protease papain. In addition, in anticipation of using this compound in rodent efficacy models, its stability in PBS and human plasma, together with mouse blood and tissue (lung, spleen and liver) levels following intraperitoneal administration of a single dose are reported.
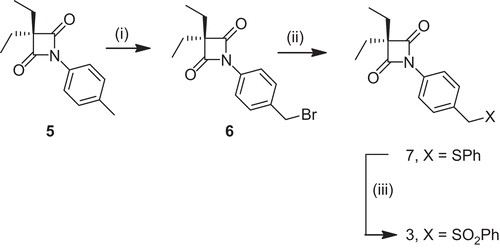
Scheme 1. Synthesis of 4-oxo-β-lactam 5.
Materials and Methods
General
Melting points were determined using a Kofler camera Bock Monoscope M (Frankfurt, Germany) and uncorrected. Infrared spectra were recorded using a Nicolet Impact 400 FTIR spectrophotometer (Nicolet Analytical Instruments, Madison, WI, USA). NMR spectra were recorded using a Brucker 400 Ultra-Shield (Brucker ARX 400 spectrometer, 400 MHz, Bruker Instruments, Billerica, MA, USA) spectrometer in CDCl3 solution; chemical shifts, δ, are expressed in ppm relative to tetramethylsilane as the internal standard, and the coupling constants, J, are expressed in Hz. Low-resolution mass spectra were recorded using an HP5988A spectrometer, by RIAIDT, University of Santiago de Compostela, Spain. Elemental analyses were performed by Medac, Brunel Science Centre, Egham, UK. Spectrophotometric assays were performed using a Shimadzu UV-2100 PC instrument (iMed.UL, Lisbon). TLC was performed using Merck grade silica gel 60 F254 aluminium plates. Preparative column chromatography was performed using silica gel 60 from Merck (70-230 mesh ASTM). Solvents and buffer materials for enzyme assays were of analytical reagent grade. Cathepsin G (EC.3.4.21.20), MeOSuc-Ala-A-Ala-Pro-Val-p-NA and Suc-Ala-Ala-Pro-Phe-p-NA were purchased from Sigma (Portugal), while HLE (EC 3.4.21.37) and proteinase 3 (EC.3.4.21.76) were purchased from Calbiochem (Portugal).
3,3-Diethyl-1-[4-(phenylsulphonylmethyl)phenyl]azetidine-2,4-dione (3)
A mixture of 3,3-diethyl-1-(4-tolyl)azetidine-2,4-dione (5, ) [13] (0.346 mmol), N-bromosuccinimide (NBS) (0.346 mmol) and benzoyl peroxide (0.0346 mmol) in tetrachloromethane was heated under reflux for 12h, the reaction being monitored by TLC. Benzoic acid was removed by filtration and the solvent removed under reduced pressure to yield 1-(4-(bromomethyl)phenyl)-3,3-diethylazetidine-2,4-dione, 6 [Citation14]. Thioether 7 was prepared by adding triethylamine(2.2 mmol) to a solution of 6 (2 mmol) and thiophenol (2.2 mmol) in dry tetrahydrofuran (THF) (5 mL). The reaction was stirred at room temperature, being monitored by TLC, on completion the triethylamine hydrochloride was removed by filtration and the solvent removed under reduced pressure [Citation14]. 3,3-Diethyl-1-[4-(phenylsulphonylmethyl)phenyl]azetidine-2,4-dione, 3, was prepared by slowly adding 3-chloroperbenzoic acid (MCPBA, 0.382 mmol) to a cold solution of the thioether 7 [14] (0.153 mmol, 51.9 mg) in DCM (10 mL). The reaction mixture was warmed to room temperature and monitored by TLC. Upon completion of the reaction (ca. 40 min), the solvent was removed under reduced pressure, the crude product was purified by column chromatography and then recrystallised from DCM-hexane to yield a white solid (47.4 mg, 83%), mp 143–145°C, νmax (film) 1856, 1737, 1315, 1153 cm−1; δ 1H-NMR 1.08 (6H, t, J = 7.6), 1.88 (4H, q, J =7.6), 4.32 (2H, s), 7.16 (2H, d, J = 8), 7.52 (2H, t, J = 8), 7.66 (1H, t, J = 8), 7.71 (2H, dd, J = 8, 1.6), 7.79 (2H, d, J = 8); δ 13C-NMR 9.24, 23.9, 62.3, 72.3, 119.1, 126.4, 128.6, 129.1, 131.8, 134, 134.2, 137.8, 172; EI-MS 230.15 (M-SO2Ar)+ (22.19%), 132.05 (C7H6NCO)+ (100%); Anal. calcd. for C20H21NO4S: C, 64.67; H, 5.7; N, 3.77; S, 8.63; found C, 64.43; H, 5.94; N, 3.56; S, 8.31.
Enzyme Kinetics
The rates of inhibition of HLE, cathepsin G and proteinase 3 by 3 were determined by the progress curve method [Citation14]. The inhibition of the HLE was studied at 25°C by continuously monitoring the absorbance at 410 nm for 20 min of a solution prepared by mixing 10 μL of HLE stock solution (2 μM in 0.05M acetate buffer, pH 5.5) with a solution containing 10 μL of 5 in DMSO, 20 μL of MeOSuc-Ala-Ala-Pro-Val-p-NA (50 mM in DMSO) and 960 μL of 0.1M pH 7.2 HEPES buffer. Control assays, in which the inhibitor was omitted, ran linearly. Inhibition of HLE was extremely rapid even at very low inhibitor concentrations (i.e. < 10[E]). In these experimental conditions, progress curves at different inhibitor concentrations were fitted to Equation 1, which accounted for the tight-binding nature of the inhibition [Citation23].
where A is the absorbance at 410 nm (related to the concentration of 4-nitroaniline formed upon hydrolysis of the MeOSuc-Ala-Ala-Pro-Val-p-NA through an extinction coefficient of 8250 M−1 cm−1), A0 is the absorbance at time t = 0, vi and vs are the initial and final rates of product formation, kobs is the pseudo-first-order rate constant for the approach to the steady-state and γ is defined by
Similarly, inhibition of cathepsin G and proteinase 3 by 3 was studied using respectively Suc-Ala-Ala-Pro-Phe-p-NA and MeOSuc-Ala-Ala-Pro-Val-p-NA as substrates. Inhibition was observed at inhibitor concentrations > 10[E] and thus, progress curves were fitted to Equation 3. The inhibitory activity of 3, towards papain (1mg/ml) was investigated by the incubation method, ([Citation3] = 50 μM in the incubation mixture), using the described procedures [Citation14].
Stability studies
The hydrolysis of 4-oxo-β-lactam 3 was studied using an HPLC system comprising a Merck Hitachi L-7100 pump (Merck, Darmstadt, Germany), a Shimadzu SPD-6AV detector (Shimadzu Co., Japan), a manual sample injection module with a 20 µL loop, and a Merck LiChroCART® 250-4 RP8 (5 µm) column. The mobile phase was acetonitrile/water (65:35) and the column effluent was monitored at 254 nm with a flow rate of 1 mL min−1. For the pH 7.4 hydrolysis, isotonic phosphate buffer solution (PBS), a 15 µL aliquot of a 10−2 M stock solution of the 4-oxo-β-lactam 3 in acetonitrile was added to 2.5 mL of PBS at 37°C. At regular intervals, samples of the reaction mixture were analysed by HPLC.
Human plasma was obtained from the pooled, heparinised blood of healthy donors, was frozen and stored at −20°C prior to use. For the hydrolysis experiments, the compounds were incubated at 37°C in human plasma that had been diluted to 80% (v/v) with pH 7.4 PBS. At appropriate intervals, aliquots of 100 µL were added to 200 µL of acetonitrile to both quench the reaction and precipitate the plasma proteins. These samples were centrifuged and the supernatant analysed by HPLC for the presence of the initial compound.
Ex vivo studies
Male NMRI mice (20–22 g) were obtained from Charles River (Barcelona, Spain). The animals were kept under standard conditions and fed ad libitum. All of the experimental procedures were carried out with the permission of the local laboratory committee, and in accordance with internationally accepted principles. Each group of mice received a single 30 mg kg−1 dose of 3 given by intraperitoneal (ip) injection and three mice were used per time point. At appropriate time intervals, blood was collected into heparinised tubes and stored at −30°C and the mice were euthanised. The lung, spleen and liver of the mice were removed and stored at −70°C. The mice which did not receive the administration of 3 were used as a control group. Determination of 3 in mice blood and tissues was performed by HPLC as described below.
Chromatographic system.
The HPLC system consisted of a 32 Katarat Software (Beckman Instruments, Fullerton, CA, USA), a Midas Spark 1.1 autoinjector (Spark, Emmen, The Netherlands) and a Diode-Array 168 detector (Beckman Instruments). The detector wavelength was set to 280 nm. The analytical column was a LiChroCART® (250-4.6) Purospher® Star RP-8 (5µm) (Merck). The mobile phase consisted of 0.05M potassium dihydrogen phosphate and 0.05M sodium acetate (pH adjusted to 4 with acetic acid) - acetonitrile (53:47, v/v) employing a flow rate of 1 mL min−1 at 25°C, adapted from Gaspar et al. [Citation24]. The retention time of 3 was approximately 5.2 min when analysing blood samples and 5 min when analysing organ samples.
Extraction of 3 from blood and tissues
The levels of compound 3 in blood and tissues were determined by HPLC after an extraction procedure according to Gaspar et al, [Citation24]. Briefly, 500 µL of blood were mixed with 250 µL of the mobile phase described above and extracted twice with 1 mL of a DCM:isooctane mixture (2:3, v/v) under stirring (15 min), followed by centrifugation at 1200 × g for 10 min using a GPR centrifuge (Beckman Instruments). Liver, lung and spleen tissues were thawed and aliquots of about 100 mg were weighed out for each sample, mixed with 500 µL of buffer and extracted twice with 2300 µL of the DCM: isooctane mixture by mechanical shaking for 30 min at room temperature, followed by centrifugation at 1200 × g for 10 min. The organic extracts were pooled and evaporated to dryness under nitrogen. The residue was dissolved in 1500 µL of mobile phase, filtered and then injected into the HPLC system. To determine the efficiency of the extraction procedures, a known amount of 3 was added to blood, solid tissues were removed from the mice that had not received 3 and then submitted to the above-mentioned extraction protocol.
Results
Chemistry
The 4-Oxo-β-lactam 3 was synthesised according to , starting with 3,3-diethyl-N-(4-methyl)phenyl azetidine-2,4-dione 5 [Citation13], which was converted into the bromomethyl derivative 6 using NBS [Citation14]. Compound 6 was then reacted with thiophenol in the presence of triethylamine to give the phenylthiomethyl derivative 7 [Citation14]. Oxidation of 7 with m-chloroperbenzoic acid (MCPBA) gave 3 in an 83% yield.
Inhibition Kinetics
Compound 3 was found to be a very potent inhibitor of HLE and the progress curves for the enzyme catalysed hydrolysis of MeOSuc-Ala-Ala-Pro-Val-p-NA and these are shown in . These data were analysed accordingly to slow-tight binding inhibition kinetics by non-linear regression analysis, using Equation 1. The resulting kobs values present a linear dependence with inhibitor concentration (), consistent with the simple time-dependent inhibition via the slow association mechanism depicted in , ]25] where kobs has the relationship
Figure 2. Progress curves for the slow-binding inhibition of HLE by 4-oxo-β-lactam 3. Reaction conditions: [HLE] = 20 nM, [MeOSuc-Ala-Ala-Pro-Val-p-NA] = 1 mM, 0.1 M HEPES buffer, pH 7.2, 25 °C. [Citation3]: (a) 0; (b) 6.25; (c) 12.5; (d) 25; (e) 50; (f) 100 nM.
![Figure 2. Progress curves for the slow-binding inhibition of HLE by 4-oxo-β-lactam 3. Reaction conditions: [HLE] = 20 nM, [MeOSuc-Ala-Ala-Pro-Val-p-NA] = 1 mM, 0.1 M HEPES buffer, pH 7.2, 25 °C. [Citation3]: (a) 0; (b) 6.25; (c) 12.5; (d) 25; (e) 50; (f) 100 nM.](/cms/asset/2a2d75cb-e09a-41cf-999f-a5e9a214997e/ienz_a_486794_f0003_b.gif)
Scheme 2. Kinetic model for acyl-enzyme inhibition of HLE by 4-oxo-β-lactam 3 in the presence of substrate.
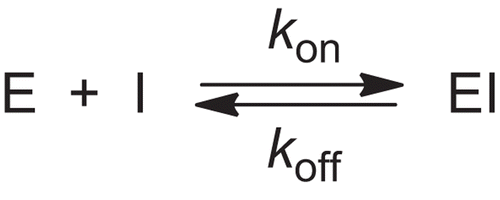
Here kon is the second-order rate constant for the formation of the enzyme-inhibitor complex, EI, and koff is the first-order rate for the decomposition of EI. The magnitude of kon is indicative of the inhibitory potency. The steady-state dissociation constant of the enzyme-inhibitor complex, Ki was calculated using the steady-state velocity, vs, together with v0, and fitting them by non-linear regression to Equation 5 [Citation26], while koff was calculated from Equation 6:
The plot of the steady-state rates, vs, versus [Citation3] for the inhibition of HLE is presented in . An extremely rapid HLE inactivation was observed at very low concentrations of the 4-oxo-β-lactam 3, giving kon = (1.46 ± 0.07) × 106 M−1s−1, Ki = (0.63 ± 0.09) nM, and koff = 9.22 × 10−4 s−1 (), indicating that 3 is a very efficient inhibitor.
The 4-oxo-β-lactam 3 also inhibited the human neutrophil serine proteases cathepsin G and proteinase-3, although with significantly lower activities compared with the HLE inhibition. The second order rate constant for cathepsin G inhibition was 41.4 M−1s−1, while that for proteinase-3 was 4.99 × 103 M−1s−1 (). However, compound 3 was found to be inactive against papain, a cysteine protease.
Table 1. Summary of enzyme inhibition at 25°C and stability kinetic data at 37°C, for 4-oxo-β-lactam 3.
Figure 3. A) Effect of inhibitor concentration on the onset of inhibition of HLE by 4-oxo-β-lactam 3. The values of kobs were obtained as an average of at least duplicate assays from fits to Equation 1 of the data shown in ; B) Plot of the steady-state rates, vs, versus [Citation3] for the inhibition of HLE. The data were obtained from fits of the curves shown in . The solid line was drawn using the best-fit parameters from a fit according to Equation 5.
![Figure 3. A) Effect of inhibitor concentration on the onset of inhibition of HLE by 4-oxo-β-lactam 3. The values of kobs were obtained as an average of at least duplicate assays from fits to Equation 1 of the data shown in Figure 2; B) Plot of the steady-state rates, vs, versus [Citation3] for the inhibition of HLE. The data were obtained from fits of the curves shown in Figure 2. The solid line was drawn using the best-fit parameters from a fit according to Equation 5.](/cms/asset/a44843a2-7c93-4378-adc7-4cd7148ea252/ienz_a_486794_f0005_b.gif)
Stability studies in aqueous buffer and human plasma
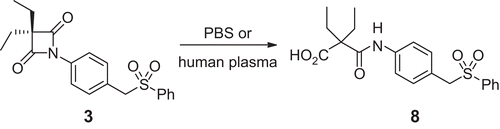
The half-lives of 3 are (1.04 ± 0.25) h in PBS and (0.11 ± 0.03) h in 80% human plasma at 37°C (). For both plasma and PBS the product of hydrolysis was found to be 2-[4-(phenylsulphonylmethyl)phenylcarbamoyl]-2-ethylbutanoic acid 8 ().
Scheme 3. Hydrolysis of 4-oxo-β-lactam 3 in pH 7.4 buffer and 80% human plasma.
Ex vivo studies
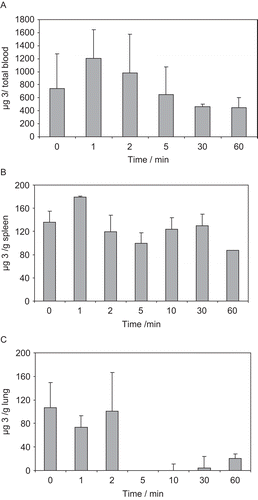
show the mean total blood, spleen and lung concentration-time profiles after ip administration of a 30 mg kg−1 dose of 3 to mice. Inhibitor 3 was not detected in mouse liver. From the concentration-time profile data presented in , the following Cmax values of 3 were determined: 1207 ± 440 μg/total blood, 179 ± 30 μg/g spleen and 106 ± 440 μg/g lung.
Figure 4. Mean ± sd pharmacokinetic profile in A) total mice blood, B) mice spleen and C) mice lung, after administration of a ip 30 mg/kg dosage of 4-oxo-β-lactam 3.
Discussion
The 4-oxo-β-lactams are acylating agents that have been reported as selective and efficient inhibitors of HLE. The sulphone derivative 3 emerged as a very potent HLE inhibitor, with a kon value of 1.46 × 106 M−1s−1, which is one order of magnitude higher than the second-order rate constant for HLE inactivation by Merck’s cephalosporin sulphone 4, (, kon = 1.61 × 105 M−1s−1) [27] and of the same order of magnitude as that of the monocyclic β-lactam HLE inhibitor L-694,458 (3.78 × 106 M−1s−1) [Citation28], which has been reported to be in Phase II clinical trials for cystic fibrosis, juvenile rheumatoid arthritis and emphysema [Citation29].
The 4-oxo-β-lactam 3 inhibited all three neutrophil serine proteases tested, being particularly potent towards HLE, with a HLE/PR3/cathepsin G inhibition ratio of 3.5 × 104/120/1. Since proteinase 3 and HLE are closely related proteases (57% homology [Citation30], presenting similar preferences for molecular recognition at the S1 binding pocket, it is not surprising to observe inhibition of both these enzymes. However, poor inhibition of cathepsin G may be the result of reduced interactions at S1, due to a chymotrypsin and trypsin-like preference for accepting aromatic and positively charged side chains at P1 [Citation30].
Interestingly, the sulphone derivative 3 was found to be a slightly more potent inhibitor of HLE than its sulphide precursor 7 and previously tested against this enzyme (kon = 1.17 × 106 M−1s−1) [Citation14]. In addition, while sulphide 7 was reported to be inactive against cathepsin G and a modest inhibitor of proteinase 3 (kon = 122 M−1s−1) [Citation14], its sulphone counterpart 3 inhibited both enzymes, giving kon = 41.4 M−1s−1 and about 5 × 103 M−1s−1, respectively. The significant increases in the rate of proteinase 3 acylation by nearly 40-fold may be a result of a better interaction of the phenylsulphonyl substituent rather than the corresponding sulphide in the polar S1’ environment of this enzyme. Indeed, the inhibition of these three neutrophil serine proteases by 3 may be an advantage for therapeutics, since all three enzymes are involved in inflammatory disorders. The fact that 3 did not inhibit the cystein protease papain is in good agreement with similar findings obtained for 4-oxo-β-lactam derivatives inhibitors of HLE [Citation14], suggesting a high degree of selectivity towards the serine proteases.
In the present study, compound 3 was found to be rapidly hydrolysed in both pH 7.4 phosphate buffer (PBS) and plasma, in line with the previously reported chemical stability of cephalosporin sulphones (e.g. half-lives of 3 and 4 in PBS pH 7.4 are about 1 h) [Citation31]. The half-life for the hydrolysis of 4-oxo-β-lactam 3 in human plasma is about 10 times lower than in PBS, indicating that 3 is very susceptible to hydrolysis catalysed by plasma enzymes, such as esterases. In both cases the product of hydrolysis was a result of 4-oxo-β-lactam ring opening, without departure of the phenylsulphinic acid.
The concentration-time profiles presented in indicate that inhibitor 3 distributes into spleen and lungs, which are well-perfused organs. In contrast, compound 3 was not detected in the liver, which suggests extensive metabolisation by liver enzymes. shows that the concentration of 3 in mice blood starts decreasing about 2 minutes after ip administration of a 30 mg kg−1 dose, but with the inhibitor still being detected 60 min after administration. There was a more constant level of the 4-oxo-β-lactam 3 in the spleen, with a second peak level at 10 min after injection (), which might result from an erratic pattern of the inhibitor’s concentration that is seen in this organ from animal to animal. The level of inhibitor in mice lung, the desired site of action, decreases sharply 2 min after injection, being almost completely eliminated from this tissue within 5 min (). This result is consistent with either reaction of 3 with mice elastase or a rapid elimination. Overall, the results herein presented compare well with the reference β-lactam L-694,458, which was reported to have a half-life of 1.8 h in rat blood after a 5 mg kg−1 dose given iv, and to be degraded by a pathway believed to involve esterases [Citation32].
Conclusions
The 4-oxo-β-lactam 3 was found be a potent tight-binding irreversible inhibitor of HLE. The presence of a phenylsulphonyl substituent increases the intrinsic chemical reactivity of the 4-oxo-β-lactam, when compared to its sulphide counterpart. Compound 3 poorly inhibited cathepsin G and moderately inhibited proteinase 3, in contrast to its sulphide analogue. Experimental results suggest that 3 is exceedingly reactive, being susceptible to off-target reactions, particularly by plasma and liver enzymes. Structural modification could provide interesting derivatives retaining the high inhibitory properties and with an enhanced hydrolytic stability as potential drug candidates.
Declarations of interest
This work was funded by Fundação para a Ciência e Tecnologia (Foundation for Science and Technology) (FCT, Portugal) through the project PTDC/QUI/64056/2006. J.M. acknowledges the FCT for the PhD grant SFRH/BD/17534/2004.
References
- Bode W, Meyer E, Powers JC. Human-leukocyte and porcine pancreatic elastase - X-ray crystal-structures, mechanism, substrate-specificity, and mechanism-based inhibitors. Biochemistry 1989;28:1951–1963.
- Barnes PJ. Chronic obstructive pulmonary disease: A growing but neglected global epidemic. PLos Med 2007;4:779–780.
- Chua F, Laurent GJ. Neutrophil elastase: mediator of extracellular matrix destruction and accumulation. Proc Am Thorac Soc 2006;3:424–427.
- Lee WL, P. Downey G. Leukocyte elastase physiological functions and role in acute lung injury. Am J Respir Crit Care Med 2001;164:896–904.
- Moraes TJ, Chow CW, Downey GP. Proteases and lung injury. Crit Care Med 2002;13:S189–S194.
- World Health Report 2000 statistical annex. World Health Organization. 2000 [cited January 2009]; Available from: http://www.who.int/whr/2000/en/statistics.htm.
- Roughley PJ, Barrett AJ. The degradation of cartilage proteoglycans by tissue proteinases proteoglycan structure and its susceptibility to proteolysis. Biochem J 1977;167:629–637.
- Pham CTN. Neutrophil serine proteases: specific regulators of inflammation. Nature Rev Immunol 2006;6:541–550.
- Barnes PJ, Hansel TT. Prospects for new drugs for chronic obstructive pulmonary disease. The Lancet 2004;364:985–996.
- Abbenante G, Fairlie DP. Protease inhibitors in the clinic. Med Chem 2005;1:71–104.
- Malhotra S, Man SFP, Sin DD. Emerging drugs for the treatment of chronic obstructive pulmonary disease. Exp Opinion Emerging Drugs 2006;11:275–291.
- Ohbayashi H. Current synthetic inhibitors of human neutrophil elastase. Exp Opin Ther Pat 2002;12:65–84.
- Mulchande J, Guedes RC, Tsang WY, Page MI, Moreira R, Iley J. Azetidine-2,4-diones (4-Oxo-b-lactams) as scaffolds for designing elastase inhibitors. J Med Chem 2008;51:1783–1790.
- Mulchande J, Oliveira R, Carrasco M, Gouveia L, Guedes RC, Iley J, Moreira R. Azetidine-2,4-diones (4-oxo-b-lactams) are potent and selective inhibitors of human leukocyte elastase J Med Chem 2010;53:241–253.
- Log P value calculated using molinspiration. [cited November 2009]; Available from: http://www.molinspiration.com
- Groutas WC, Epp JB, Venkataraman R, Kuang RZ, Truong TM, McClenahan JJ, Prakash O. Design, synthesis, and in vitro inhibitory activity toward human leukocyte elastase, cathepsin G, and proteinase 3 of saccharin-derived sulfones and congeners. Bioorg Med Chem 1996;4:1393–1400.
- Groutas WC, Kuang R, Venkataraman R, Epp JB, Ruan S, Prakash O. Structure-based design of a general class of mechanism-based inhibitors of the serine proteinases employing a novel amino acid-derived heterocyclic scaffold. Biochemistry 1997;36:4739–4750.
- Groutas WC, Kuang RZ, Ruan SM, Epp JB, Venkataraman R, Truong TM. Potent and specific inhibition of human leukocyte elastase, cathepsin g and proteinase 3 by sulfone derivatives employing the 1,2,5-thiadiazolidin-3-one 1,1 dioxide scaffold. Bioorg Med Chem 1998;6:661–671.
- He S, Kuang RZ, Venkataraman R, Tu J, Truong TM, Chan HK, Groutas WC. Potent inhibition of serine proteases by heterocyclic sulfide derivatives of 1,2,5-thiadiazolidin-3-one 1,1 dioxide. Bioorg Med Chem 2000;8:1713–1717.
- Li Y, Yang Q, Dou D, Alliston KR, Groutas WC. Inactivation of human neutrophil elastase by 1,2,5-thiadiazolidin-3-one 1,1 dioxide-based sulfonamides. Bioorg Med Chem 2008;16:692–698.
- Li Y, Dou DF, He GJ, Lushington GH, Groutas WC. Mechanism-based inhibitors of serine proteases with high selectivity through optimization of S ‘ subsite binding. Bioorg Med Chem 2009;17:3536–3542.
- Mulchande J, Martins L, Moreira R, Archer M, Oliveira TF, Iley J. The efficiency of C-4 substituents in activating the b-lactam scaffold towards serine proteases and hydroxide ion. Org Biomol Chem 2007;5:2617–2626.
- Copeland RA. Evaluation of Enzyme Inhibitors in Drug Discovery: A Guide for Medicinal Chemists and Pharmacologists New Jersey: John Wiley and Son, 2005. pp. 141–177.
- Gaspar MM, Cruz A, Penha AE, Reymao J, Sousa AC, Eleuterio CV, Domingues SA, Fraga AG, Filho AL. Rifabutin encapsulated in liposomes exhibits increased therapeutic activity in a model of disseminated tuberculosis. Int J Antimicrob Agents 2008;31:37–45.
- Morrison JF, Walsh CT. The behavior and significance of slow-binding enzyme inhibitors. Adv Enzymol 1988;61:201–301.
- Gutschow M, Neumann U. Novel thieno[2,3-d][1,3]oxazin-4-ones as inhibitors of human leukocyte elastase. J Med Chem 1998;41:1729–1740.
- Doherty JB, Ashe BM, Argenbright LW, Barker PL, Bonney RJ, Chandler GO, Dahlgren ME, Dorn CP, Finke PE. Cephalosporin antibiotics can be modified to inhibit human-leukocyte elastase. Nature 1986;322:192–194.
- Finke PE, Dorn CP, Kissinger AL, Shah SK, Ball RG, Chabin R, Davie P, Dellea PS, Doherty JB. Orally-Active, Intracellular Inhibitors of Human-Leukocyte Elastase (Hle). 22nd National Meeting of the American Chemical Society; 1995 Aug; Chicago; 1995: 83–MEDI.
- Ilies MA, Scozzafava A, Supuran CT. Therapeutic applications of serine protease inhibitors. Expert Opin Ther Pat 2002;12:1181–1214.
- Korkmaz B, Moreau T, Gauthier F. Neutrophil elastase, proteinase 3 and cathepsin G: physicochemical properties, activity and physiopathological functions. Biochimie 2008;90:227–242.
- Alpegiani M, Bissolino P, Corigli R, Delnero S, Perrone E, Rizzo V, Sacchi N, Cassinelli G, Franceschi G, Baici A.Cephem sulfones as inactivators of human-leukocyte elastase .5. 7-Alpha-methoxy-1,1-dioxocephem-4-ketone and 7-alpha-chloro-1,1-dioxocephem-4-ketone. J Med Chem 1994;37:4003–4019.
- Vincent SH, Painter SK, LufferAtlas D, Karanam BV, McGowan E, Cioffe C, Doss G, Chiu SHL.Orally active inhibitors of human leukocyte elastase .2. Disposition of L-694,458 in rats and rhesus monkeys. Drug Metab Dispos 1997;25:932–939.