Abstract
The CB1 receptor belongs to the G-protein-coupled receptor superfamily. CB1 antagonism has been considered as a new therapeutic target for the treatment of obesity. In this study, we report the synthesis and in vitro binding affinity assay of some 1,5-diarylpyrazole scaffold compounds. The binding results showed that some of the target compounds had an excellent potency toward the CB1 receptor with IC50 values lying at the nanomole level.
Introduction
Two subtypes of the cannabinoid receptor, namely CB1 and CB2, have been cloned and were pharmacologically characterised in the early 1990s [Citation1,Citation2]. Both CB1 and CB2 belong to the G-protein-coupled receptor superfamily (GPCRs). The CB1 receptor is mainly located within the central nervous system (CNS) and at the presynaptic nerve terminals including the limbic system (amygdala, hippocampus), hypothalamus, cerebral cortex, cerebellum, basal ganglia and is also expressed in the periphery in the gut, eye, testis and bladder. The CB2 receptor is almost exclusively expressed in the cells of the immune system such as B-cells, NK cells and monocytes [Citation3]. It has been shown that the CB1 receptor, especially in the limbic system-hypothalamus axis cannabinoid, has an important role in the treatment of obesity, which has become a major health problem for many postindustrial societies [Citation4,Citation5]. Obesity poses a major health risk for serious diet-related chronic disease, for instance hypertension, type 2 diabetes, cardiovascular disease and stroke, and some cancers [Citation6]. It has been proved that the CB1 antagonist is effective for the treatment of obesity, metabolic disorders [Citation7,Citation8] and drug abuse, including alcoholism, nicotine dependence and heroin addiction [Citation9]. All these findings make the CB1 receptor an interesting target for possible drug research, which require the development of a more potent antagonist.
To date, several antagonists of CB1, including rimonabant and AVE1625 (), have been developed and some of them are currently being investigated for clinical use [Citation10]. The 1,5-diarylpyrazole derivative rimonabant (SR141716), launched by Sanofi-Aventis, is the first therapeutically relevant CB1 receptor antagonist in the European market for the treatment of obesity and metabolic disorders [Citation11,Citation12]. Moreover, the ability of rimonabant to improve memory acquisition and consolidation has been shown in the mouse [Citation13] and it has also been found that rimonabant can prolong spatial memory duration cognitive deficits associated with neurodegenerative disorders in combination with donepezil in the rat [Citation14]. A combination of a CB1 antagonist with a dopaminergic neurotransmission enhancer has been reported for the treatment of Parkinson disease [Citation15]. Many research groups are still searching for a novel CB1 antagonist with improved physicochemical properties or reduced psychiatric adverse effects, such as the depression or anxiety caused by rimonabant [Citation16]. The purpose of this study was to design and synthesise a set of compounds based on the 1,5-diarylpyrazole template while focusing on various substitutions on the 1,5-diphenylpyrazole scaffold to examine the structure–activity relationships (SARs).
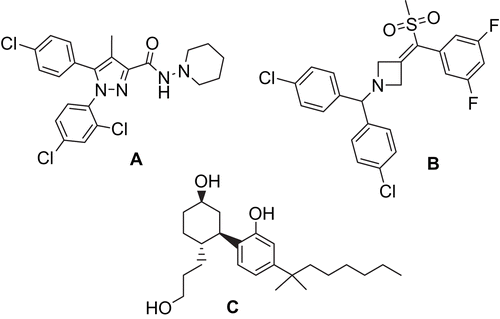
Figure 1. The structure of rimonabant (A), AVE1625 (B) and CP55940 (C).
Methods
Chemistry
Unless otherwise noted, materials were obtained from commercial suppliers and used without further purification. All the solvents had been distilled before use. All reactions were monitored by thin-layer chromatography on 0.25 mm silica gel plates (60GF-254) and visualised with UV light. Column chromatography was performed on silica gel (200–300 mesh). ESI-MS was determined on an Aglient-1100 series LC/MSD trap spectrometer. 1H-NMR spectra was obtained on a Bruker-400. The chemical shifts were expressed in δ values (parts per million) relative to tetramethylsilane (TMS) as an internal standard. Any significant 1H-NMR data were reported in the following order: multiplicity (s, singlet; d, doublet; t, triplet; m, multiplet) number of protons.
[[1-(4-Chlorophenyl)-1-propenyl]oxy]trimethylsilane (2)
Under a nitrogen atmosphere, Et3N (133 mmol) and chlorotrimethylsilane (133 mmol) were added to a solution of 4-chloropropiophenone (66.5 mmol) in acetonitrile (25 mL), and then a solution of sodium iodide (99.7 mmol) in acetonitrile (100 mL) was further added at room temperature. The reaction mixture was stirred for 5 h at room temperature. The acetonitrile was removed under reduced pressure and 150 mL of toluene was added to the solid residual. Then 50 mL of solvent was distilled under reduced pressure to drive the residual acetonitrile off. The inorganic material was extracted with 100 mL of iced water. The organic phase was washed with 100 mL of iced water and dried over MgSO4. The toluene was removed under reduced pressure, the residual was recrystallised from MeOH to afford 14.8 g of oil, yield: 93.6%. 1H-NMR (DMSO-d6): δ 0.21(s, 9H), 1.91(d, 3H), 5.32(q, 1H), 7.36–7.47(m, 4H); ESI-MS: m/z [M]+ 241.3.
5-(4-Chlorophenyl)-4-methyl-2,3-furandione (3)
Oxalyl chloride (30.2 mmol) was slowly added to a solution of compound 2 (60.3 mmol) in methyltertiarybutyl ether (50 mL). The reaction mixture was stirred for 5 h at room temperature. The precipitate was filtered, washed with methyl t-butyl ether, and recrystallised from MeOH to afford the target compound as yellow solid 6.4 g, yield: 47.6%. 1H-NMR (DMSO-d6): δ 1.89(s, 3H), 7.36–7.47(m, 4H); ESI-MS: m/z [M]+ 223.1.
4-(4-Chlorophenyl)-3-methyl-2,4-dioxobutyric acid ethyl ester (4)
Compound 3 (46 mmol) was dissolved in ethanol and stirred for 5 h at room temperature. After completion of the reaction, the ethanol was removed under reduced pressure. The residual was recrystallised from MeOH to afford 12.1 g of white solid, yield: 98.1%. 1H-NMR (CDCl3): δ 1.32(t, J = 7.1 Hz, 3H), 1.47(d, J = 7.2 Hz, 3H), 4.29(q, J = 7.1 Hz, 2H), 5.04(q, J = 7.2 Hz, 1H), 7.5–7.97(m, 4H); 13C-NMR (CDCl3): δ 12.66, 13.84, 50.91, 62.93, 129.29, 130.15, 133.43, 140.41, 160.36, 190.23, 196.88; ESI-MS: m/z [M]+. 268.8.
4-(4-Chlorophenyl)-2-[(2,4-dichlorophenyl)-hydrazono]-3-methyl-4-oxobutyric acid ethyl ester (5)
Compound 4 (11.9 mmol) was dissolved in ethanol (20 mL), and added to a solution of 2,4-dichlorophenylhydrazine hydrochloride (11.9 mmol) and Et3N (11.9 mmol) in ethanol (30 mL). The reaction mixture was stirred for 20 h at room temperature. The precipitate was filtered, washed with water and ethanol, and recrystallised from MeOH to afford the target compound as a white solid 3.1 g, yield: 61.9%. 1H-NMR (CDCl3): δ 12.4(s, 1H), 7.91(d, J = 8.6 Hz, 2H), 7.47–7.42(m, 3H), 7.29(d, J = 2.3 Hz, 1H), 7.17(dd, J = 10.9 Hz, 2.1 Hz, 1H), 4.71(q, J = 20.9 Hz, 1H), 4.28–4.19(m, 2H), 1.48(d, J = 7 Hz, 3H), 1.18(t, J = 14.2, 3H); 13C-NMR (CDCl3): 198.99, 162.3, 139.32, 138.64, 134.69, 130.18, 129.8, 129.01, 128.89, 128.03, 126.47, 119.16, 115.21, 61.48, 44.89, 15.05, 13.94; ESI-MS: m/z [M]+ 427.
5-(4-Chlorophenyl)-1-(2,4-dichlorophenyl)-4-methyl-1H-pyrazole-3-carboxylic acid ethyl ester (6)
Compound 5 (4.5 mmol) was dissolved in acetic acid (20 mL) and heated under reflux for 24 h. The reaction mixture was poured into cold water (30 mL) and extracted with EtOAc (3 × 20 mL). The combined extracts were washed with water, saturated aqueous sodium bicarbonate, and brine, dried over anhydrous MgSO4, then filtered. The target compound was recrystallised from n-hexane/EtOAc as a white solid 1.78 g, yield: 95.7%. 1H-NMR(DMSO-d6): δ 7.79(d, J = 2.2 Hz, 1H), 7.74(d, J = 8.5 Hz, 1H), 7.59(dd, J = 8.5 Hz, 2.2 Hz, 1H), 7.47(d, J = 8.5 Hz, 2H), 7.25(d, J = 8.4 Hz, 2H), 4.33(q, J = 21.3 Hz, 2H), 2.3(s, 3H), 1.32(t, J = 14.2 Hz, 3H); 13C-NMR(DMSO-d6): δ 162.57, 143.03, 142.55, 136.13, 135.71, 134.4, 132.38, 132.21, 131.82, 130.12, 129.26, 128.87, 127.35, 118.53, 60.83, 14.65, 9.85. ESI-MS: m/z [M]+: 409.2.
[5-(4-Chlorophenyl)-1-(2,4-dichlorophenyl) -4-methyl-1H-pyrazole-3-yl]methanol (7)
An aliquot of LiAlH4 (50 mmol) was slowly added to a solution of compound 6 (20 mmol) in THF (250 mL) below 0°C. After the mixture was stirred at the same temperature for 15 min, EtOAc (15 mL) was added to the mixture, and then saturated aqueous ammonium chloride was added. The insoluble material was filtered off and washed with methyltertiarybutyl ether (3 × 30 mL). The organic layer was washed successively with 5% HCl and saturated brine, dried over anhydrous MgSO4, filtered, and evaporated. The target compound was recrystallised from n-hexane/EtOAc as a white solid 6.69 g, yield: 91%. 1H-NMR(CDCl3): δ 2.17 (s, 3H), 2.86 (s, 1H), 4.79 (s, 2H), 7.07–7.44 (m, 7H); 13C-NMR(CDCl3): δ 8.29, 57.62, 113.84, 127.82, 127.99, 128.83, 130.19, 130.52, 130.69, 132.97, 134.49, 135.38, 136.31, 141.92, 152.28; ESI-MS: m/z [M]+: 367.7.
3-(Bromomethyl)-5-(4-chlorophenyl)-1-(2,4-dichlorophenyl)-4-methyl-1H-pyrazole (8)
Compound 7 (10 mmol) was dissolved in 100 mL of benzene and added dropwise to a solution of phosphorus tribromide (50 mmol) in benzene (100 mL). The mixture was heated under reflux for 1 h. After cooling to room temperature, water (100 mL) was added. The organic materials were extracted with ether, washed with 10% NaHCO3, dried over anhydrous MgSO4, filtered, and evaporated then recrystallised from n-hexane to afford the target compound as a white solid 3.7 g, yield: 86.1%. 1H-NMR(CDCl3): δ 2.17 (s, 3H), 4.59 (s, 2H), 7.07–7.41 (m, 7H); 13C-NMR(CDCl3): δ 8.54, 24.01, 114.77, 127.76, 127.83, 128.87, 130.21, 130.57, 130.68, 132.97, 134.67, 135.54, 136.17, 142.13, 148.94; ESI-MS: m/z [M]+: 430.8.
5-(4-Chlorophenyl)-1-(2,4-dichlorophenyl)-4-methyl-1H-pyrazole-3-carboxaldehyde (9)
Compound 7 (4.6 mmol) was dissolved in CH2Cl2 (40 mL) and slowly added to a solution of CrO3 (34 mmol) in pyridine (68 mmol) and CH2Cl2 (45 mL) below 0°C. The reaction mixture was stirred for 0.5 h below 0°C. After completion of the reaction, the insoluble material was filtered off with diatomite and washed with CH2Cl2. The organic layer was evaporated under reduced pressure. The residual was dissolved in ether, and washed successively with 10% NaOH, 5% HCl, and saturated brine, then dried over anhydrous MgSO4, filtered, evaporated and recrystallised from n-hexane/ether to afford the target compound as a white solid 0.7 g, yield: 40.1%. 1H-NMR(CDCl3): δ 2.29 (s, 3H), 7.27–7.82 (m, 7H), 10.9 (s, 1H); 13C-NMR(CDCl3): δ 9.09, 116.99, 126.89, 128.96, 129.3, 130.25, 131.74, 132.05, 132.29, 134.57, 135.89, 136.02, 143.61, 149.52, 188.44; ESI-MS: m/z [M]+: 365.7.
5-(4-Chlorophenyl)-1-(2,4-dichlorophenyl)-4-methyl-1H-pyrazole-3-carboxylic acid (10a)
Compound 6 (4.34 mmol) was added to a solution of KOH (8.68 mmol) in ethanol (16 mL) and H2O (16 mL). The mixture was heated under reflux for 3 h. The cooling reaction mixture was then poured into water (30 mL) and acidified with 10% HCl. The precipitate was filtered, washed with water, and recrystallised from MeOH to afford the target compound as a white solid 1.58 g, yield: 95.7%.1H-NMR(DMSO-d6): δ 12.97(s, 1H), 7.78(d, J = 2.2 Hz, 2H), 7.72(d, J = 12.1 Hz, 1H), 7.59(dd, J = 8.5 Hz, 2.2 Hz, 2H), 7.46(d, J = 8.5 Hz, 1H), 7.24(d, J = 8.4 Hz, 1H), 2.24(s, 3H); 13C-NMR(DMSO-d6): δ 164.11, 143.35, 142.87, 136.25, 135.59, 134.29, 132.41, 132.19, 131.79, 130.11, 129.23, 128.84, 127.56, 118.41, 9.92; ESI-MS: m/z [M]+: 381.1.
5-(4-Chlorophenyl)-4-methyl-1-(pyridine-2-yl)-1H-pyrazole-3-carboxylic acid (10b)
The title compound was prepared as described for compound 10a. Flash column chromatography: (CHCl3/MeOH, 1/2), white solid, yield: 67.5%. 1H-NMR(DMSO-d6): δ12.53 (br s, 1H), 8.26 (dd, J = 4.8 Hz, 1 Hz, 1H), 8.00–7.98 (m, 1H), 7.71 (d, J = 22.6 Hz, 1H), 7.46–7.38 (m, 3H), 7.25–7.23 (m, 2H), 2.2 (s, 3H); 13C-NMR(DMSO-d6): δ 164.16, 152.07, 148.47, 143.21, 141.14, 139.7, 133.57, 131.93, 129.23, 128.81, 124.06, 119.96, 119.38, 9.74; ESI-MS: m/z [M]+: 314.2.
5-(4-Chlorophenyl)-4-methyl-1-[4-(trifluoromethyl)phenyl]-1H-pyrazole-3-carboxylic acid (10c)
The title compound was prepared as described for compound 10a. Flash column chromatography: (CHCl3/MeOH, 10/1), white solid, yield: 57.8%. 1H-NMR(CDCl3) δ 13.04(s, 1H), 7.8(d, J = 8.6 Hz, 2H), 7.53(d, J = 8.9 Hz, 2H), 7.47(d, J = 8.4 Hz, 2H), 7.32–7.29(dd, J = 8.5 Hz, 1.9 Hz, 2H), 2.21(s, 3H). ESI-MS: m/z [M]+: 379.3.
General procedure for preparing carboxamide
5-(4-Chlorophenyl)-1-(2,4-dichlorophenyl)-4-methyl-N-(4-methylpiperazin-1-yl)-1H-pyrazole-3- carboxamide (12a)
A solution of compound 10a (1.13 mmol) and SOCl2 (4.14 mmol) in toluene (10 mL) was refluxed for 3 h. The solvent was evaporated under reduced pressure, and the residual was then redissolved in toluene (20 mL) and evaporated to yield the crude acyl chlorides as a solid. A solution of the above carboxylic chloride in CH2Cl2 (5 mL) was added dropwise to a solution of 1-amino-4-methylpiperazine (1.92 mmol) and Et3N (1.92 mmol) in CH2Cl2 (5 mL) below 0°C. After stirring at room temperature for 3 h, the reaction mixture was added to brine and extracted with CH2Cl2 (3 × 15 mL). The combined extracts were washed with brine, dried over anhydrous MgSO4, filtered and evaporated. The final product was recrystallised from MeOH to afford carboxamide as a white solid in 50% yield. 1H-NMR(CDCl3) δ 7.66(s, 1H), 7.46(s, 1H), 7.34–7.32(m, 4H), 7.1–7.08(m, 2H), 2.99(br s, 4H), 2.66(br s, 4H), 2.39(s, 3H), 2.35(s, 3H); 13C-NMR(CDCl3) δ 160.09, 144.18, 136.07, 135.84, 134.96, 132.97, 130.8, 130.57, 130.35, 128.90, 127.93, 127.12, 118.32, 55.54, 54.4, 45.76, 9.3. ESI-MS: m/z [M]+: 478.4.
5-(4-Chlorophenyl)-1-(2,4-dichlorophenyl)-4-methyl-N-[3-(4-morpholinyl)propyl]-1H-pyrazole -3-carboxamide (12b)
Starting with compound 10a and N-(3-aminopropyl)morpholine, then purified by flash column chromatography(CHCl3/MeOH, 8/1) to afford the titled compound as a white solid in 63.4% yield. 1H-NMR(CDCl3) δ 8.12(t, J = 10.8 Hz, 1H), 7.43(d, J = 1.7 Hz, 1H), 7.33–7.28(m, 4H), 7.09–7.07(m, 2H), 3.65–3.62(m, 4H), 3.53(q, J = 18.1 Hz, 2H), 2.49(t, J = 12.7 Hz, 2H), 2.44(br s, 4H), 2.39(s, 3H), 1.81–1.74(m, 2H); 13C-NMR(CDCl3) δ 162.68, 145.27, 142.94, 136.03, 135.93, 134.81, 132.95, 130.81, 130.5, 130.26, 128.86, 127.86, 127.31, 117.55, 66.74, 57.97, 53.77, 38.78, 25.25, 9.44. ESI-MS: m/z [M]+: 507.4.
5-(4-Chlorophenyl)-1-(2,4-dichlorophenyl)-4-methyl-N-[3-(N, N-diethylamino) propyl]-1H -pyrazol-3-carboxamide (12c)
Starting with compound 10a and N,N-diethyl-1,3-propanediamine, then purified with flash column chromatography(CHCl3/MeOH, 8/1) to afford the titled compound as a white solid in 56.4% yield. 1H-NMR(CDCl3) δ 7.45–7.42(m, 2H), 7.32–7.3(m, 4H), 7.08–7.06(m, 2H), 3.57–3.56(m, 2H), 3.15–3.14(m, 6H), 2.35(s, 3H), 2.21–2.17(m, 2H), 1.41–1.38(m, 6H); 13C-NMR(CDCl3) δ 163.43 144.51, 143.15, 136.02, 135.76, 134.96, 132.71, 130.79, 130.6, 130.25, 128.9, 128, 127.07, 117.58, 49.44, 46.55, 36.24, 24.35, 9.37, 8.55. ESI-MS: m/z [M]+: 493.4.
5-(4-chlorophenyl)-1-(2,4-dichlorophenyl)-4-methyl-N-(4-methylcyclohexyl)-1H-pyrazol -3-carboxamide (12d)
Starting with compound 10a and 4-methylcyclohexylamine, then purified with flash column chromatography (n-hexane/AcOEt, 6/1) to afford the titled compound as a white solid in 63% yield. 1H-NMR(CDCl3) δ 7.95(s, 1H), 7.42(d, J = 1.3 Hz, 1H), 7.3–7.27(m, 4H), 7.07–7.03(m, 3H), 4.19–4.17(m, 1H), 2.37(s, 3H), 1.78–1.75(m, 2H), 1.71–1.67(m, 1H), 1.65–1.6(m, 3H), 1.59–1.24(m, 2H), 0.93(d, J = 6.4 Hz, 3H); 13C-NMR(CDCl3) δ 161.95, 145.26, 142.95, 136.06, 135.86, 134.83, 133.02, 130.84, 130.64, 130.31, 128.86, 127.83, 127.4 117.68, 45.26, 30.32, 30.14, 29.33, 20.9, 9.46. ESI-MS: m/z [M]+: 476.1.
5-(4-Chlorophenyl)-1-(2,4-dichlorophenyl)-4-methyl-N-(3-(pyrrolidin-1-yl)propyl)-1H-pyrazole-3-carboxamide (12e)
Starting with compound 10a and N-(3-aminopropyl)tetrahydropyrrole, then purified with flash column chromatography(CHCl3/MeOH, 8/1) to afford the titled compound as a white solid in 23.1% yield. 1H-NMR(CDCl3) δ 8.03(t, J = 10.4 Hz, 1H), 7.41(t. J = 3.1 Hz, 1H), 7.31–7.29(m, 4H), 7.07–7.05(m, 2H), 3.55–3.5(m, 2H), 2.61(t, J = 13.4 Hz, 2H), 2.51–2.5(m, 4H), 2.38(s, 3H), 1.83–1.72(m, 2H), 1.67–1.66(m, 4H); 13C-NMR(CDCl3) δ 162.7, 145.37, 142.84, 136.13, 135.81, 134.79, 133.03, 130.81, 130.53, 130.2, 128.84, 127.75, 127.41, 117.55, 54.97, 54.1, 38.6, 27.96, 23.39, 9.43. ESI-MS: m/z [M]+: 491.4.
5-(4-Chlorophenyl)-1-(2,4-dichlorophenyl)-4-methyl-N-(4-(piperidin-1-yl)butyl)-1H-pyrazole-3-carboxamide (12f)
Starting with compound 10a and 4-(1-piperidyl)-1-butylamine, then purified with flash column chromatography (CHCl3/MeOH, 6/1) to afford the titled compound as a white solid in 31.6% yield. 1H-NMR(CDCl3) δ 7.42(t, J = 2.3 Hz, 1H), 7.31–7.28(m, 4H), 7.14(s, 1H), 7.07–7.06(m, 2H), 3.47–3.42(m, 2H), 2.61–2.55(m, 6H), 2.37(s, 3H), 1.73–1.71(m, 6H), 1.99–1.63(m, 2H), 1.49(m, 2H); 13C-NMR(CDCl3) δ 162.82, 144.97, 143.01, 135.92, 135.91, 134.88, 132.91, 130.8, 130.58, 130.27, 128.87, 127.89, 127.24, 117.61, 58.18, 54.06, 38.42, 27.59, 24.69, 23.64, 23.11, 9.41. ESI-MS: m/z [M]+: 519.3.
5-(4-Chlorophenyl)-1-(2,4-dichlorophenyl)-4-methyl-N-[2-(7-methoxynaphthalen-2-yl)ethyl] -1H-pyrazol-3-carboxamide (12g)
Starting with compound 10a and 2-(7-methoxynaphthalen-2-yl)ethanamine, then recrystallised from EtOAc to afford the titled compound as a white solid in 42.9% yield. 1H-NMR(CDCl3) δ 7.74(d, J = 9 Hz, 1H), 7.63(m, 1H), 7.51(m, 1H), 7.41(d, J = 2.2 Hz, 1H), 7.33(m, 1H), 7.31–7.26(m, 5H), 7.22(m, 1H), 7.21(m, 1H), 7.14(m, 1H), 7.06(m, 1H), 3.94(s, 3H), 3.78(q, J = 14.5 Hz, 2H), 3.36(t, J = 14.8 Hz, 2H), 2.4(s, 3H); 13C-NMR(CDCl3) δ 162.85, 157.97, 145.03, 143.04, 135.94, 134.95, 133.89, 133.27, 132.98, 130.82, 130.48, 130.32, 130.21, 129.34, 128.92, 127.85, 127.26, 127.17, 126.97, 123.24, 118.32, 117.68, 102.5, 55.46, 39.78, 33.5, 9.42. ESI-MS: m/z [M]+: 564.2.
5-(4-Chlorophenyl)-4-methyl-1-(pyridin-2-yl)-N-(piperidin-1-yl) -1H-pyrazole-3-carboxamide (12h)
Starting with compound 10b and 1-aminopiperidin, then purified with flash column chromatography (petroleum ether/EtOAc, 5/1), and recrystallised from petroleum ether/acetone to afford the titled compound as a white solid in 40.4% yield. 1H-NMR(DMSO-d6) δ 9.12(s, 1H), 8.25(dd, J = 4.7 Hz, 0.8 Hz, 1H), 8.01–7.79(m, 1H), 7.77(d, J = 8.2 Hz, 1H), 7.44(d, J = 8.4 Hz, 2H), 7.39–7.36(m, 1H), 7.22(d, J = 8.4 Hz, 2H), 2.82(t, J = 10.3 Hz, 4H), 2.17(s, 3H), 1.61–1.59(m, 4H), 1.36–1.34(m, 2H); 13C-NMR(DMSO-d6) δ 159.95, 152.02, 148.43, 144.91, 140.98, 139.53, 133.5, 131.88, 129.35 128.81, 123.83, 119.45, 118.45, 55.82, 25.86, 9.3. ESI-MS: m/z [M]+: 396.3.
5-(4-Chlorophenyl)-4-methyl-1-(pyridin-2-yl)-N-(azepan-1-yl)-1H-pyrazole-3-carboxamide (12i)
Starting with compound 10b and 1-aminoazepan, then purified with flash column chromatography (petroleum ether/EtOAc, 3/1), and recrystallised from petroleum ether/EtOAc(5/1) to afford the titled compound as a white solid in 17.8% yield. 1H-NMR(CDCl3) δ 8.34(dd, J = 4.8 Hz, 1.2 Hz, 1H), 8.21(s, 1H), 7.75–7.74(m, 1H), 7.38–7.32(m, 3H), 7.27–7.24(m, 1H), 7.13–7.1(m, 2H), 3.17(t, J = 11.1 Hz, 4H), 2.34(s, 3H), 1.79–1.76(m, 4H), 1.67–1.64(m, 4H); 13C-NMR(CDCl3) δ 160.36, 151.8, 148.57, 144.4, 141.3, 138.3, 134.44, 131.15, 128.64, 122.85, 119.87, 118.7, 58.4, 26.99, 26.51, 9.22. ESI-MS: m/z [M]+: 410.2.
5-(4-Chlorophenyl)-4-methyl-1-(pyridin-2-yl)-N-(4-methylcyclohexyl)-1H-pyrazole-3-carboxamide (12j)
Starting with compound 10b and 1-aminoazepan, then purified with flash column chromatography (petroleum ether/acetone, 5/1), and recrystallised from petroleum ether/acetone (1/1) to afford the titled compound as a white solid in 41.5% yield. 1H-NMR(CDCl3) δ 8.34(dd, J = 4.8 Hz, 1.5 Hz, 1H), 7.71(q, J = 9.6 Hz, 1H), 7.37(d, J = 8.1 Hz, 1H), 7.32(d, J = 4.4 Hz, 2H), 7.23–7.2(m, 1H), 7.16(d, J = 8.3 Hz, 2H), 6.94(d, J = 8.4 Hz, 1H), 3.96–3.86(m, 1H), 2.35(s, 3H), 2.07–2.05(m, 2H), 1.76–1.73(m, 2H), 1.37–1.27(m, 3H), 1.14–1.08(m, 2H), 0.90(d, J = 6.5 Hz, 3H); 13C-NMR(CDCl3) δ 161.91, 151.79, 148.54, 145.16, 141.36, 138.26, 134.37, 131.15, 130.88, 128.73, 128.6, 128.27, 122.8, 119.41, 118.74, 48.14, 33.97, 33.16, 31.99, 22.22, 9.32. ESI-MS: m/z [M]+: 409.2.
5-(4-Chlorophenyl)-4-methyl-1-[4-(trifluoromethyl)phenyl]-N-(piperidin-1-yl)-1H-pyrazole-3 –carboxamide (12k)
Starting with compound 10c and 1-aminopiperidin, then purified with flash column chromatography (petroleum ether/EtOAc, 4/1), and recrystallised from petroleum ether/EtOAc (4/1) to afford the titled compound as a white solid in 35.6% yield. 1H-NMR(CDCl3) δ 7.73(s, 1H), 7.59(d, J = 8.5 Hz, 2H), 7.4–7.35(m, 4H), 7.12–7.09(m, 2H), 2.90(t, J = 9.8 Hz, 4H), 2.35(s, 3H), 1.8–1.74(m, 4H), 1.48–1.45(m, 2H); 13C-NMR(CDCl3) δ 159.84, 144.43, 142.06, 141.04, 135.19, 131.21, 129.75, 129.34, 127.7, 126.19, 124.87, 122.27, 120.04, 57.1, 25.47, 23.36, 9.21. ESI-MS: m/z [M]+: 463.3.
5-(4-Chlorophenyl)-4-methyl-1-[4-(trifluoromethyl)phenyl]-N-(azepan-1-yl)-1H-pyrazole-3- carboxamide (12l)
Starting with compound 10c and 1-aminoazepan, then purified with flash column chromatography (petroleum ether/EtOAc, 3/1) to afford the titled compound as a white solid in 39.8% yield. 1H-NMR(CDCl3) δ 8.16(s, 1H), 7.57(d, J = 8.5 Hz, 2H), 7.37–7.32(m, 4H), 7.08(d, J = 10.7 Hz, 2H), 3.16(t, J = 11.1 Hz, 4H), 2.32(s, 3H), 1.75–1.73(m, 4H), 1.65–1.63(m, 4H); 13C-NMR(CDCl3) δ 160.2, 144.45, 142.04, 140.99, 135.17, 131.21, 129.85, 129.3, 129.25, 127.71, 126.17, 124.84, 122.28, 119.95, 58.34, 26.95, 26.44, 9.23. ESI-MS: m/z [M]+: 477.2.
5-(4-Chlorophenyl)-4-methyl-1-[4-(trifluoromethyl)phenyl]-N-(4-methylcyclohexyl)-1H-pyrazole-3-carboxamide (12m)
Starting with compound 10c and 4-methylcyclohexanamine, then purified with flash column chromatography (petroleum ether/EtOAc, 6/1), and recrystallised from petroleum to afford the titled compound as a white solid in 42.6% yield. 1H-NMR(CDCl3) δ 7.58(d, J = 8.6 Hz, 2H), 7.39–7.35(m, 4H), 7.12–7.09(m, 2H), 6.86(d, J = 8.4 Hz, 1H), 3.95–3.86(m, 1H), 2.35(s, 3H), 2.09–2.05(m, 2H), 1.77–1.74(m, 2H), 1.33–1.23(m, 3H), 1.16–1.09(m, 2H), 0.91(d, J = 6.5 Hz, 3H); 13C-NMR(CDCl3) δ 161.77, 145.23, 142.12, 141.11, 135.14, 131.24, 129.85, 129.25, 127.77, 126.19, 124.75, 122.3, 119.57, 48.13, 33.95, 33.23, 32.01, 22.19, 9.34. ESI-MS: m/z [M]+: 476.1.
[2-[(5-(4-Chlorophenyl)-1-(2,4-dichlorophenyl)-4-methyl-1H-pyrazol-3-yl]methylthio]acetic acid (13a)
EtONa (1.53 mmol) was added to a solution of mercaptoacetic acid (0.767 mmol) in MeOH (5 mL), then an aliquot of compound 8 (0.767 mmol) in THF (5 mL) was added to the solution. The mixture was heated under reflux for 1.5 h, acidified to pH 4 by the dropwise addition of a solution of HCl in MeOH and the solvent was removed under reduced pressure. Solutions of CHCl3 (10 mL) and H2O (10 mL) were added to the solid residual. The organic phase was dried over MgSO4, the solvent was evaporated and the residual was purified by flash column chromatography (CHCl3/MeOH, 10/1) to afford a a white solid in 52.8% yield. 1H-NMR(CDCl3): δ 2.1 (s, 3H), 3.24 (s, 2H), 3.92(s, 2H), 7.04–7.37(m, 7H), 11.24 (s, 1H); 13C-NMR(CDCl3): δ 8.42, 27.09, 29.69, 33.01, 114.1, 127.62, 127.91, 128.85, 130.18, 130.6, 130.83, 133.12, 134.74, 135.62, 135.64, 143.36, 149.06, 174.04; ESI-MS: m/z [M]+: 441.8.
[2-[(5-(4-Chlorophenyl)-1-(2,4-dichlorophenyl)-4-methyl-1H-pyrazol-3-yl]methylthio]propionic acid (13b)
The title compound was prepared as described for compound 13a. Flash column chromatography: (n-hexane/AcOEt, 7/1), white solid, yield: 49.6%. 1H-NMR(DMSO-d6): δ 1.37 (d, J = 7.1 Hz, 3H), 2.09 (s, 3H), 3.51(q, J = 7.1 Hz, 1H), 3.91 (d, J = 13.3 Hz, 1H), 3.95 (d, J = 13.3 Hz, 1H), 7.19–7.77(m, 7H), 11.51 (s, 1H); 13C-NMR(DMSO-d6): δ 8.64, 18.05, 26.81, 41.77, 113.96, 128.51, 128.71, 129.16, 130.08, 131.32, 132.14, 132.65, 133.72, 134.79, 136.75, 141.53, 149.29, 174.61; ESI-MS: m/z [M]+: 455.9.
[2-Bis-[[5-(4-chlorophenyl)-1-(2,4-dichlorophenyl)-4-methyl-1H-pyrazol-3-yl]methyl]am ino] acetic acid (13c)
An aliquot of EtONa (1.51 mmol) was added to the solution of glycine methyl ester hydrochloride (0.757 mmol) in MeOH (20 mL) and compound 8 (0.697 mmol) was added to the solution. The mixture was heated under reflux for 1.5 h. and acidified to pH 6 by the dropwise addition of a solution of HCl in MeOH. The solvent was removed under reduced pressure and 20 mL of H2O was added to the solid residual. The precipitate was filtered and purified by flash column chromatography (n-hexane/AcOEt, 4/1) to afford a white solid which was added to the solution of KOH (0.528 mmol) in MeOH (10 mL). The mixture was heated under reflux for 1 h and acidified to pH 4 by the dropwise addition 10% HCl. The precipitate was filtered and recrystallised from MeOH to afford the target compound as a white solid in 65.4% yield. 1H-NMR(DMSO-d6): δ 1.99 (s, 6H), 3.43 (s, 2H), 3.91(s, 4H), 7.14 (d, J = 8.3 Hz, 4H), 7.4 (d, J = 8.3 Hz, 4H), 7.48–7.72(m, 6H), 10.53 (s, 1H); 13C-NMR(DMSO-d6): δ 8.41, 49.08, 53.17, 115.06, 128.67, 128.71, 129.15, 130.09, 131.27, 132.13, 132.68, 133.66, 134.78, 136.85, 141.51, 150.02, 172.41; ESI-MS: m/z [M]+: 774.7.
[2-[5-(4-Chlorophenyl)-1-(2,4-dichlorophenyl)-4-methyl-1H-pyrazol-3-yl]methyl]diethyl malonate (13d)
An aliquot of NaH (1.88 mmol) was added to the solution of diethyl malonate (1.88 mmol) in THF (25 mL) and compound 8 (1.25 mmol) was added to the solution. The mixture was heated under reflux for 3 h. The solvent was removed under reduced pressure. Solutions of CHCl3 (20 mL) and H2O (20 mL) were added to the solid residual. The organic phase was dried over MgSO4 and the solvent was evaporated and the residual was purified by flash column chromatography (n-hexane/AcOEt, 10/1) to afford a white solid in 40.8% yield. 1H-NMR(CDCl3): δ 1.27 (t, J = 7.1 Hz, 6H), 2.1 (s, 3H), 3.31(d, J = 7.8 Hz, 2H), 4.07 (t, J = 7.8 Hz, 1H), 4.2 (q, J = 7.1 Hz, 4H), 7.04–7.41(m, 7H); 13C-NMR(CDCl3): δ 8.39, 14.05, 25.75, 50.96, 61.39, 113.88, 127.62, 128.35, 128.71, 128.83, 130.11, 130.52, 133.06, 134.26, 134.99, 136.63, 141.18, 149.61, 169.12; ESI-MS: m/z [M]+: 509.9.
[2-[(5-(4-Chlorophenyl)-1-(2,4-dichlorophenyl)-4-methyl-1H-pyrazol-3-yl]methoxy]methyl acetate (14a)
An aliquot of methyl chloroacetate (1.50 mmol) was added to a solution of compound 7 (1.36 mmol) and NaH (2.04 mmol) in THF (15 mL). The mixture was heated under reflux for 3 h and the solvent was removed under reduced pressure. Solutions of CHCl3 (15 mL) and H2O (15 mL) were added to the solid residual. The organic phase was dried over MgSO4, the solvent was evaporated and the residual was purified by flash column chromatography(n-hexane/AcOEt, 2/1) to afford the target compound as a white solid in 55.7% yield. 1H-NMR(CDCl3): δ 2.22 (s, 3H), 3.81(s, 3H), 4.24(s, 2H), 4.79(s,2H), 7.09–7.45(m, 7H); 13C-NMR(CDCl3): δ 8.33, 51.84, 65.68, 66.95, 115.21, 127.79, 127.99, 128.82, 130.19, 130.54, 130.66, 133.07, 134.51, 136.34, 141.98, 148.79, 170.77; ESI-MS: m/z [M]+: 439.4.
[2-[(5-(4-Chlorophenyl)-1-(2,4-dichlorophenyl)-4-methyl-1H-pyrazol-3-yl]methoxy]methyl propionate (14b)
The title compound was prepared as described for compound 14a. Flash column chromatography: (n-hexane/AcOEt, 5/1), white solid, yield: 47.3%. 1H-NMR(CDCl3): δ 1.48 (d, J = 6.9 Hz, 3H), 2.21(s, 3H), 3.78(s, 3H), 4.21(q, J = 6.9 Hz, 1H), 4.62(d, J = 11.4 Hz, 1H), 4.81(d, J = 11.4 Hz, 1H), 7.21–7.64(m, 7H); 13C-NMR(CDCl3): δ 8.32, 18.65, 51.88, 64.66, 73.86, 127.76, 128.1, 128.78, 128.83, 130.14, 130.53, 130.71, 133, 134.62, 135.29, 136.4, 141.83, 149.12, 173.53; ESI-MS: m/z [M]+: 453.9.
[2-[(5-(4-Chlorophenyl)-1-(2,4-dichlorophenyl)-4-methyl-1H-pyrazol-3-yl]methoxy]acetic acid (14c)
A stirring mixture of compound 14a (0.6 mmol), KOH (2.7 mmol) and MeOH (15 mL) was heated under reflux for 1 h. After cooling, the mixture was poured into H2O (15 mL) and acidified to pH 4 by the dropwise addition of a 10% HCl solution. The precipitate was filtered and recystallised from ethanol to afford the title compound as a white solid in 38.4% yield. 1H-NMR(CDCl3): δ 2.18 (s, 3H), 4.22 (s, 2H), 4.81(s, 2H), 7.09–7.43 (m, 7H), 10.85(s, 1H); 13C-NMR(CDCl3): δ 8.25, 65.29, 66.83, 114.98, 127.61, 127.89, 128.87, 130.21, 130.58, 130.74, 133.05, 134.74, 135.69, 135.81, 142.32, 148.66, 173.77; ESI-MS: m/z [M]+: 425.8.
[2-[(5-(4-Chlorophenyl)-1-(2,4-dichlorophenyl)-4-methyl-1H-pyrazol-3-yl]methoxy]propionic acid (14d)
The title compound was prepared as described for compound 14c in 60.2% yield. 1H-NMR(DMSO-d6): δ 1.35 (d, J = 6.8 Hz, 3H), 2.13 (s, 3H), 4.1(q, J = 6.8 Hz, 1H), 4.50(d, J = 11.1 Hz, 1H), 4.69(d, J = 11.1 Hz, 1H), 7.02–7.38 (m, 7H), 11.05(s, 1H); 13C-NMR(DMSO-d6): δ 8.47, 18.9, 64.09, 73.75, 115.04, 128.53, 128.71, 129.15, 130.06, 131.32, 132.17, 132.52, 133.75, 134.86, 136.75, 141.6, 149.44; ESI-MS: m/z [M]+: 439.8.
[2-[(5-(4-Chlorophenyl)-1-(2,4-dichlorophenyl)-4-methyl-1H-pyrazol-3-yl]methoxy]butyric acid (14e)
Compound 7 (0.82 mmol) and NaH (0.98 mmol) were dissolved in THF (15 mL) and an aliquot of methyl 2-bromobutyrate (77 mL) was added to the solution. The mixture was heated under reflux for 3.5 h and the solvent was removed under reduced pressure. An aliquot of CHCl3 (15 mL) and H2O (15 mL) were added to the solid residual. The organic phase was dried over MgSO4, the solvent was evaporated and the residual was purified by flash column chromatography (n-hexane/AcOEt, 6/1) to afford a white solid. The white solid and KOH (5.36 mmol) was dissolved in MeOH (20 mL). Then the mixture was heated under reflux for 1 h. After cooling, the mixture was poured into H2O (15 mL) and acidified to pH 4 by the dropwise addition of a 10% HCl solution. The precipitate was filtered and recystallised from ethanol to afford the title compound as a white solid in 52.6% yield. 1H-NMR(DMSO-d6): δ 0.92 (t, J = 7.4 Hz, 3H), 1.62–1.78 (m, 2H), 2.13(s, 3H), 3.9(t, J = 6 Hz, 1H), 4.44(d, J = 11.2 Hz, 1H), 4.7(d, J = 11.2 Hz, 1H), 7.19–7.77(m, 7H), 11.15(s, 1H); 13C-NMR(DMSO-d6): δ 8.55, 10.25, 26.04, 64.24, 79.16, 115.06, 128.56, 128.73, 129.16, 130.07, 131.32, 132.19, 132.5, 133.71, 134.83, 136.76, 141.58, 149.62, 174.07; ESI-MS: m/z [M]+: 453.9.
[2-[(5-(4-Chlorophenyl)-1-(2,4-dichlorophenyl)-4-methyl-1H-pyrazol-3-yl]methylamino]acetic acid (15a)
Glycine ethyl ester hydrochloride (0.76 mmol) and Et3N (0.76 mmol) were added to a solution of compound 9 (0.76 mmol) in CH2Cl2 (15 mL). Dry MgSO4 (41.7 mmol) was added to the solution and then KBH4 was added. The mixture was heated under reflux for 4 h. The insoluble material was filtered off and H2O (15 mL) was added to the filtrate. The organic phase was dried over MgSO4 and the solvent was evaporated. The residual was purified by flash column chromatography (n-hexane/AcOEt, 5/1) to afford a white solid which was added to the solution of KOH (5.36 mmol) in MeOH (20 mL). The mixture was heated under reflux for 1 h. The solvent was evaporated and the residual was acidified to pH 4 by the dropwise addition 5% HCl. The precipitate was filtered to afford the target compound as a white solid in 53.8% yield. 1H-NMR(DMSO-d6): δ 2.09 (s, 4H), 3.32(s, 2H), 4.04 (s, 2H), 7.21 (d, J = 8.3 Hz, 2H), 7.47 (d, J = 8.3 Hz, 2H), 7.55–7.79(m, 3H), 11.21(s, 1H); 13C-NMR(DMSO-d6): δ 8.48, 43.33, 49.62, 114.58, 128.38, 128.79, 129.21, 130.12, 131.32, 132.13, 132.49, 133.82, 134.93, 136.67, 141.62, 147.96, 170.1; ESI-MS: m/z [M]+: 424.9.
CB1 receptor binding assay (in vitro)
The target compounds were evaluated using a binding assay towards the CB1 receptor expressed on the membranes of Chinese hamster ovarian (CHO) cells. Rimonabant was used as the positive control and DMSO was used as the blank control. The potent synthetic cannabinoid agonist, [3H]-CP55940, was used as a radioligand (). The CHO cells were incubated in 96-well microtiter plates. Various concentrations of the synthesised cannabinoid ligands were added and incubated for 10 min at 37°C. Then the [3H]-CP55940 was added at a final concentration of 1.7 nM and incubated for another 30 min at 37°C. The cells were lysed and scintillation fluid was added. The radioactivity was quantitated by liquid scintillation spectrometry [Citation17]. There were three parallels in the tests. The half maximal inhibitory concentration (IC50) was determined by nonlinear regression analysis of the concentration/binding data.
Results and discussion
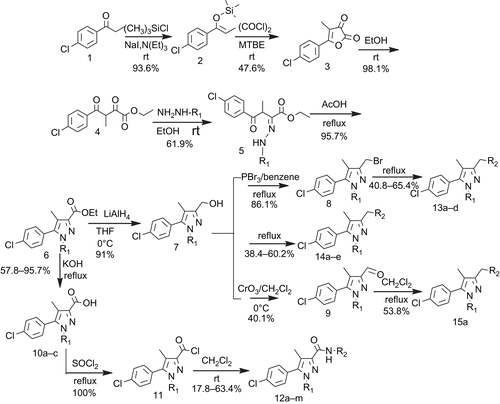
The target compounds were synthesised via the route shown in . The readily available 4-chloropropiophenone (compound 1) was condensed with chlorotrimethylsilane, yielding compound 2 which was reacted with oxalyl chloride to prepare compound 3 [Citation18,Citation19]. Alcoholysis of compound 3 with ethanol produced compound 4 which was coupled with 2,4-dichlorophenylhydrazine hydrochloride to prepare compound 5 and then reacted with acetic acid to form the important intermediate compound 6. The reduction and hydrolysation of compound 6 yielded compounds 7 and 10 respectively [Citation20]. Compound 10 was treated with SOCl2 to produce the acyl chlorides (compound 11) firstly and then reacted with various amino derivatives respectively to prepare the target carboxamids (12a–m) [Citation21,Citation22]. Compound 7 was substituted to afford the target compounds 13a–d, 14a–e, and was oxidised with CrO3 to yield compound 9 which was treated with glycine ethyl ester hydrochloride and KBH4 to afford the target compound 15a [Citation23,Citation24].
Figure 2. Scheme for the synthesis of the target compounds.
All the binding results have been summarised in . The binding results showed that some of the target compounds displayed excellent potency toward the CB1 receptor with IC50 values lying at the nanomole level. Compound 12d (IC50 = 4.31 nM) was the most potent of all the target compounds, and exhibited a potency almost equivalent to rimonabant (IC50 = 4.72 nM). Comparing 12a–m, we could confirm that the introduction of a pyridin-2-yl to the pyrazole resulted in significant decrease in binding affinity at the CB1 receptor, while the introduction of a 4-(trifluoromethyl)phenyl group resulted in a mild decrease in the CB1 receptor binding affinity. For example, the 4-methylcyclohexyl derivatives, 12j and 12m, proved to lose binding affinity compared with compound 12d. This suggested that the substituent of 2,4-dichlorophenyl would help to increase the binding affinity with the CB1 receptor. The compounds with no amide group (13a–d, 14a–e, 15a) showed a low binding affinity compared to the compounds with an amide group (12a–m). The binding affinity loss may in part be attributed to the deficiency of the carbonyl group which could form hydrogen-bonding, as the crucial receptor-ligand interaction, with the Lys192-Asp366 residue of the CB1 receptor. For rimonabant and its analogues, three principal structural requirements are essential for displaying a potent CB1 receptor binding activity: (a) a 2,4-dichlorophenyl substituent at position 1, (b) a para-substituted phenyl ring at position 5 and (c) a carboxamide group at position 3 of the pyrazole ring.
Table 1. The in vitro binding affinities for target compounds and Rimonabant.
Conclusions
In summary, we have described the synthesis and the CB1 receptor binding assay study of a novel series of 1,5-diarylpyrazole scaffold derivatives. The preliminary biological results showed that compound 12d displayed excellent potency toward the CB1 receptor which exhibited almost equivalent potency to rimonabant. This information provides the basis for the synthesis of 1,5-diarylpyrazoles endowed with an improved affinity toward the CB1 receptor.
Declaration of interest
The work was supported by Science and Technology Research Project of Jiangxi Provincial Educational Department (grant no. GJJ09076), Natural Science Foundation of Jiangxi Province, China (grant no. 0620074) and Key Technologies R & D Program of Jiangxi (grant no. 20061B0100400).
References
- Matsuda LA, Lolait SJ, Brownstein MJ, Young AC, Bonner TI. Structure of a cannabinoid receptor and functional expression of the cloned cDNA. Nature 1990;346:561–564.
- Munro S, Thomas KL, Abu-Shaar M. Molecular characterization of a peripheral receptor for cannabinoids. Nature 1993;365:61–65.
- Howlett AC, Barth F, Bonner TI, Cabral G, Casellas P, Devane WA, Felder CC, Herkenham M, Mackie K, Martin BR, Mechoulam R, Pertwee RG. International Union of Pharmacology. XXVII. Classification of Cannabinoid Receptors. Pharmacol Rev 2002;54:161–202.
- Lange JH, Kruse CG. Recent advances in CB1 cannabinoid receptor antagonists. Curr Opin Drug Discov Dev 2004;7:498–506.
- Xie S, Furjanic MA, Ferrara JJ, McAndrew NR, Ardino EL, Ngondara A, Bernstein Y, Thomas KJ, Kim E, Walker JM, Nagar S, Ward SJ, Raffa RB. The endocannabinoid system and rimonabant: a new drug with a novel mechanism of action involving cannabinoid CB1 receptor antagonism-or inverse agonism-as potential obesity treatment and other therapeutic use. J Clin Pharm Ther 2007;32:209–231.
- Patel PN, Pathak R. Rimonabant: a novel selective cannabinoid-1 receptor antagonist for treatment of obesity. Am J Health Syst Pharm 2007;64:481–489.
- Lange JH, Kruse CG. Keynote review: Medicinal chemistry strategies to CB1 cannabinoid receptor antagonists. Drug Discov Today 2005;10:693–702.
- Kakafika AI, Mikhailidis DP, Karagiannis A, Athyros VG. The role of endocannabinoid system blockade in the treatment of the metabolic syndrome. J Clin Pharmacol 2007;47:642–652.
- Le Foll B, Goldberg SR. Cannabinoid CB1 receptor antagonists as promising new medications for drug dependence. J Pharmacol Exp Ther 2005;312:875–883.
- Song KS, Lee SH, Chun HJ, Kim JY, Jung ME, Ahn K, Kim SU, Kim J, Lee J. Design, synthesis and biological evaluation of piperazine analogues as CB1 cannabinoid receptor ligands. Bioorg Med Chem 2008;16:4035–4051.
- Rinaldi-Carmona M, Barth F, Héaulme M, Shire D, Calandra B, Congy C, Martinez S, Maruani J, Néliat G, Caput D. SR141716A, a potent and selective antagonist of the brain cannabinoid receptor. FEBS Lett 1994;350:240–244.
- Carai MA, Colombo G, Gessa GL. Rimonabant: the first therapeutically relevant cannabinoid antagonist. Life Sci 2005;77:2339–2350.
- Takahashi RN, Pamplona FA, Fernandes MS. The cannabinoid antagonist SR141716A facilitates memory acquisition and consolidation in the mouse elevated T-maze. Neurosci Lett 2005;380:270–275.
- Wise LE, Iredale PA, Stokes RJ, Lichtman AH. Combination of rimonabant and donepezil prolongs spatial memory duration. Neuropsychopharmacology 2007;32:1805–1812.
- Benavides J, Boccio D, Henin Y, Piot-Grosjean O. U.S. Patent 7105504, 2006.
- Boström J, Berggren K, Elebring T, Greasley PJ, Wilstermann M. Scaffold hopping, synthesis and structure-activity relationships of 5,6-diaryl-pyrazine-2-amide derivatives: a novel series of CB1 receptor antagonists. Bioorg Med Chem 2007;15:4077–4084.
- Menozzi G, Fossa P, Cichero E, Spallarossa A, Ranise A, Mosti L. Rational design, synthesis and biological evaluation of new 1,5-diarylpyrazole derivatives as CB1 receptor antagonists, structurally related to rimonabant. Eur J Med Chem 2008;43:2627–2638.
- Barth F, Casellas P, Congy C, Martinez S, Rinaldi M, Anne-Archard G. U.S. Patent 5624941, 1997.
- Schweizer EE, Hayes JE, Lee KJ, Rheingold AL. Reactions of azines. 10. Synthesis of 4H, 6H-pyrazolo[1,5-c]oxazol-4-ylidenes,4H-pyrrolo[1,2-b]pyrazole-4-ones,and/or 4H, 8H-pyrazolo [1,5-c][1,3]oxazepin-4-ones. J Org Chem 1987;52:1324–1332.
- Hurst D, Umejiego U, Lynch D, Seltzman H, Hyatt S, Roche M, McAllister S, Fleischer D, Kapur A, Abood M, Shi S, Jones J, Lewis D, Reggio P. Biarylpyrazole inverse agonists at the cannabinoid CB1 receptor: importance of the C-3 carboxamide oxygen/lysine3.28(192) interaction. J Med Chem 2006;49:5969–5987.
- Lan R, Liu Q, Fan P, Lin S, Fernando SR, McCallion D, Pertwee R, Makriyannis A. Structure-activity relationships of pyrazole derivatives as cannabinoid receptor antagonists. J Med Chem 1999;42:769–776.
- Francisco ME, Seltzman HH, Gilliam AF, Mitchell RA, Rider SL, Pertwee RG, Stevenson LA, Thomas BF. Synthesis and structure-activity relationships of amide and hydrazide analogues of the cannabinoid CB(1) receptor antagonist N-(piperidinyl)- 5-(4-chlorophenyl)-1-(2,4-dichlorophenyl)-4-methyl-1H-pyrazole-3-carboxamide (SR141716). J Med Chem 2002;45:2708–2719.
- William V. Murray, Susan K. Hadden, Michael P. Wachter. Synthesis of 3-(1,5-diphenyl-3-pyrazolyl)aryl propanoates. J Heterocycl Chem 1990;27:1933–1940.
- Zimmer H, Amer A. Some Novel Observations on the Reaction of 1-Hydrazinophthalazine with Polycarbonyl Compounds. Heterocycles 1987;26:1177–1182.