Abstract
We extended our previous studies based on the design of 1-[(1H-indol-5-ylmethyl)amino]-2-phenyl-3-(1H-1,2,4-triazol-1-yl)propan-2-ols as antifungal agents toward the identification of new indol-3-ylmethylamino derivatives. The majority of these compounds exhibited antifungal activity against a Candida albicans strain (minimum inhibitory concentrations ranging from 199.0 to 381.0 ng/mL) suggesting an inhibition of 14α-demethylase by sterol analysis studies, but are weaker inhibitors compared to their indol-5-ylmethylamino analogs.
Introduction
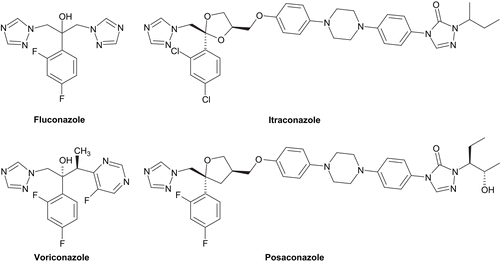
During the last few decades, fungi have become a major threat to many hospitalized patients particularly for severely immunocompromised hosts who are highly susceptible to systemic fungal infections, most often caused by Candida and Aspergillus speciesCitation1. Currently, four main chemical families which have globally three cellular targets in fungal cells are used for the treatment of invasive fungal infections: polyenes, pyrimidine analogues, azoles, and the echinocandins. Among them, azoles are the largest class of antifungal agents in clinical use (). They target mainly P450-dependent lanosterol 14α-demethylase (CYP51), encoded by the ERG11 gene, a key enzyme in the fungal ergosterol biosynthesis. This inhibition leads to the depletion of ergosterol in the cell membrane and accumulation of toxic intermediate sterols, causing increased membrane permeability and inhibition of fungal growth. Voriconazole, itraconazole, and posaconazole have good activity against most filamentous fungi, in contrast to fluconazole, whose activity is largely limited to yeastCitation2,Citation3. However, despite the arrival of new effective drugs such as voriconazole and posaconazole, some therapeutic problems remain, in particular variable drug bioavailability, some toxicity, lack of either oral or intravenous preparations, significant drug interactions for some agents and development of resistance or breakthrough infectionsCitation4, thus creating a need for new antifungal agents.
Figure 1. Structures of some triazoles used in clinic.
CYP51 family members were known to be membrane bound and much more difficult to crystallize. Therefore, the rational design of new antifungal agents was done by molecular modelling studies. Several three-dimensional (3D) models of CYP51 from Candida albicans and Aspergillus fumigatus were constructed on the basis of the crystal coordinates of water-soluble CYP51 family member from Mycobacterium tuberculosisCitation5,Citation6 and interactions with azoles have been explored by flexible molecular docking. In particular, the receptor-based pharmacophore model published by Sheng et al. was established to guide the rational optimization of the azole antifungal agents within the channel 2 oriented to the FG loop and highlighted the importance of Tyr118 and Ser378 residues of C. albicans sterol 14α-demethylase P450 in the stabilization of the inhibitorsCitation7. Tyr118, a highly conserved residue in CYP51 family, may be a potential site for the π–π interaction with the inhibitor, while Ser378, a conserved amino acid across the fungal CYP51 enzymes, could form specific hydrogen-bonding interaction. The role of the Tyr118 residue in determining azole susceptibility was also validated by site-directed mutagenesisCitation8,Citation9.
We reported earlier the synthesis and the structure-activity relationships (SARs) of a novel series of antifungal agents, based on an indole scaffold (I, ). These indol-5-ylmethylamino compounds exhibited potent minimum inhibitory concentrations (MICs; < 65 ng/mL) against a C. albicans strain whereas no activity was demonstrated for the A. fumigatus strainCitation10. Interestingly, the best compound (X = F, R = H, R1 = Boc, R3 = H, MIC80 = 3.0 ng/mL) retained all the main characteristics mentioned above (an H-bond acceptor substituent positioned on an aromatic moiety).
Scheme 1. General structures of synthesized compounds.
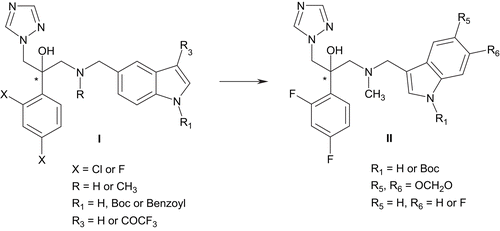
In this paper, considering necessity to develop azoles with improved profile and due to a limited range of variations at the indole moiety, we extended the structural modification to a series of indol-3-ylmethylamino analogs (II, ). We focussed first on the introduction of a N-methyl group in the linker since in a precedent series an emergence of activity was observed on a A. fumigatus strainCitation11, but also on the effect of the introduction of electronegative functions such as a N-Boc protective group, a methylenedioxy substituent and a fluorine atom, at positions 1, 5, and/or 6 of the indole ring. We hypothesized that the use of these easily available substituents should alter the physical properties and binding characteristics of the studied compounds by acting as appropriate hydrogen-bond acceptors into the active site.
Materials and methods
Chemistry
Instrumentation. Melting points were determined using an Electrothermal IA9300 digital melting point apparatus and reported uncorrected. Infrared (IR) spectra were obtained in KBr pellets using a Perkin–Elmer Paragon FTIR 1000 PC spectrometer (Beaconsfield Bucks, England). Only the most significant absorption bands have been reported. Citation1H and Citation13C nuclear magnetic resonance (NMR) spectra were recorded on a Bruker AC250 (250 MHz) or on Bruker Avance 400 spectrometer (400 MHz). Chemical shifts are expressed as δ values (ppm) relative to tetramethylsilane as internal standard (in NMR description, s = singlet, d = doublet, t = triplet, m = multiplet and b = broad). Coupling constants J are given in Hz. Electrospray mass spectrometric analysis was performed on an Esquire-LC Ion Trap System mass spectrometer. Elemental analyses were performed by CEISAM UMR CNRS 6230 Laboratory (Nantes, France) and found to be within a range of ± 0.4% of theoretical values. All reactions were monitored by thin-layer chromatography using 0.2 mm-thick silica gel plates 60F-254 (5735 Merck). Column chromatography was carried out using silica gel 60 (70–230 Mesh, ASTM, Merck). Chemicals and solvents used were commercially available. Compound 4 was prepared according to reported methodCitation10.
General procedure for the preparation of compounds 2a-c
To a three-necked round flask was introduced anhydrous N,N-dimethylformamide (1 mL, 12.92 mmol) at 0°C followed by slow addition of phosphorus oxychloride (0.190 mL, 2.05 mmol). Solution was mixed at 0°C for 40 min. A solution of an appropriate indole 1a-c (1.86 mmol) in 1 mL of N,N-dimethylformamide was slowly added maintaining the temperature below 10°C. The mixture was stirred at this temperature for 40 min and then at 35°C for additional 40 min. Pilled ice was added to the flask and a solution of sodium hydroxide (931 mg, 23.27 mmol dissolved in 2.5 mL of water) was introduced dropwise. Solution was vigorously stirred during the addition, then heated to 100°C for 30 min and left to reach room temperature. Mixture was diluted with water (100 mL) and product was extracted with ethyl acetate (3 × 30 mL). Organics layers were combined, dried over anhydrous sodium sulphate, filtered and concentrated in vacuo.
5H-1,3-dioxolo[4,5-f]indole-7-carbaldehyde (2a). Yield 80%; light brown powder; mp = 217–218°C; IR (KBr) cm−1: 3195 (N-H), 1632 (C=O), 1442 (C=C), 1179 (C-O); 1H NMR [dimethylsulfoxide d6 (DMSO-d6)]: δ ppm 6.05 (s, 2H), 7.07 (s, 1H), 7.50 (s, 1H), 8.11 (d, 1H, 3J = 2.7 Hz), 9.87 (s, 1H), 11.97 (bs, 1H).
1H-indole-3-carbaldehyde (2b). Yield 87%; white powder; mp = 195–196°C; IR (KBr) cm−1: 3167 (N-H), 1634 (C=O), 1463, 1514 (C=C); 1H NMR (DMSO-d6): δ ppm 7.25–7.29 (m, 2H), 7.54 (d, 1H, 3J = 7.0 Hz), 8.12 (dd, 1H, 3J = 8.2 Hz, 4J = 1.8 Hz), 8.32 (s, 1H), 9.97 (s, 1H), 12.17 (bs, 1H).
6-fluoro-1H-indole-3-carbaldehyde (2c). Yield 85%; light brown powder; mp = 173–174°C; IR (KBr) cm−1: 3463 (N-H), 1638 (C=O), 1444, 1530 (C=C), 1148 (C-F); 1H NMR (DMSO-d6): δ ppm 7.08–7.17 (m, 1H), 7.35 (dd, 1H, 3JH-F = 9.7 Hz, 4JH-H = 2.1 Hz), 8.10 (dd, 1H, 3JH-H = 8.5 Hz, 4JH-F = 5.5 Hz), 8.34 (s, 1H), 9.95 (s, 1H), 12.21 (bs, 1H).
General procedure for the preparation of compounds 3a-c
To a solution of an appropriate indolecarbaldehyde 2a-c (1.32 mmol) in 4 mL of acetonitrile was added di-tert-butyl dicarbonate (374 mg, 1.71 mmol) followed by addition of 4-dimethylaminopyridine (DMAP; 16 mg, 0.13 mmol). The mixture was stirred at room temperature for 2 h. Water was added (20 mL) and product was extracted with dichloromethane (3 × 20 mL). Organics layers were combined, dried over anhydrous sodium sulfate, filtered and concentrated in vacuo.
5-(tert-butoxycarbonyl)-1,3-dioxolo[4,5-f]indole-7-carbaldehyde (3a). Yield 94%; light brown powder; mp = 149–150°C; IR (KBr) cm−1: 1740 (C=O), 1670 (C=O), 1563 (C=C); 1H NMR (DMSO-d6): δ ppm 1.68 (s, 9H), 6.13 (s, 2H), 7.54 (s, 1H), 7.60 (s, 1H), 8.52 (s, 1H), 10.02 (s, 1H).
1-(tert-butoxycarbonyl)-1H-indole-3-carbaldehyde (3b). Yield 98%; white powder; mp = 125–126°C; IR (KBr) cm−1: 1745 (C=O), 1679 (C=O), 1454, 1559 (C=C), 1363 (C-N), 1154 (C-O); 1H NMR (DMSO-d6): δ ppm 1.71 (s, 9H), 7.39–7.53 (m, 2H), 8.18 (m, 2H), 8.71 (s, 1H), 10.12 (s, 1H).
1-(tert-butoxycarbonyl)-6-fluoro-1H-indole-3-carbaldehyde (3c). Yield 99%; light yellow powder; mp = 136–137°C; IR (KBr) cm−1: 1746 (C=O), 1670 (C=O), 1554 (C=C), 1157 (C-F); 1H NMR (DMSO-d6): δ ppm 1.70 (s, 9H), 7.27–7.35 (m, 1H), 7.86 (dd, 1H, 3JH-F = 10.3 Hz, 4JH-H = 2.1 Hz), 8.17 (dd, 1H, 3JH-H = 8.8 Hz, 4JH-F = 5.8 Hz), 8.71 (s, 1H), 10.10 (s, 1H).
General procedure for the preparation of compounds 5a-c
To a solution of 4 (278 mg, 1.04 mmol) in 5 mL of methanol and 0.1 mL of acetic acid was added the corresponding N-Boc indoles 3a-c (1.04 mmol) at room temperature under argon. Then sodium cyanoborohydride (78 mg, 1.24 mmol) was added and the mixture was stirred for 24 h. Mixture was diluted with water (20 mL) and product was extracted with dichloromethane (3 × 20 mL). Organic layers were combined, washed with saturated sodium bicarbonate (20 mL), dried over anhydrous sodium sulfate, filtered and concentrated in vacuo. The residue was purified on silica gel column chromatography (dichloromethane and dichloromethane/ethanol, 99:1).
1-({[5-(tert-butoxycarbonyl)-1,3-dioxolo[4,5-f]indol-7-yl]methyl}methylamino)-2-(2,4-difluorophenyl)-3-(1H-1,2,4-triazol-1-yl)propan-2-ol (5a). Yield 59%; white powder; mp = 108–109°C; IR (KBr) cm−1: 3445 (O-H), 1729 (C=O), 1406, 1466, 1620 (C=C and C=N), 1288 (C-O), 1163 (C-F); 1H NMR (DMSO-d6): δ ppm 1.64 (s, 9H), 2.11 (s, 3H), 2.69 (d, 1H, 2J = 13.8 Hz), 3.14 (d, 1H, 2J = 13.8 Hz), 3.45 (d, 1H, 2J = 13.2 Hz), 3.64 (d, 1H, 2J = 13.2 Hz), 4.48 (d, 1H, 2J = 14.0 Hz), 4.54 (d, 1H, 2J = 14.0 Hz), 5.79 (s, 1H), 6.04 (d, 2H, 3J = 3.6 Hz), 6.69 (s, 1H), 6.96 (ddd, 1H, 3JH-F = 3JH-H = 8.4 Hz, 4JH-H = 2.4 Hz), 7.08 (ddd, 1H, 3JH-F = 3J′H-F = 9.2 Hz, 4JH-H = 2.4 Hz), 7.30 (s, 1H), 7.43 (ddd, 1H, 3JH-H = 8.4 Hz, 4JH-F = 4J′H-F = 6.8 Hz), 7.50 (s, 1H), 7.79 (s, 1H), 8.28 (s, 1H); 13C NMR (DMSO-d6): δ ppm 27.8 (3C, CH3), 43.9 (CH3), 53.7 (CH2), 56.2 (CH2), 63.1 (CH2), 75.3 (Cq), 83.8 (Cq), 96.0 (CH), 99.3 (CH), 101.2 (CH2), 103.8 (CH), 110.9 (CH), 118.4 (CH), 123.1 (Cq), 124.2 (Cq), 126.3 (Cq), 130.0 (CH), 130.1 (Cq), 143.9 (Cq), 145.0 (CH), 145.8 (Cq), 149.1 (C=O), 150.6 (CH), (CF not visible); MS (ES+) m/z 542.3 (M + H)+. Anal. Calcd. for C27H29F2N5O5: C, 59.88; H, 5.40; N, 12.93; found: C, 59.69; H, 5.45; N, 12.67%.
1-{[(1-tert-butoxycarbonylindol-3-yl)methyl]methylamino}-2-(2,4-difluorophenyl)-3-(1H-1,2,4-triazol-1-yl)propan-2-ol (5b). Yield 47%; white powder; mp = 120–121°C; IR (KBr) cm−1: 3446 (O-H), 1733 (C=O), 1451, 1497, 1621 (C=C and C=N), 1262 (C-O), 1154 (C-F); 1H NMR (DMSO-d6): δ ppm 1.66 (s, 9H), 2.12 (s, 3H), 2.75 (d, 1H, 2J = 13.6 Hz), 3.15 (d, 1H, 2J = 13.6 Hz), 3.56 (d, 1H, 2J = 13.4 Hz), 3.71 (d, 1H, 2J = 13.4 Hz), 4.49 (d, 1H, 2J = 14.2 Hz), 4.59 (d, 1H, 2J = 14.2 Hz), 5.78 (s, 1H), 6.97 (ddd, 1H, 3JH-F = 3JH-H = 8.4 Hz, 4JH-H = 2.4 Hz), 7.11–7.16 (m, 2H), 7.28–7.32 (m, 2H), 7.40–7.46 (m, 2H), 7.79 (s, 1H), 8.02 (d, 1H, 3J = 8.4 Hz), 8.29 (s, 1H); 13C NMR (DMSO-d6): δ ppm 27.9 (3C, CH3), 43.9 (CH3), 53.5 (CH2), 56.2 (CH2), 63.2 (CH2), 75.2 (Cq), 83.8 (Cq), 103.8 (CH), 110.8 (CH), 114.7 (CH), 118.2 (Cq), 120.2 (CH), 122.4 (CH), 124.4 (CH), 124.5 (CH), 126.3 (Cq), 130.1 (Cq), 130.2 (CH), 135.1 (Cq), 145.0 (CH), 149.2 (C=O), 150.6 (CH), (CF not visible); MS (ES+) m/z 498.1 (M + H)+. Anal. Calcd. for C26H29F2N5O3: C, 62.77; H, 5.88; N, 14.08; found: C, 62.62; H, 5.91; N, 13.79%.
1-{[(1-tert-butoxycarbonyl-6-fluoroindol-3-yl)methyl]methylamino}-2-(2,4-difluorophenyl)-3-(1H-1,2,4-triazol-1-yl)propan-2-ol (5c). Yield 68%; white powder; mp = 149–150°C; IR (KBr) cm−1: 3444 (O-H), 1734 (C=O), 1494, 1616 (C=C and C=N), 1274 (C-O), 1157 (C-F); 1H NMR (DMSO-d6): δ ppm 1.66 (s, 9H), 2.12 (s, 3H), 2.73 (d, 1H, 2J = 13.6 Hz), 3.15 (d, 1H, 2J = 13.6 Hz), 3.54 (d, 1H, 2J = 13.4 Hz), 3.70 (d, 1H, 2J = 13.4 Hz), 4.48 (d, 1H, 2J = 14.3 Hz), 4.54 (d, 1H, 2J = 14.3 Hz), 5.79 (s, 1H), 6.97–7.02 (m, 2H), 7.11 (ddd, 1H, 3JH-F = 3J′H-F = 9.2 Hz, 4JH-H = 2.4 Hz), 7.28 (dd, 1H, 3JH-H = 8.8 Hz, 4JH-F = 5.6 Hz), 7.40–7.46 (m, 2H), 7.73 (dd, 1H, 3JH-F = 10.4 Hz, 4JH-H = 1.6 Hz), 7.79 (s, 1H), 8.29 (s, 1H); 13C NMR (DMSO-d6): δ ppm 27.8 (3C, CH3), 44.0 (CH3), 53.5 (CH2), 56.2 (CH2), 63.1 (CH2), 75.3 (Cq), 84.3 (Cq), 101.6 (CH), 103.8 (CH), 110.3 (CH), 110.9 (CH), 118.2 (Cq), 121.4 (CH), 124.9 (CH), 126.3 (Cq), 126.7 (Cq), 130.1 (CH), 135.2 (Cq), 145.0 (CH), 149.0 (C=O), 150.6 (CH), 160.2 (CF), (CF not visible); MS (ES+) m/z 516.1 (M + H)+. Anal. Calcd. for C26H28F3N5O3: C, 60.58; H, 5.47; N, 13.58; found: C, 60.12; H, 5.48; N, 13.23%.
General procedure for the preparation of compounds 6a-c
To a solution of an appropriate azole 5a-c (0.54 mmol) in 10 mL of dichloromethane was added zinc bromide (724 mg, 3.21 mmol) at room temperature under argon. The solution was stirred 24 h. Then water (55 mL) was added and the mixture was stirred for 2 h. Product was extracted with dichloromethane (3 × 30 mL), organic layers were combined, dried over anhydrous sodium sulfate, filtered and concentrated in vacuo. The residue was purified on silica gel column chromatography (dichloromethane and dichloromethane/ethanol, 99:1).
1-{[(5H-1,3-dioxolo[4,5-f]indol-7-yl)methyl]methylamino}-2-(2,4-difluorophenyl)-3-(1H-1,2,4-triazol-1-yl)propan-2-ol (6a). Yield 21%; grey powder; mp = 137–138°C; IR (KBr) cm−1: 3422 (O-H and N-H), 1472, 1503, 1619 (C=C and C=N), 1312 (C-O), 1185 (C-F); 1H NMR (DMSO-d6): δ ppm 2.09 (s, 3H), 2.78 (d, 1H, 2J = 13.7 Hz), 3.04 (d, 1H, 2J = 13.7 Hz), 3.53 (d, 1H, 2J = 13.2 Hz), 3.60 (d, 1H, 2J = 13.2 Hz), 4.48 (d, 1H, 2J = 14.6 Hz), 4.53 (d, 1H, 2J = 14.6 Hz), 5.72 (s, 1H), 5.93 (s, 2H), 6.71 (s, 1H), 6.85 (s, 1H), 6.99 (m, 2H), 7.13 (ddd, 1H, 3JH-F = 3J′H-F = 9.2 Hz, 4JH-H = 2.4 Hz), 7.43 (ddd, 1H, 3JH-H = 8.4 Hz, 4JH-F = 4J′H-F = 6.8 Hz), 7.77 (s, 1H), 8.29 (s, 1H), 10.67 (bs, 1H); 13C NMR (DMSO-d6): δ ppm 43.7 (CH3), 54.3 (CH2), 56.2 (CH2), 62.6 (CH2), 74.9 (Cq), 92.1 (CH), 97.8 (CH), 100.1 (CH2), 103.7 (CH), 110.5 (CH), 111.9 (Cq), 121.3 (Cq), 123.2 (CH), 126.4 (Cq), 130.1 (CH), 131.2 (Cq), 141.8 (Cq), 143.9 (Cq), 144.3 (CH), 150.6 (CH), (CF not visible); MS (ES+) m/z 442.2 (M + H)+. Anal. Calcd. for C22H21F2N5O3: C, 59.86; H, 4.80; N, 15.86; found: C, 59.59; H, 4.79; N, 15.58%.
2-(2,4-difluorophenyl)-1-[(1H-indol-3-ylmethyl)methylamino]-3-(1H-1,2,4-triazol-1-yl)-propan-2-ol (6b). Yield 64%; white powder; mp = 73–74°C; IR (KBr) cm−1: 3435 (O-H and N-H), 1492, 1625 (C=C and C=N), 1183 (C-F); 1H NMR (DMSO-d6): δ ppm 2.11 (s, 3H), 2.84 (d, 1H, 2J = 13.6 Hz), 3.04 (d, 1H, 2J = 13.6 Hz), 3.64 (d, 1H, 2J = 13.2 Hz), 3.68 (d, 1H, 2J = 13.2 Hz), 4.48 (d, 1H, 2J = 14.3 Hz), 4.53 (d, 1H, 2J = 14.3 Hz), 5.72 (s, 1H), 6.93 (dd, 1H, 3J = 3J′ = 7.6 Hz), 6.98 (ddd, 1H, 3JH-F = 3JH-H = 8.4 Hz, 4JH-H = 2.4 Hz), 7.07 (dd, 1H, 3J = 3J′ = 7.6 Hz), 7.13–7.19 (m, 2H), 7.33 (m, 2H), 7.43 (ddd, 1H, 3JH-H = 8.4 Hz, 4JH-F = 4J′H-F = 6.8 Hz), 7.76 (s, 1H), 8.28 (s, 1H), 10.89 (bs, 1H); 13C NMR (DMSO-d6): δ ppm 43.7 (CH3), 54.2 (CH2), 56.2 (CH2), 63.0 (CH2), 74.9 (Cq), 103.9 (CH), 110.8 (CH), 111.4 (CH), 111.6 (Cq), 118.5 (CH), 119.1 (CH), 121.1 (CH), 124.8 (CH), 126.7 (Cq), 127.5 (Cq), 130.0 (CH), 136.5 (Cq), 145.0 (CH), 150.6 (CH), (CF not visible); MS (ES+) m/z 398.3 (M + H)+. Anal. Calcd. for C21H21F2N5O: C, 63.47; H, 5.33; N, 17.62; found: C, 63.25; H, 5.48; N, 17.48%.
2-(2,4-difluorophenyl)-1-[(6-fluoro-1H-indol-3-ylmethyl)methylamino]-3-(1H-1,2,4-triazol-1-yl)-propan-2-ol (6c). Yield 76%; white powder; mp = 77–78°C; IR (KBr) cm−1: 3419 (O-H and N-H), 1456, 1503, 1622 (C=C and C=N), 1275 (C-N), 1137 (C-F); 1H NMR (DMSO-d6): δ ppm 2.11 (s, 3H), 2.80 (d, 1H, 2J = 13.6 Hz), 3.05 (d, 1H, 2J = 13.6 Hz), 3.60 (d, 1H, 2J = 13.2 Hz), 3.66 (d, 1H, 2J = 13.2 Hz), 4.47 (d, 1H, 2J = 14.0 Hz), 4.53 (d, 1H, 2J = 14.0 Hz), 5.75 (s, 1H), 6.77 (m, 1H), 6.98 (ddd, 1H, 3JH-F = 3JH-H = 8.4 Hz, 4JH-H = 2.4 Hz), 7.11 (dd, 1H, 3JH-F = 10.4 Hz, 4JH-H = 2.4 Hz), 7.13–7.18 (m, 2H), 7.26 (dd, 1H, 3JH-H = 8.8 Hz, 4JH-F = 5.6 Hz), 7.43 (ddd, 1H, 3JH-H = 8.4 Hz, 4JH-F = 4J′H-F = 6.8 Hz), 7.76 (s, 1H), 8.29 (s, 1H), 10.95 (bs, 1H); 13C NMR (DMSO-d6): δ ppm 43.7 (CH3), 54.1 (CH2), 56.2 (CH2), 63.0 (CH2), 75.0 (Cq), 97.3 (CH), 103.7 (CH), 106.8 (CH), 110.8 (CH), 111.9 (Cq), 120.2 (CH), 124.3 (Cq), 125.3 (CH), 126.5 (Cq), 130.1 (CH), 136.3 (Cq), 145.0 (CH), 150.6 (CH), (CF not visible); MS (ES+) m/z 416.4 (M + H)+. Anal. Calcd. for C21H20F3N5O: C, 60.72; H, 4.85; N, 16.86; found: C, 60.42; H, 5.01; N, 16.56%.
Antifungal activity
All these compounds were screened for their antifungal activity against C. albicans CA98001 and A. fumigatus AF98003 strains. Inhibition growth was measured as previously described12. Fluconazole and itraconazole were used as positive controls. The MIC80 values (in ng/mL) are presented in .
Table 1. In vitro antifungal activity of indol-3-ylmethylamino derivatives.
Sterol extraction and analysis
To study sterol synthesis, C. albicans CA98001 cells were incubated in 50 mL Sabouraud broth medium (Sigma–Aldrich, Munich, Germany) during 18 h at 35°C with stirring. Compound 5c was introduced into culture medium before incubation. Cells were collected by centrifugation at 1500g. Pellet was suspended in 3 mL of saponification medium (25 g of KOH, 36 mL of distilled water and brought to 100 mL with 100% ethanol). Then suspension was vortexed for 1 min and incubated at 80°C for 60 min. Sterols were then extracted by addition of a mixture of 1 mL of distilled water and 4 mL of n-hexane (Merck, Darmstadt, Germany). Hexane extract was then evaporated.
Samples were derivatized with 100 µL of silylating mixture (Fluka, Saint Quentin Fallavier, France) at room temperature for 30 min, evaporated and diluted in 500 µL of n-hexane. Two microliters were injected into a gas chromatograph (model 6890N, Agilent Technologies, Palo Alto, CA) coupled with a quadrupole mass spectrometer (5973i, Agilent Technologies, Palo Alto, CA). Analyses were carried out in a splitless mode with helium as carrier gas (constant rate of 1.2 mL/min) and the injector temperature was 250°C. The transfer line between gas chromatograph and mass spectrometer was operated at 290°C and the EI source was held at 280°C. The gas chromatography (GC) capillary column was an HP-5MS (30 m × 0.25 mm ID, 0.25 µm film thickness, Agilent Technologies). The GC oven was programmed as follows: initial temperature 150°C, hold for 0.5 min, ramp to 280°C at 40°C/min, and ramp to 300°C at 5°C/min, hold for 6 min. Sterols of interest were identified by their mass spectrum. In view to study the treatment influence on sterol abundance, the area under curve (AUC) of each peak was used to calculate a ratio: sterol AUC/sum of sterols AUC.
Homology model of CYP51-C. albicans
Molecular modelling studies were performed using SYBYL software version 8.0Citation13 running on a dell precision T3400 workstation. The structure of CYP51 from M. tuberculosis complexed with fluconazole (PDB code, 1EA1)Citation14 was used as the template for the construction of our house-made model of CYP51-C. albicansCitation15. Multiple alignment of the CYP51-M. tuberculosis sequence with those of CYP51-C. albicans (PIR code, P10613) and CYP51-A. fumigatus (PIR code, Q9P8R0) was performed using ClustalWCitation16. This alignment was further checked by comparing a secondary structure elements prediction for CYP51-C. albicans, obtained through the PSIPRED protein structure prediction serverCitation17, with the experimental secondary structure assignments for CYP51-M. tuberculosis deduced from the PDB file. The 3D model of CYP51-C. albicans was then constructed by the Nest program from the protein structure modelling package JACKALCitation18. The resulting model was further subjected to an energy minimization using Powell’s method available in Maximin2 procedure with the MMFF94 force field and a dielectric constant of 4.0 until the gradient value reached 0.1 kcal/mol/Å. Energy minimization was started with the core side chains, then the core main chains. During the optimization procedure, the model quality was assessed periodically by Verify-3DCitation19 and Ramachandran plotsCitation20. If present, improper geometries were manually corrected and the structure minimized again with the above procedure. Haeme and fluconazole extracted from the CYP51-M. tuberculosis structure was added to the model, and residues neighbouring haeme were minimized to avoid steric conflicts. As in the crystal structure of CYP51 from M. tuberculosis, the entrance of channel 2 is closed from the surface by interaction between the A′ helix and FG loopCitation14, modifications of some residues were applied to leave enough space in the pocket for the binding of azoles with extended side chains, according to Xiao et alCitation5.
Docking of azoles
The 3D structures of azoles were constructed using the standard sketch procedure of SYBYL and their geometries were subsequently optimized using the Tripos force field including the electrostatic term calculated from Gasteiger and Huckel atomic charges. Powell’s method available in Maximin2 procedure was used for energy minimization until the gradient value was smaller than 0.001 kcal/mol/Å. Flexible docking of azoles into the enzyme active site was performed using GOLD softwareCitation21. A distance constraint was applied from N-4 of the triazole ring to the haeme iron (2.0 < d < 2.4 Å). For each compound, the most stable docking model was selected according to the best scored conformation predicted by the GoldScore scoring function.
Results and discussion
Chemistry
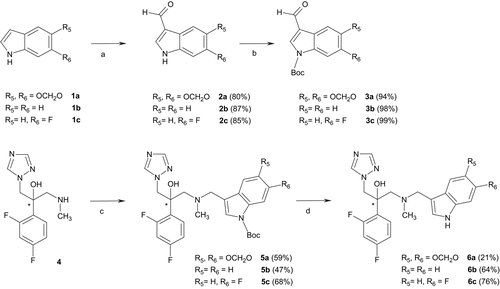
outlines the synthesis of compounds 5a-c and 6a-c. Indoles 1b and 1c were commercially available whereas 5H-1,3-dioxolo[4,5-f]indole 1a was prepared in three steps from heliotropinCitation22. Vilsmeier–Haack reaction under standard conditions followed by N-Boc protectionCitation23 with di-tert-butyl dicarbonate and 4-dimethylaminopyridine (DMAP) in acetonitrile afforded derivatives 3a-c. Products 5a-c were synthesized from previously describedCitation10 key intermediate 4 by reductive amination with 3a-c. Finally, removal of the Boc protective group with zinc bromide in methylene chloride led to compounds 6a-cCitation24.
Scheme 2. Preparation of targeted compounds 5a-c and 6a-c. Reagents and conditions: (a) (i) POCl3, DMF, 0°C to 35°C, (ii) NaOH, 100°C; (b) Boc2O, DMAP, CH3CN, room temperature, 2 h; (c) 3, NaBH3CN, AcOH/MeOH 2% v/v, room temperature, 24 h; (d) ZnBr2, CH2Cl2, room temperature, 24 h.
Antifungal activity
On C. albicans strain, except for compound 5a, our compounds exhibited high antifungal activity with MIC values ranging from 199.0 to 381.0 ng/mL, comparable to that of fluconazole (). Compound 5a, bearing a N-Boc protective group on the indole moiety and an additional bulky methylenedioxy group showed a decrease in activity (MIC80 > 541.0 ng/mL) leading to the assumption that steric factors could affect the biological result. It is noteworthy that compared to their previously described indol-5-ylmethylamino analogs (I, )Citation10, both compounds 5b and 6b are weaker inhibitors with MIC values approximatively 7-fold less active, confirming that isomerization from the 5- to the 3-position of the indole ring is more sensitive to steric and/or electrostatic effects. Furthermore, the degree of inhibition was not significantly influenced neither by compound 6a, or compound 6c, with only H-bond acceptor substituents at positions 5 and/or 6 of the indole ring. Once again, all these compounds were ineffective against the A. fumigatus strain.
Sterol analysis
In view to confirm the mechanism of action of this series of compounds, we have investigated the inhibition of ergosterol biosynthesis after treatment by compound 5c. As shown in , compound 5c strongly inhibited ergosterol biosynthesis while lanosterol accumulation did occurred. At concentrations of 0.25 and 0.5 µg/ mL, surrounding the MIC (0.381 µg/mL), this effect was maximal. About 50% of ergosterol biosynthesis inhibition was obtained with a lower concentration of 0.125 µg/mL. These results show that inhibition of ergosterol biosynthesis by compound 5c is dose-dependent and reached his maximum at the MIC concentration. Otherwise, methylated sterols at the C14 position did also appeared (14-methylfecosterol, 14-methylepisterol, eburicol and 14-methyl-3,6-diol) in a dose-dependent manner. Accumulation of lanosterol and production of 14-methylated sterols are typically a result of a 14α-demethylase blockage by triazole derivatives. Depletion of ergosterol, accumulation of lanosterol and production of 14-methylated sterols lead to membrane instability and cell death. Thus, antifungal activity of compound 5c is due to inhibition of ergosterol biosynthesis by inhibition of 14α-demethylase, suggesting that all these compounds act in a similar fashion.
Table 2. Effect of compound 5c on the sterol composition of Candida albicans CA98001.
SAR study
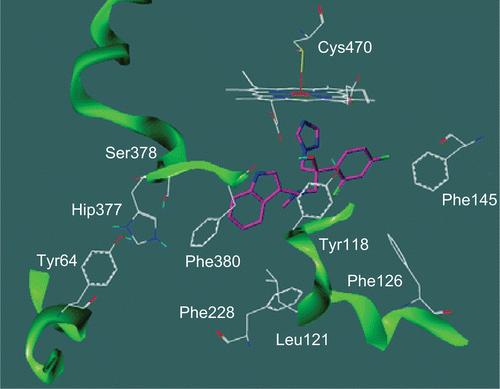
In attempts to understand the observed SARs, we performed the docking of one of the most active compound (6b, MIC = 258.0 ng/mL) in our model of CYP51-C. albicans (). Interestingly, the indole nitrogen is in close proximity to the backbone and the propionate side chain of the haeme, suggesting that sterically encumbering compounds would be expected to encounter unfavourable steric interactions with the enzyme. The reported weak activities for compound 5a bearing a N-Boc protective group at the 1-position of the indole ring are consistent with that expectation. Although compounds 5b-c share the same group, we can speculate that the lack of bulky substituents at positions 5- and 6- of the indole ring allows greater flexibility into the active site. Moreover, the decrease of activity observed for compound 6b compared to its indol-5-ylmethylamino analog could be explained by the ability of the benzo and pyrrolo portions of indole ring to undergo optimal stacking interactions with the phenol moiety of key residue Tyr118 and/or the class-specific residue Phe380.
Figure 2. Proposed binding solution of compound (S)-6b (magenta) in the supposed active site pocket (channel 2) of CYP51-Candida albicans. Hip377 is the protonated form of histidine residue.
The complete lack of activity against A. fumigatus strain may be due to steric hindrance within the active site induced by the bulky indole ring or a lack of permeability of these compounds inside this filamentous pathogen. However, only results on the isolated CYP51 could confirm our hypotheses. In addition, despite an explicit information about the enzyme binding site of pathogenic fungi, the successful design of an optimum inhibitor using 3D models of CYP51 seems to be difficult. Recently, many crystal structures of CYP51 from human and Trypanosomatidae in complex with azole antifungal inhibitors revealed profound conformational changes both in organization of the active site cavity and in the substrate access channel location which is different from that observed in M. tuberculosis CYP51Citation25,Citation26. Therefore, particular attention must be given to the choice of the target in a structure-based design strategy, keeping in mind as the ideal azole antifungals are those which react strongly with fungal cytochrome P450s and have weak or no activities against the mammalian enzyme variants.
Conclusion
We studied the utility of original indole-containing azole compounds as antifungal agents. These molecules exhibited antifungal activity against a C. albicans strain by inhibition of 14α-demethylase but were ineffective against a A. fumigatus strain. However, in order to develop broad-spectrum compounds, the choice of a side chain bearing a bulky and rigid indole moiety attached to the C-3 of antifungal agents does not seem to be useful scaffold for further optimization.
Declaration of interest
The authors report no conflicts of interest.
References
- Farowski F, Vehreschild JJ, Cornely OA. Posaconazole: a next-generation triazole antifungal. Future Microbiol 2007; 2:231–243.
- Sanglard D, Coste A, Ferrari S. Antifungal drug resistance mechanisms in fungal pathogens from the perspective of transcriptional gene regulation. FEMS Yeast Res 2009; 9:1029–1050.
- Sheehan DJ, Hitchcock CA, Sibley CM. Current and emerging azole antifungal agents. Clin Microbiol Rev 1999; 12:40–79.
- Pasqualotto AC, Denning DW. New and emerging treatments for fungal infections. J Antimicrob Chemother 2008; 61 Suppl 1:i19–i30.
- Xiao L, Madison V, Chau AS, Loebenberg D, Palermo RE, McNicholas PM. Three-dimensional models of wild-type and mutated forms of cytochrome P450 14alpha-sterol demethylases from Aspergillus fumigatus and Candida albicans provide insights into posaconazole binding. Antimicrob Agents Chemother 2004; 48:568–574.
- Sheng C, Zhang W, Zhang M, Song Y, Ji H, Zhu J, Yao J, Yu J, Yang S, Zhou Y, Zhu J, Lu J. Homology modeling of lanosterol 14alpha-demethylase of Candida albicans and Aspergillus fumigatus and insights into the enzyme-substrate Interactions. J Biomol Struct Dyn 2004; 22:91–99.
- Sheng C, Zhang W, Ji H, Zhang M, Song Y, Xu H, Zhu J, Miao Z, Jiang Q, Yao J, Zhou Y, Zhu J, Lü J. Structure-based optimization of azole antifungal agents by CoMFA, CoMSIA, and molecular docking. J Med Chem 2006; 49:2512–2525.
- Chen SH, Sheng CQ, Xu XH, Jiang YY, Zhang WN, He C. Identification of Y118 amino acid residue in Candida albicans sterol 14alpha-demethylase associated with the enzyme activity and selective antifungal activity of azole analogues. Biol Pharm Bull 2007; 30:1246–1253.
- Sheng C, Chen S, Ji H, Dong G, Che X, Wang W, Miao Z, Yao J, Lü J, Guo W, Zhang W. Evolutionary trace analysis of CYP51 family: implication for site-directed mutagenesis and novel antifungal drug design. J Mol Model 2010; 16:279–284.
- Guillon R, Giraud F, Logé C, Le Borgne M, Picot C, Pagniez F, Le Pape P. Design of new antifungal agents: synthesis and evaluation of 1-[(1H-indol-5-ylmethyl)amino]-2-phenyl-3-(1H-1,2,4-triazol-1-yl)propan-2-ols. Bioorg Med Chem Lett 2009; 19:5833–5836.
- Giraud F, Logé C, Pagniez F, Crepin D, Le Pape P, Le Borgne M. Design, synthesis, and evaluation of 1-(N-benzylamino)-2-phenyl-3-(1H-1,2,4-triazol-1-yl)propan-2-ols as antifungal agents. Bioorg Med Chem Lett 2008; 18:1820–1824.
- Pagniez F, Le Pape P. New fluorometric screening test for possible antifungal drugs. J Mycol Med 2001;11:73–78.
- Tripos International. SYBYL 8.0. 1699 South Hanley Road, St. Louis, MO 63144, USA.
- Podust LM, Poulos TL, Waterman MR. Crystal structure of cytochrome P450 14α-sterol demethylase (CYP51) from Mycobacterium tuberculosis in complex with azole inhibitors. Proc Natl Acad Sci USA 2001;98:3068–73.
- Giraud F. PhD Thesis. Université de Nantes, Nantes Atlantique Universités, France, October 2007.
- Smith TF, Waterman MS. Identification of common molecular subsequences. J Mol Biol 1981;47:195–197.
- Jones DT. Protein secondary structure prediction based on position-specific scoring matrices. J Mol Biol 1999; 292:195–202.
- Petrey D, Xiang Z, Tang CL, Xie L, Gimpelev M, Mitros T, Soto CS, Goldsmith-Fischman S, Kernytsky A, Schlessinger A, Koh IY, Alexov E, Honig B. Using multiple structure alignments, fast model building, and energetic analysis in fold recognition and homology modeling. Proteins 2003; 53 Suppl 6:430–435.
- Lüthy R, Bowie JU, Eisenberg D. Assessment of protein models with three-dimensional profiles. Nature 1992; 356:83–85.
- Laskowski RA, MacArthur MW, Moss DS, Thornton JM. PROCHECK—a program to check the stereochemical quality of protein structures. J App Cryst 1993;26:283–91. http://www.ebi.ac.uk/thornton-srv/software/PROCHECK/
- Jones G, Willett P, Glen RC, Leach AR, Taylor R. Development and validation of a genetic algorithm for flexible docking. J Mol Biol 1997; 267:727–748.
- Marchi I, Rebelo AR, Rosa AFF, Maiochi RA. Synthesis and evaluation of the plant growth regulator property of indolic compounds derived from safrole. Quim Nova 2007;30:763–67.
- Davies JR, Kane PD, Moody CJ, Slawin AM. Control of competing N-H insertion and Wolff rearrangement in dirhodium(II)-catalyzed reactions of 3-indolyl diazoketoesters. synthesis of a potential precursor to the marine 5-(3-indolyl)oxazole martefragin A. J Org Chem 2005; 70:5840–5851.
- Ku JM, Jeong BS, Jew SS, Park HG. Enantioselective synthesis of (-)-cis-clavicipitic acid. J Org Chem 2007; 72:8115–8118.
- Lepesheva GI, Park HW, Hargrove TY, Vanhollebeke B, Wawrzak Z, Harp JM, Sundaramoorthy M, Nes WD, Pays E, Chaudhuri M, Villalta F, Waterman MR. Crystal structures of Trypanosoma brucei sterol 14alpha-demethylase and implications for selective treatment of human infections. J Biol Chem 2010; 285:1773–1780.
- Strushkevich N, Usanov SA, Park HW. Structural basis of human CYP51 inhibition by antifungal azoles. J Mol Biol 2010; 397:1067–1078.