Abstract
In this work, we studied the behaviours of urease in the presence of l-ascorbic acid (AA) and dehydroascorbic acid (DHA) in different conditions. The inactivations of urease were carried out in an unbuffered and buffered system. We show that in the unbuffered system AA inactivated urease in a biphasic manner by denaturation brought about by AA-lowered pH. Further, we show that in the buffered system neither AA nor DHA themselves are inhibitors of urease. The inhibitory action of AA and DHA was revealed in the presence of Fe3+ ions and most importantly, unlike reported in the literature, it was found to be primarily mediated by H2O2. The resulting inhibition by DHA-Fe3+ consisted of enzyme thiol oxidation and its effectiveness grew with increasing pH. The results may shed light on the roles of AA in therapies applied in ureolytic bacteria infections, notably those with Helicobacter pylori.
Introduction
Ureases are enzymes that catalyze the hydrolysis of urea, ammonia, and carbonic acid being the final products of the reactionCitation1. The consequential characteristics of their action are the product ammonia and the resulting increase in pH. The enzymes are widely distributed in nature among plants, fungi, and bacteria, some of the latter having profound medical implications. These result from infections of the gastrointestinal and urinary tracts where bacterial ureases serve as a virulence factor, giving rise to peptic ulcer disease, gastric cancer and hepatic coma in the former, and to kidney stone formation, catheter encrustation and pyelonephritis in the latter. The primary ureolytic bacterium infecting the intestinal tract is Helicobacter pylori, whereas bacteria infecting the urinary tracts are chiefly Proteus mirabilis and Ureaplasma urealyticum. The pathological conditions provoked by the bacteria are presently cured with antibiotics, but another strategy to combat the bacteria that have recently emerged is to incapacitate urease with use of inhibitorsCitation2.
Although with different protein structures, ureases share most of general, kinetic and inhibitory features. A salient feature common to all ureases is the presence of nickel(II) ions in the active site, essential for catalytic activity. Another important feature is the presence of multiple cysteinyl residues in the enzymes bearing thiol groupsCitation3. These are susceptible to a variety of reactionsCitation3–8, which can result in enzyme inhibition. Most importantly, the contention that ureases have a conserved active site and consequently the same mechanism of catalysis, allows the generalization of the results over all ureases independent of their origin.
Ureases are inhibited by a number of compoundsCitation1, including thiolsCitation5, hydroxamic acidsCitation9, amides and esters of phosphoric acidCitation10, phosphinic and thiophosphinic acidsCitation11, boric and boronic acidsCitation12,Citation13, phosphateCitation14, heavy metal ionsCitation8, bismuth compoundsCitation6 and quinonesCitation7. Due to their toxicity, however, only few of the compounds classify as therapeutic agents under physiological conditions. In this context, compounds of natural origin may be of invaluable service, and one such compound contemplated as having such potential is l-ascorbic acid (AA) (vitamin C). The knowledge of AA inhibition of urease is of importance for clarifying the roles the acid plays in ureolytic bacteria infections and in the bacteria eradication, especially in Helicobacter pylori infection of the stomach, where vitamin C has been shown to be intricately implicated.
Helicobacter pylori, a gram-negative bacterium, typically colonizes the mucosal lining of the stomachCitation15–17. Urease that the bacterium synthesizes in large quantities (10–15% protein) is central to its survival in the hostile acidic conditions, which is on account of the pH raised by the ammonia resulting from urea hydrolysis. The concomitant damage it incurs to the host tissue gives rise to chronic active gastritis, peptic ulcer disease and gastric cancer. The roles and implications of AA in the infection have been reported to include the following. In healthy individuals the concentration of the acid in gastric juice is in the range 50–400 μM, whereas with Helicobacter pylori infection it drastically falls below 50 μM, frequently to undetectable levels, which is due to the impairement of the intragastric secretionCitation18. Consequent to this decrease, the protective effect of vitamin C that consists of scavenging reactive oxygen and nitrogen species produced in very high quantities during Helicobacter pylori infection, is diminished, and this is believed to be responsible for the observed increase in gastric cancer riskCitation19. To counteract this risk, a high intake of dietery vitamin C is highly recommendableCitation20. Furthermore, it was demonstrated that high doses of vitamin C afforded to reduce colonization of the bacterium in the gastric mucosaCitation21 or even to eradicate the bacteriumCitation22. AA was also shown in vitro to inhibit Helicobacter pylori growthCitation23 and urease activityCitation24, the latter hypothetically by reducing Ni2+ ions in the enzyme active siteCitation25. Noteworthily, it has now been estimated that at least half the world’s population are infected with Helicobacter pylori. Even though in most individuals the infection is asymptomatic, ~10–20% of the infected will utlimately develop peptic ulcer disease and 1%, gastric cancer; hence the importance of urease inhibition studies.
Chemical properties of AA
AA, the formula of which is C6H8O6 (), is a sugar acid with redox properties. As an acid it shows strength comparable with the lower monocarboxylic acids, its pKa for dissociation of the first hydrogen ion being 4.1 and for the second, 11.8Citation26. As a redox agent by contrast, AA exhibits a dual natureCitation27,Citation28. First, it is best known for being an antioxidant with powerful reducing properties. In vivo the properties are exploited at the cellular level for scavenging reactive oxygen species (ROS), including superoxide O2•–, hydroxyl •OH and hydrogen peroxide H2O2, thereby providing protection to biomolecules against oxidative damage. During the process (), AA is initially oxidized to ascorbyl radical and further to its first chemically stable oxidized form, dehydroascorbic acid (DHA). This can be reduced back to AA or be irreversibly hydrolyzed to 2,3-diketogulonic acid, which then is converted primarily to oxalic and threonic acids among other numerous five or less carbon speciesCitation29. Second, AA also has prooxidant properties, which are revealed when the acid is oxidized in reactions catalyzed by transition metal ions, notably cupric and ferricCitation27,Citation28,Citation30. It is because the metal ion-catalyzed oxidation promotes the generation of ROS, paradoxically the same AA is known to destroy. The model currently adopted for the process assumes that on its oxidation AA reduces the metal ions Men+ → Me(n–1)+, which is accompanied by the generation of H2O2. Further, the reduced metal ions react with H2O2 driving the formation of hydroxyl radicals •OH via the Fenton reaction: Me(n–1)+ + H2O2 → Men+ + OH− + •OHCitation29,Citation31. The resulting ROS bring about inactivation of biomolecules by oxidation.
Scheme 1. Oxidation of L-ascorbic acid
Urease activity and AA
Intriguingly, there is no consensus of opinion whether at all, and if yes, how AA interacts with ureases. In the early works the acid was reported to be a strong urease inhibitor (IC50 = 5.7 μM) with the action prevented by cysteineCitation32. Later it was suggested that the inhibition was due to a reaction of DHA with the enzyme thiol groupsCitation33. DHA, however, was later proved not to be an inhibitor, and instead it was proposed that the inhibition was provoked by the presence of traces of Cu2+ ions, which when reduced by AA, formed an inhibitory product Cu2OCitation34. In refs. Citation32–33, the buffers applied were not specified. A subsequent study performed in 0.7 M phosphate buffer pH 6.8Citation35, showed that neither ascorbic (up to 1.1 mM), nor dehydroascorbic or 2,3-diketogulonic acid themselves were urease inhibitors, but that ascorbic and 2,3-diketogulonic acid inhibited the enzyme in the presence of Cu2+ (15.6 μM). The inhibition by AA was ascribed to the reduction of Cu2+ → Cu+, and the involvement of Cu+ in the inhibition could not be ruled out. Both cysteine and glutathione protected the enzyme. In contrast to these findings, in a more recent study of Helicobacter pylori urease by electron microscopy and electrophoresisCitation24, AA was found to completely inhibit the enzyme at a concentration of 2.8 mM within 30 min, and the authors suggested a DHA-mediated mechanism. In another recent work, where a Citation13C-NMR screening for urease inhibitors was presentedCitation36, the inhibition of urease by AA in 70 mM phosphate buffer pH 6.5 was determined to be noncompetitive with Ki = 16.7 mM (IC50 = 16.7 mM). The reduction of Ni2+ ions at urease active site by AA or a covalent modification of the protein by DHA were hypothesized as mechanisms of this inhibition.
In view of these conflicting data, the aim of this work was to present an overview of behaviours of urease in the presence AA in different conditions to resolve the question of AA inactivation of the enzyme. Given the properties of AA, the inactivation was studied in an unbuffered and buffered system. In the unbuffered system the inactivation was compared with that by acetate buffer of low pH. In the buffered system on the other hand, the inactivation was studied at pH 7.2 in the absence and presence of Fe3+ ions, and was further extended into the inactivation study by DHA, also in the absence and presence of Fe3+ ions, the latter at different pHs. Being a much weaker inhibitor of ureaseCitation37, Fe3+ ions were chosen as a catalyst in place of commonly applied Cu2+ ions to avoid their interference with the activity of urease. The urease inactivation results were correlated with UV-vis spectral kinetic data recorded for AA and DHA conversion.
Materials and methods
Materials
Urease (from jack beans, type III, nominal activity 45 units/mg solid), urea (for Molecular Biology), d,l-dithiothreitol (DTT), catalase (from bovine liver, activity 1340 units/mg solid), superoxide dismutase (SOD) (from bovine erythrocytes, activity 3780 units/mg solid), AA and DHA were from Sigma. Phosphate buffers 200 mM with pHs 6.2, 6.7, 7.2, 7.8, and 8.2 were prepared by adjusting pH of phosphoric acid with a NaOH solution. Ultrapure, deionized water obtained from a Simplicity 185 Millipore water purification system (resistivity 18.2 MΩ cm) was used throughout. The measurements were performed at ambient temperature (23 ± 1°C) in a lab protected from the sunlight. pH of the solutions was measured with a pH-meter CX-731, Elmetron, Poland, and UV-vis spectra were recorded with a Shimadzu UV-2100 spectrometer. For the spectra the solutions had AA and DHA prepared at concentrations 0.05 mM and 0.71 mM, respectively. Of note, AA and DHA solutions were always prepared fresh immediately prior to the measurements.
Standard urease activity assay
The standard urease assay mixture contained 100 mM urea in 20 mM phosphate buffer pH 7.2, 1 mM EDTA, its volume being 20 mL. The reactions were initiated by the addition of small aliquots of the enzyme-containing (0.16 mg) solution. The assay was run for 5 min and the enzyme activity was determined by measuring the concentration of the ammonia released. For that samples were withdrawn from the reaction mixtures and the ammonia was determined by the colorimetric phenol-hypochlorite methodCitation38.
Urease inactivations and residual activity measurements
Urease was dissolved in a given solution at a concentration of 0.8 mg/mL. Equal volumes of the solutions of urease and of an inactivating agent were mixed. The mixtures were incubated with occasional stirring. During the incubations, at time intervals 0.4 mL aliquots were transferred into the standard assay mixtures for enzyme residual activity (RA) determinations. The residual activities were normalized to the measured activity of the control sample stored at the same conditions, and the reported values are the means of at least three independent experiments. All the inactivation measurements were done under aerobic conditions.
For the inactivation of urease by AA and DHA in a buffered system, 200 mM phosphate buffer was chosen as the one capable of providing buffering to the chosen concentration of AA and DHA (10 mM). For the inactivations in the presence of Fe3+ ions, a submaximal noninhibitory concentration of 10 μM Fe3+ was applied (IC50,Fe3+ = 50 μM)Citation37.
Inactivation by AA in an unbuffered system
Urease solution was prepared in 20 mM phosphate buffer, pH 7.2, 1 mM EDTA, and AA in water. The concentration of AA in the incubation mixtures varied between 34.2 and 45.0 mM. By contrast, for the inactivations by low pHs in acetate buffer, urease was prepared in water with 1 mM EDTA. The enzyme was incubated with 20 mM acetate buffer, 1 mM EDTA, of pHs between 4.18 and 4.34.
Inactivation by AA in a buffered system in the absence and presence of Fe3+ ions
For the inactivation study in the absence of Fe3+ions, urease and AA solutions were prepared in 200 mM phosphate buffer, pH 7.2, 1 mM EDTA, the incubating concentration of AA being 10 mM. By contrast, for the inactivation study in the presence of Fe3+ ions, EDTA was omitted, and Fe3+ ions were added upon mixing the urease and AA solutions, to form an incubating concentration of 10 μM.
Inactivation by DHA in a buffered system in the absence and presence of Fe3+ ions
The same protocol as presented above for AA was used for the inactivation study of urease by 10 mM DHA at pH 7.2 in the absence of Fe3+ ions. The inactivation study in the presence of Fe3+ ions on the other hand, was performed at pHs 6.2, 6.7, 7.2, 7.8, and 8.2. For this study urease and DHA solutions were prepared in 200 mM phosphate buffer of respective pHs without EDTA. The incubating concentration of DHA was always 10 mM. Fe3+ ions were added upon mixing urease and DHA, to have an incubating concentration of 10 μM.
Urease protection
In the urease protection experiments, in the final incubation mixture of urease with protectors and inactivators, the concentration of urease was always 0.4 mg/ mL (like in the inactivation experiments). Following the pre-incubation with a protector, an inactivator was added and the mixtures were further incubated for the time that was required to obtain a submaximal inactivation observed in the absence of a protector, corresponding to a RA of about 5–10%.
For the protection studies with DTT, urease was pre-incubated for 10 min with excess DTT (5 mM), followed by mixing with an inactivator. Upon incubation of the mixture the urease activity was assayed.
For the protection experiments with catalase and SOD, few crystals of the enzymes were added to the initial urease solutions, followed by the addition of an inactivator. The mixtures were incubated and urease had its activity determined.
Reactivation of the inactivated urease
Reactivation was carried out with DTT. For that urease was incubated with an inactivator for different time intervals, after which the incubation mixtures were further incubated with excess DTT (2 mM). The activity recovery of the enzyme was measured over time.
Results and discussion
Inactivation of urease by AA in an unbuffered system
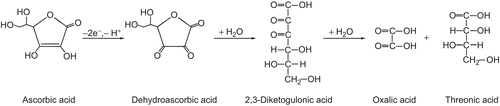
The unbuffered conditions were established where urease, initially dissolved in dilute phosphate buffer pH 7.2, was exposed to the action of different concentrations of AA. The results are shown in in the form of semi-logarithmic plots of RA versus time of enzyme incubation. They demonstrate that the loss of urease activity followed time- and concentration-dependent inactivation.
Figure 1. Time courses of urease inactivation (semilogarithmic plots of residual activity (RA) versus incubation time) by: (A) AA in unbuffered system, and (B) acetate buffer. The RA values are reported as percent of the control activity.
To account for this loss of activity, it was reasonable to assume that the principal factor behind it was not AA itself but the acidic conditions created by the acid. Compared to other acids studied as urease inhibitors, such as acetohydroxamic (pKa = 8.7) and boric (pKa1 = 9.24) that are weak acids and could be investigated in dilute buffersCitation9,Citation12,Citation14, AA is a relatively strong acid (pKa1 = 4.1)Citation26 easily lowering the pH. Indeed, the pH measured in the studied solutions had the acidic values between 3.52 and 3.66 (indicated in ), that are the values lower than ~4.5 below which urease is known to irreversibly lose activityCitation39. To ascertain this observation we repeated the AA inactivation measurements in urease solutions prepared at different initial pH from the range 6.2–8.2. The inactivation always occurred at the same low pHs, requiring in each case slightly lower or higher amounts of AA relative to pH 7.2, to reach the inactivating values.
Further, to check if the AA-generated inactivation of urease was purely pH-dependent, we performed the inactivation of urease by low pHs in 20 mM acetate buffer. The inactivation time courses are shown in . They are similar to those of the AA-induced inactivation, but intriguingly, the same amount of inactivation as in AA was brought about here by higher pHs from the range 4.18–4.34.
The semi-logarithmic plots in demonstrate that in both cases the inactivation consisted of two phases, an initial fast phase followed by a slow phase, consistent with a bi-exponential function of timeCitation40:
where the contributions of the slow phases a to the overall process were estimated to be 63% in the AA-system and slightly higher, 76%, in the acetate buffer.
In addition to the kinetic studies, we also observed that both in the AA and acetate buffer inactivation, the presence of DTT neither protected urease nor restored its activity after inactivation, thereby excluding the involvement of enzyme thiol groups in the process. This test was done because, due to different reactivities of the groups, thiol targeted inactivation of urease often shows as bi-phasicCitation40,Citation41.
Similar to our results, a bi-phasic inactivation by pH 4.5 and 4.8 was observed for pigeonpea urease in 50 mM Tris-acetate buffer at 37°CCitation42. The contributions of both phases, however, were found ~50% and, unlike in the present study, the inactivation was ascribed to the modification of enzyme thiol groups. In another work, where the role of EDTA in promoting the inactivation at low pH was examined, which incidentally is an effect that we did not observe at zero and 1 mM EDTA, a mono-exponential inactivation by pH 3.5–4 was suggested for jack bean urease in 0.1 M acetate buffer at 0°CCitation43.
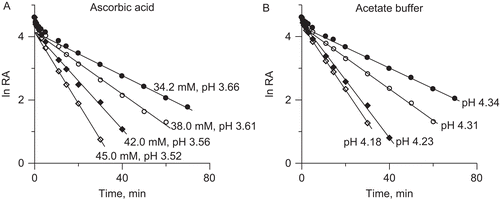
From the observations collected it follows that in the unbuffered system AA inactivated urease in a manner similar to that of acetate buffer, primarily by denaturation of the enzyme by low pH. This pH, however, was found lower in the AA solutions than in the acetate buffer. A similar effect was reported for Helicobacter pylori ureaseCitation44, where the HCl unbuffered system inactivated the enzyme at lower pHs than citrate buffer. presents the dose-response curves for 20 min incubation observed in both studied systems, with the pH50 corresponding to 50% reduction in enzyme activity, being 3.8 for AA and 4.4 for acetate buffer. Given different compositions of the inactivating solutions it might be viewed that at least in part responsible for this difference was the ionic strength of the solutions. This for the AA solutions was calculated to be around twice that for the acetate buffer, for instance for 45 mM AA, I = 0.016 M, whereas for the acetate buffer with pH 4.18, I = 0.007 M. Importantly, higher ionic strengths are known for being conducive to the refolding of proteins under acidic conditions. Among various proteinsCitation45, this effect was also reported for jack bean ureaseCitation46 where the presence of salts was found to induce the formation of the refolded urease intermediate with the stability nearly equal to that of the native protein against denaturation. It may therefore be partly for this reason that the AA solutions required lower pHs than the acetate buffer to inactivate urease to the same extent. There may also be other reasons, which, however, cannot be resolved with the present results.
Figure 2. Dose-response curves for 20 min incubation observed in: (A) AA unbuffered solutions, and (B) acetate buffer.
Given the observations that to counteract the acidity of AA, comparatively concentrated buffers are required, e.g. 200 mM phosphate for 10 mM AA, the inactivation of urease observed in some previous reports, most likely was denaturation rather than the true inhibition by AACitation24,Citation36.
Inactivation of urease by AA in a buffered system in the absence and presence of Fe3+ ions
Having established that in the unbuffered systems AA inactivates urease by denaturation consequent on the lowering of pH, to further examine if AA itself is a urease inhibitor, we chose the conditions that could assure the buffering to the AA-urease mixtures over prolonged incubation times. Such conditions were provided by 200 mM phosphate buffer. This buffer will be further used in all experiments with AA and DHA.
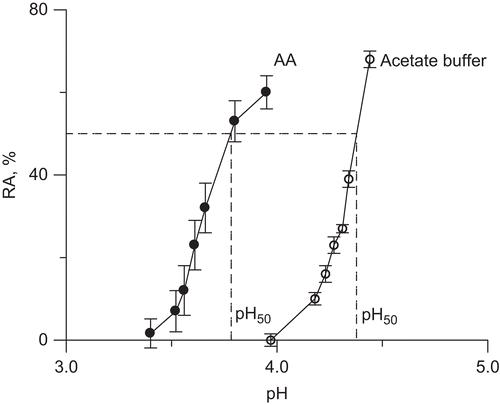
The results of the inactivation study of urease by 10 mM AA in the buffered system pH 7.2, 1 mM EDTA, are presented in . AA appeared to be a very weak urease inhibitor, whose slow inhibitory action developed not sooner than after ∼10 h of incubation, the half-life of inactivation, t50, being 90 h. The results are strongly suggestive that it is not AA itself that provokes the inactivation, but a product of its conversion, and that the product is also a weak inhibitor. To see if this is DHA, an immediate product of AA oxidation, we recorded the inactivation time course of urease by DHA in the same conditions (). The time course shows an onset of inactivation at 3 h incubation, and a half-life of 56 h, proving that to inactivate urease DHA must also undergo conversion. Importantly, the inactivations by AA and DHA performed in the presence of catalase showed that in these very long processes catalase afforded some protection of urease activity from the loss. This observation is consequential in that it supports the participation of H2O2 in the process that, although non-metallic conditions were assured by using ultra-pure deionized water and EDTA, could be generated in the mixtures by the presence of some trace transition metal ions.
Figure 3. Time courses of urease inactivation (residual activity (RA) versus incubation time) by 10 mM AA and DHA in buffered system (200 mM phosphate buffer pH 7.2) in the absence and presence of 10 μM Fe3+ ions. The RA values are reported as percent of the control activity.
The observed kinetics of urease inactivation was correlated with the kinetics of AA and DHA conversion recorded in UV-vis spectra between 500 and 200 nm. AA is known to show a well defined absorption band at 265 nm at neutral pHs, corresponding to ascorbateCitation26,Citation47,Citation48, whereas DHA has little absorbance above 220 nmCitation47,Citation48. The absorbance change at 265 nm is, therefore, often employed to monitor the oxidation of AA and the reduction of DHA. The latter, however, may be disturbed, which is on account of the enigmatic properties and functions of DHA, much less known than those of AACitation29,Citation47,Citation48. The chemical activity of DHA and related products that form from DHA are still not well defined, and effectively, it is never clear what exact species are present in experiments where DHA has been added. It was reported that in such cases the numerous products included AA produced in significant amount by reduction of DHA, and also other acids resulting from 2,3-diketogulonic acid, in addition to oxalic and threonic acids, also lyxonic and xylonic acids produced by decarboxylation, and importantly, l-erythroascorbic acid, a product of a spontaneous conversion of DHACitation47. An important feature of l-erythroascorbic acid is that like AA it absorbs light at 265 nm. Thus the presence of the above products may significantly interfere with DHA-related experiments, which is especially true if the studies depend on changes in absorbance at 265 nm and in pH. This may account for the disturbances seen in this study for instance in where the absorbance at 265 nm recorded for DHA conversion in 200 mM phosphate buffer pH 7.2 did not level off at zero after 40 h, and also in where a decrease in pH recorded for DHA conversion in the presence of Fe3+ in 20 mM phosphate buffer, although slower, was bigger in value at pH below 7 than above 7.
Figure 4. Representative UV-vis spectra of: (A) AA, and (B) DHA during their conversions in 200 mM phosphate buffer pH 7.2, 1 mM EDTA. The insets present kinetic curves of the change in absorbance at 265 nm over time.
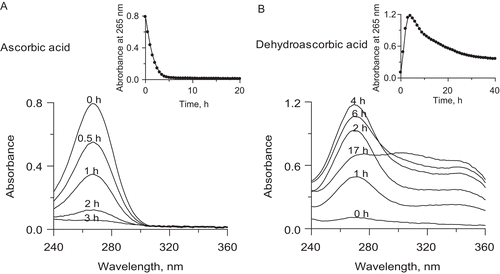
The representative UV spectra of AA and DHA recorded during their conversions in 200 mM phosphate buffer pH 7.2, are presented in , and the corresponding kinetic curves of the change in absorbance at 265 nm over time, in the insets. shows that AA disappeared from the solution by auto-oxidation during 5 h, which corresponds to the first initial time interval with no impact on urease. In the same conditions DHA also underwent degradation (). The 265 nm band first rose during 4 h, corresponding to the subsequent time interval with no impact on urease, to later decrease, and it was within this time that the inactivation of urease began (). It may therefore be suggested that to inactivate urease, it takes AA overall as much time as needed for its oxidation to DHA and for DHA to be converted to further products, altogether ~10 h. The correlations prove that neither AA nor DHA are the direct inactivators of urease and that the slow inactivation of the enzyme is provoked by a common product generated in the course of AA oxidation downstream of DHA, possibly ROS, H2O2 included.
To check this possibility we performed the same inactivations in the presence of Fe3+ ions. A concentration of 10 μM Fe3+ was chosen as submaximal noninhibitory for ureaseCitation37. As can be seen from , the addition of Fe3+ considerably speeded up the reactions, the half-lives of the inactivations being now 18 h and 2 h for AA-Fe3+ and DHA-Fe3+ systems, respectively. In a separate experiment we also observed that the activity of urease was protected by catalase practically entirely (98 %) from the inactivation by AA-Fe3+ and in 82% by DHA-Fe3+, this being strongly supportive of the involvement of ROS in the process, notably H2O2Citation27,Citation28,Citation30. To clarify the mechanism of these inactivations we further examined the quickest system, urease-DHA-Fe3+.
Inactivation of urease by DHA in the presence of Fe3+ ions
The inactivation of urease in 200 mM phosphate buffer by 10 mM DHA in the presence of 10 μM Fe3+ was studied at five pHs from the range 6.2–8.2. The results are shown in , where RA of urease is plotted against incubation time. The inactivation rate appeared to be pH-dependent, the higher was the pH the faster was the process. All the inactivation curves are sigmoidal in shape. The presence of an initial lag phase before the inflection point in the curves implies a built-up of an inhibitory species with the true inactivation of the enzyme occurring after the inflection point. When presented in semi-logarithmic plots, ln RA versus time (), the time courses show that after the inflection point the inactivations obeyed a mono-exponential decay, from which the rate constants kobs were obtained. The rate constants progressively increased at increasing pH values, as presented .
Figure 5. Urease inactivation by 10 mM DHA in the presence of 10 μM Fe3+ in 200 mM phosphate buffer at pHs in the range 6.2–8.2; (A) time courses RA versus time, and (B) semi-logarithmic plots of RA versus time. The RA values are reported as percent of the control activity.
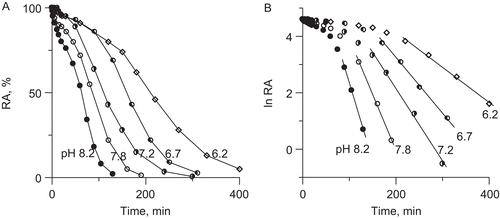
Figure 6. Effect of pH on rate constant of urease inactivation by 10 mM DHA in the presence of 10 μM Fe3+ ions.
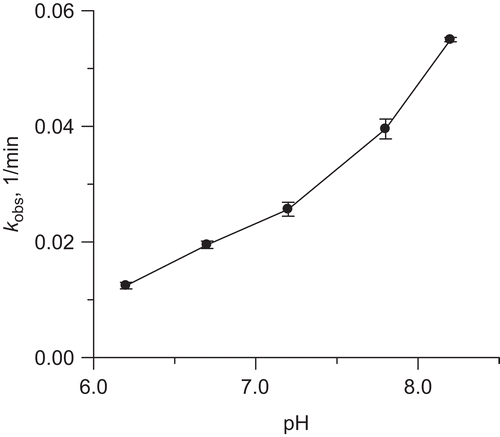
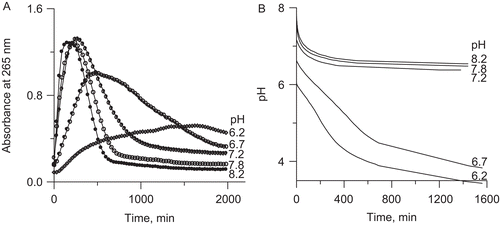
To check if this correlates with the rate of the decomposition of DHA, we monitored the DHA-Fe3+ systems at 265 nm. The results are presented in . They reveal that DHA in the presence of Fe3+ decomposed at a rate that increased with increasing pH and that the appreciable decomposition was seen at pH higher than 7. At each pH the 265 nm absorbance initially rose to further be reduced, hence indicating the formation of AA which subsequently underwent decomposition. Independently we measured the pH of the DHA-Fe3+ systems over time in 20 mM phosphate buffer selected to allow the pH to be changed in the systems. As shown in , the pH of all the solutions decreased over time consistent with the formation of acidic compounds. The decomposition was favored by higher pH values and the systems again could be grouped into those fast- and slow-decomposing above and below pH 7, respectively.
Figure 7. Kinetic curves of DHA decomposition in the presence of Fe3+ ions at different pHs in the range 6.2–8.2: (A) change in absorbance at 265 nm (27°C), and (B) change in pH in 20 mM phosphate buffer.
To further explore the mechanism of urease inactivation by DHA-Fe3+ we performed the following experiments: (i) protection of urease with catalase and SOD, (ii) protection of urease with DTT, and (iii) reactivation of the inhibited urease with DTT.
The protection with catalase, a H2O2-decomposing enzyme, and SOD, an enzyme that catalyzes the breakdown of superoxide radical anions, was done to asses if these were ROS produced from the degradation of DHA in the presence of Fe3+ that brought about the inactivation of urease. The results are presented in . It turned out that whereas SOD was hardly effective in preventing the inactivation (3–7% protection), catalase prevented the inactivation of urease by about 71–84 % in the solutions of pH between 8.2 and 6.2. Catalase and SOD alone had no effect on urease. The experiments thus suggest that for the inactivation of urease observed in the DHA-Fe3+ system, responsible are ROS, chiefly H2O2, the oxygen radicals playing a negligible role. Importantly, the inhibitory potency of H2O2 toward urease was evidenced previouslyCitation41. The inactivation by H2O2 in phosphate buffer pH 7.8 was found to be time- and concentration-dependent following a first-order kinetics with the inactivation rate constant equal to ki = 0.114 min−1. Consequently, the results of catalase and H2O2 experiments are supportive of the hypothesis that oxidation is the mechanism of urease inactivation. The oxidation is brought about mainly by the DHA-Fe3+-generated H2O2, which accounts for a specific behaviour of AA and DHA in the inhibition of urease in the presence of Fe3+ ions disparate from that in the absence of the metal ions.
The DTT protection experiments that consisted of the pre-incubation of urease with excess DTT prior to the addition of DHA and Fe3+, showed that DTT at a concentration of 5 mM afforded complete protection of urease. The action of DTT in the pre-incubation mixture apparently consisted of its covalent binding to urease thiol groups thereby protecting them from the interactions with the inactivator. This is an important observation in that it points to the thiol groups as those exclusively involved in the inactivation of the enzyme. The action of DTT could also be that of arresting the inactivating system through reducing DHA to AA and Fe3+ to Fe2+.
The reactivation of urease inhibited by DHA-Fe3+, with excess DTT, is presented in . The addition of 2 mM DTT at different times during the incubation of urease with DHA-Fe3+, was found to reactivate ∼50% of the lost activity in each case. This may mean that as a result of urease interactions with DHA-Fe3+, thiol groups in urease were oxidized in two different ways, 50% of them could be reactivated with DTT, the remaining part being modified in a way that could not be reversed by the reaction with DTT. Given the fact that thiol (-SH) groups undergo oxidation that first leads to sulfenic acid (-SOH) that can be reduced back to -SH by DTT, and further to sulfinic (-SO2H) and sulfonic acid (-SO3H) whose oxidation cannot be reversed by DTTCitation49, it seems reasonable to suppose that in the inactivation by DHA-Fe3+ 50% of urease thiol groups were oxidized to sulfenic acid and another 50%, to sulfinic and sulfonic acids.
Figure 8. Protection of urease by catalase (grey) and SOD (black) against inactivation by DHA-Fe3+.
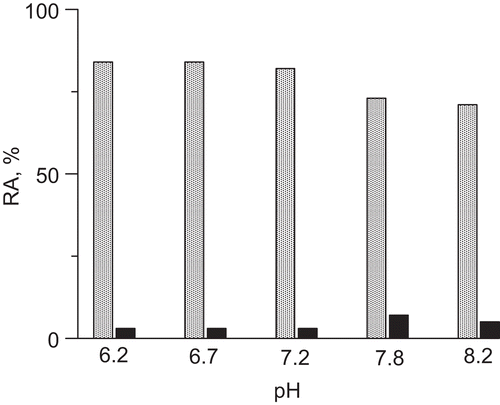
Figure 9. Reactivation of (DHA-Fe3+)-inhibited urease by DTT (pH 8.2).
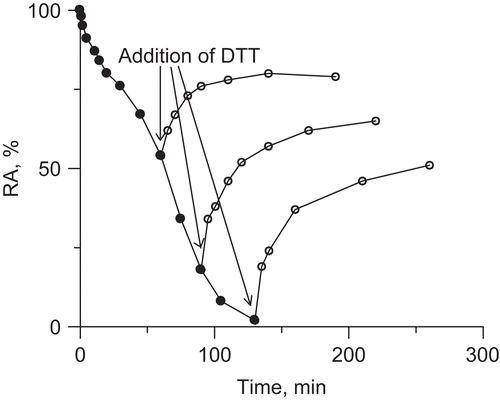
Taken together, the results of the above experiments imply that the inactivation of urease in the DHA-Fe3+ system consisted of generation of ROS, notably H2O2 that oxidized the enzyme thiol groups. Apparently, half of the groups were oxidized to sulfenic acid and the remaining half, most likely, to further thiol oxidation states. The generation of H2O2 was favored by pH values above 7.
Concluding remarks
Previous studies on the effects of AA on urease yielded conflicting results ranging from strongly inhibitoryCitation32 through moderately inhibitoryCitation24,Citation36 to entirely noninhibitoryCitation35. The discrepancies may be due to the choice of experimental conditions in which the inhibition was studied, including different unbuffered or bufferred systems, and the presence or absence of metal ions, such as Cu2+ that are known for their interference both with AA and urease. As a result, different mechanisms of AA interactions with urease were hypothesized, including DHA-modification of the proteinCitation24,Citation33,Citation36, AA-reduction of Ni2+ ions in the urease active siteCitation25,Citation36, some reactions resulting from the reduction of Cu2+ by AACitation35, and finally the reactions of Cu2+ and Cu+ with ureaseCitation34,Citation35.
In this work, we showed that in the unbuffered system AA inactivated urease by denaturation brought about by AA-lowered pH. Further we showed that in the buffered system neither AA nor DHA inhibited urease. Their inhibitory action, weak by AA and stronger by DHA, was revealed in the presence of Fe3+ ions and most importantly, unlike previously reported in the literature, it was found to be primarily mediated by H2O2. Thiol groups of urease appeared to be the target of this inhibition, as observed in some previous reportsCitation32,Citation35, but the reaction involved was determined to be the oxidation. Its effectiveness in the DHA-Fe3+-urease system was found to grow with increasing pH.
The results collected in this work clarify the behaviours of urease in the presence of AA. If they may be of practical value under physiological conditions in therapeutic suppression of ureolytic bacteria apparently requires further considerations. For instance, in the conditions of the Helicobacter pylori-infected stomach, low pH-inactivation of urease by denaturation may be limited by the fact that the pH adjacent to the bacteria in the stomach is maintained close to neutrality. The prooxidant activity of AA- and DHA systems, by contrast, to be effective requires the presence of free metal ions (Cu2+, Fe3+). These, however, in the human body primarily occur bound to proteins. Iron for instance, is essential in the human body and its overall concentration is comparatively high, but it is almost entirely complexed with proteins, such as ferritin, transferrin or hemoglobin, hence practically unavailable for the catalysis of ROS generation. Ascorbate can, however, under certain conditions release iron from some of these complexes by reduction. Then under the condition of iron availability, the locally increased pH, such as the one around Helicobacter pylori in the stomach, would facilitate the inactivation of urease. The exact biological implications of these findings and considerations, however, for urease-producing bacteria and their possible eradication remain to be established.
Declaration of interest
This work was supported by DS WCh 43 from Faculty of Chemistry of the Jagiellonian University, Krakow, Poland
References
- Krajewska B, Ureases I. Functional, kinetic and catalytic properties: a review. J Mol Catal B: Enzym 2009; 59:9–21.
- Follmer C. Ureases as a target for the treatment of gastric and urinary infections. J Clin Pathol 2010; 63:424–430.
- Riddles PW, Andrews RK, Blakeley RL, Zerner B. Jack bean urease. VI. Determination of thiol and disulfide content: Reversible inactivation of the enzyme by the blocking of the unique cysteine residue. Biochim Biophys Acta 1983; 743:115–120.
- Todd MJ, Hausinger RP. Reactivity of the essential thiol of Klebsiella aerogenes urease. Effect of pH and ligands on thiol modification. J Biol Chem 1991; 266:10260–10267.
- Benini S, Rypniewski WR, Wilson KS, Ciurli S, Mangani S. The complex of Bacillus pasteurii urease with β-marcaptoethanol from X-ray data at 1.65-Å resolution. J Biol Inorg Chem 1998; 3:268–273.
- Zhang L, Mulrooney SB, Leung AF, Zeng Y, Ko BBC, Hausinger RP, Sun H. Inhibition of urease by bismuth(III): implications for the mechanism of action of bismuth drugs. BioMetals 2006; 19:503–511.
- Zaborska W, Krajewska B, Kot M, Karcz W. Quinone-induced inhibition of urease: elucidation of its mechanisms by probing thiol groups of the enzyme. Bioorg Chem 2007; 35:233–242.
- Krajewska B. Mono- (Ag, Hg) and di- (Cu, Hg) valent metal ions effects on the activity of jack bean urease. Probing the modes of metal binding to the enzyme. J Enzyme Inhib Med Chem 2008; 23:535–542.
- Odake S, Morikawa T, Tsuchiya M, Imamura L, Kobashi K. Inhibition of Helicobacter pylori urease activity by hydroxamic acid derivatives. Biol Pharm Bull 1994; 17:1329–1332.
- Benini S, Rypniewski WR, Wilson KS, Miletti S, Ciurli S, Mangani S. A new proposal for urease mechanism based on the crystal structures of the native and inhibited enzyme from Bacillus pasteurii: why urea hydrolysis costs two nickels. Structure 1999; 7:205–216.
- Vassiliou S, Grabowiecka A, Kosikowska P, Yiotakis A, Kafarski P, Berlicki L. Design, synthesis, and evaluation of novel organophosphorus inhibitors of bacterial ureases. J Med Chem 2008; 51:5736–5744.
- Krajewska B, Zaborska W, Leszko M. Inhibition of chitosan-immobilized urease by boric acid as determined by integration methods. J Mol Catal B: Enzym 1997; 3:231–238.
- Reddy KR, Kayastha AM. Boric acid and boronic acids inhibition of pigeonpea urease. J Enzyme Inhib Med Chem 2006; 21:467–470.
- Krajewska B, Ciurli S. Jack bean (Canavalia ensiformis) urease. Probing acid-base groups of the active site by pH variation. Plant Physiol Biochem 2005; 43:651–658.
- Marshall BJ, Warren JR. Unidentified curved bacilli in the stomach of patients with gastritis and peptic ulceration. Lancet 1984; 1:1311–1315.
- Mobley HLT. (2001) Urease. In: Mobley HLT, Mendz GL, Hazell SL, editors, Helicobacter pylori: Physiology and Genetics, ASM Press, Washington, p. 179, Chapter 16.
- Crespo A, Suh B. Helicobacter pylori infection: epidemiology, pathophysiology, and therapy. Arch Pharm Res 2001; 24:485–498.
- Zhang ZW, Abdullahi M, Farthing MJG. Effect of physiological concentrations of vitamin C on gastric cancer cells and Helicobacter pylori. Gut 2002; 50:165–169.
- Drake IM, Davies MJ, Mapstone NP, Dixon MF, Schorah CJ, White KLM, Chalmers DM, Axon ATR. Ascorbic acid may protect against human gastric cancer by scavenging mucosal oxygen radicals. Carcinogenesis 1996; 17:559–562.
- Correa P, Malcom G, Schmidt B, Fontham E, Ruiz B, Bravo JC, Bravo LE, Zarama G, Realpe JL. Antioxidant micronutrients and gastric cancer. Aliment Pharmacol Ther 1998; 12 Suppl 1:73–82.
- Zhang HM, Wakisaka N, Maeda O, Yamamoto T. Vitamin C inhibits the growth of a bacterial risk factor for gastric carcinoma: Helicobacter pylori. Cancer 1997; 80:1897–1903.
- Jarosz M, Dzieniszewski J, Dabrowska-Ufniarz E, Wartanowicz M, Ziemlanski S, Reed PI. Effects of high dose vitamin C treatment on Helicobacter pylori infection and total vitamin C concentration in gastric juice. Eur J Cancer Prev 1998; 7:449–454.
- Goldie J, Jalali S, Van Zanten S, Stowe C, Hunt RH. Ascorbic acid inhibits the growth and urease activity of Campylobacter pylori. Gut 1989; 30:A1484.
- Nilius M, Bode G, Lehnhardt G, Malfertheiner P. In vitro inhibition of Helicobacter pylori urease: biochemical and ultrastructural analysis. Eur J Clin Invest 1991; 21:551–557.
- Agha A, Opekun AR, Abudayyeh S, Graham DY. Effect of different organic acids (citric, malic and ascorbic) on intragastric urease activity. Aliment Pharmacol Ther 2005; 21:1145–1148.
- Ogata Y, Kosugi Y. Ultraviolet spectra of L-ascorbic acid and cupric ascorbate complex. Tetrahedron 1970; 26:4711–4716.
- Stadtman ER. Ascorbic acid and oxidative inactivation of proteins. Am J Clin Nutr 1991; 54:1125S–1128S.
- Halliwell B. Vitamin C: poison, prophylactic or panacea? Trends Biochem Sci 1999; 24:255–259.
- Deutsch JC, Santhosh-Kumar CR. Dehydroascorbic acid undergoes hydrolysis on solubilization which can be reversed with mercaptoethanol J Chromatogr 1996; A724:271–278.
- Khan MMT, Martell AE. Metal ion and metal chelate catalyzed oxidation of ascorbic acid by molecular oxygen. I. Cupric and ferric ion catalyzed oxidation. J Am Chem Soc 1967; 89:4176–4185.
- Samuni A, Aronovitch J, Godinger D, Chevion M, Czapski G. On the cytotoxicity of vitamin C and metal ions. A site-specific Fenton mechanism. Eur J Biochem 1983; 137:119–124.
- Elson LA. Inhibition of urease activity by ascorbic acid. Nature 1943; 152:149.
- Quastel JH. Urease activity and ascorbic acid. Nature 1943; 152:215.
- Giri KV, Seshagiri R. Urease activity and ascorbic acid. Nature 1946; 153:253–254.
- Mapson LW. Inhibition of urease by copper salts in the presence of ascorbic acid and related substances. Biochem J 1946; 40:240–247.
- Chiyoda T, Iida K, Takatori K, Kajiwara M. Screening system for urease inhibitors using 13C-NMR. Chem Pharm Bull 1998; 46:718–720.
- Krajewska B. Urease immobilized on chitosan membrane. Inactivation by heavy metal ions. J Chem Tech Biotechnol 1991; 52:157–162.
- Weatherburn MW. Phenol-hypochlorite reaction for determination of ammonia. Anal Chem 1967; 39:971–974.
- Todd MJ, Hausinger RP. Purification and characterization of the nickel-containing multicomponent urease from Klebsiella aerogenes. J Biol Chem 1987; 262:5963–5967.
- Srivastava PK, Kayastha AM. Significance of sulfhydryl groups in the activity of urease from pigeonpea (Cajanus cajan L.) seeds. Plant Sci 2000; 159:149–158.
- Krajewska B, Zaborska W. Double mode of inhibition-inducing interactions of 1,4-naphthoquinone with urease: arylation versus oxidation of enzyme thiols. Bioorg Med Chem 2007; 15:4144–4151.
- Srivastava PK, Kayastha AM, Reddy RC, Grant DF. Molecular asymmetry in pigeonpea urease: pH inactivation studies. J Plant Biochem Biotechnol 2003; 12:49–51.
- Dixon NE, Gazzola C, Asher CJ, Lee DSW, Blakeley RL, Zerner B. Jack been urease (EC 3.5.1.5). II. The relationship between nickel, enzymatic activity, and the “abnormal” ultraviolet spectrum. The nickel content of jack beans. Can J Biochem 1980; 58:474–480.
- Bauerfeind P, Garner R, Dunn BE, Mobley HLT. Synthesis and activity of Helicobacter pylori urease and catalase at low pH. Gut 1997; 40:25–30.
- Goto Y, Takahashi N, Fink AL. Mechanism of acid-induced folding of proteins. Biochemistry 1990; 29:3480–3488.
- Bhowmick R, Jagannadham MV. Multiple intermediate conformations of jack bean urease at low pH: anion-induced refolding. Protein J 2006; 25:399–410.
- Jung CH, Wells WW. Spontaneous conversion of L-dehydroascorbic acid to L-ascorbic acid and L-erythroascorbic acid. Arch Biochem Biophys 1998; 355:9–14.
- Deutsch JC. Dehydroascorbic acid. J Chromatogr A 2000; 881:299–307.
- Hancock J, Desikan R, Harrison J, Bright J, Hooley R, Neill S. Doing the unexpected: proteins involved in hydrogen peroxide perception. J Exp Bot 2006; 57:1711–1718.