Abstract
Human uridine-5′-diphosphoglucuronosyltransferases (UGTs) are the major phase II metabolizing enzymes. In the present study, five human UGTs (UGT1A1, 1A4, 1A6, 2B7, and 2B10) were individually expressed and used to examine the inhibition IC50 values of 20 selective substrates and inhibitors of major cytochromes P450 (CYPs). The inhibition kinetics of UGT1A1 was also analyzed. The results showed that some compounds like α-naphthoflavone, paclitaxel, midazolam, cyclosporine A, and ketoconazole displayed strong inhibitions on UGT activities with their IC50 values in a range of 4.1–26 µM. Especially, the IC50 values were 4.1 ± 0.8 µM for ketoconazole in inhibiting UGT1A1-mediated β-estradiol-3-glucuronidation, and 4.9 ± 0.3 µM for paclitaxel towards UGT1A4-mediated midazolam-N-glucuronidation. Additionally, the IC50 values of bupropion, tolbutamide, and testosterone in inhibiting UGT-mediated metabolisms were similar with the Km values of respective CYPs. Some kinetic behaviours of UGTs were following Michaelis–Menten kinetics, while some were not.
Introduction
Cytochromes P450 (CYPs) and uridine-5′-diphosphoglucuronosyltransferases (UGTs) are the major phase I and II drug metabolism enzymes. These two super enzyme families play important roles in drug metabolism by metabolizing endogenous and exogenous compounds to generate metabolites, which are water-soluble and more easily excreted in bile and urine compared to the parent compounds. CYPs catalyze the oxidative reactions, like the hydroxylation,Citation1 which are commonly ready for phase II enzymes such as UGTs. UGTs are responsible for the glucuronic acid conjugation via hydroxyl, carboxyl, sulfuryl, carbonyl, and amino linkages.Citation2 They also can catalyze many chemicals directly without reactions of CYPs. Since the glucuronidation reactions may associate with important biological activities and severe clinical influence, it is necessary to evaluate the metabolism of UGTs. There are two subfamilies of UGT1A and UGT2B.
The glucuronide conjugation catalyzed by UGTs may enhance the biological effects of drugs. It is well known that morphine 6-O-glucuronide, the metabolite of morphine catalyzed by UGT2B7, displayed analgesic effect, approximately 600 times more effective than morphine.Citation3,Citation4 In contrast, the acyl-glucuronides of non-steroidal anti-inflammatory drugs could lead to formation of DNA and protein adducts to induce severe hepatotoxic and immunological toxicity.Citation5,Citation6 The deficiencies and polymorphism of UGTs could also relate to severe diseases, such as the Crigler–Najjar type I and II syndromes as well as the Gilbert’s syndrome, associated with genetic deficiencies of UGT1A1.Citation7,Citation8 It is well known that UGT1A1 is the major enzyme responsible for elimination of bilirubin. Phase II enzyme-mediated drug metabolisms have been paid much more attention in current study.
The glucuronide conjugation could also lead to severe pharmacokinetic drug interactions. Gemfibrizil 1-O-β glucuronide is a much more potent inhibitor to CYP2C8 compared to its parent compound, gemfibrizil (IC50 values: 24 µM for gemfibrizil vs 1.8 µM for gemfibrizil 1-O-β glucuronide).Citation9 This metabolite would mainly respond for the drug interaction risk by a 5-fold increase of cerivastatin area under the curve when they were co-administration in clinics.Citation10 Additionally, a glucuronidation metabolite of zidovudine catalyzed by UGT2B7 led to mitochondrial toxicity when patients were given high doses zidovudine (1000 mg or more per day).Citation11 Co-administration of fluconazole or valproic acid with zidovudine would increase this toxicity risk.Citation12,Citation13 Thus, drug interaction potentials caused by CYPs and/or UGTs should be paid much more attention. In addition, in vitro assays for UGT activities would help to mimic the simultaneous CYP and UGT functions in vivo.
In the present study, the genes of major human UGTs (UGT1A1, 1A4, 1A6, 2B4, 2B7, and 2B10) were cloned and they were constructed into the insect cell expression systems. The expressed microsomal fractions of each UGT were isolated and used to evaluate their kinetic parameters. The inhibition potentials of 20 selective substrates and inhibitors of CYPs were also studied with these recombinant human UGTs.
Materials and methods
Materials
β-Estradiol, β-estradiol 3-(β-d-glucuronide) sodium salt, uridine 5′-diphosphoglucuronic acid (UDPGA), alamethicin from trichodermavirvide, d-saccharic acid 1,4-lactone monohydrate, 1-naphthol, flufenamic acid, ketoconazole, testosterone, oleandomycin triacetate, α-naphthoflavone, phenacetin, furafylline, coumarin, s-mephentoin, bupropion, tolbutamide, sulfaphenazole, dextromethorphan, quinidine, cyclosporine A, omeprazole, chlorzoxazone, chlormethiazole, and diclofenac, were purchased from Sigma-Aldrich (St. Louis, MO). Midazolam maleate and paclitaxel were purchased from National Institute for the Control of Pharmaceutical and Biological Products (Beijing, PR China). Acetonitrile, methnol, and formic acid [high-performance liquid chromatography (HPLC) grade] were purchased from Dikma Technology Inc. (Lake Forest, CA). All other reagents and HPLC solvents were of highest quality commercially available.
The expression of recombinant human UGTs and the preparation of microsomes
The recombinant human UGTs were expressed in Baculovirus-Transfected insect cell system (sf9). The clones of individual UGT gene were from FulenGen Company (Guangzhou, China), and the Baculovirus was from Invitrogen (Carlsbad, CA). After the construction of Bacmids, DNA sequence analysis and Western-blot (rabbit anti-human UGT polyclonal antibody from Cypex, Dundee, Scotland, UK) were used to confirm each expressed UGT sequence matching with gene bank (data not shown). Each UGT was expressed in sf9 cells based on methods previously reported.Citation14,Citation15
Each human UGT enzyme was expressed for several times and all pooled together. After expression of each UGT, microsomes were prepared by differential centrifugation method. The total concentrations of protein were determined with the method of Lowry et al.,Citation16 using bovine serum albumin as the standard. These enzymes were stocked in the concentrations of 10–20 mg/mL. The final protein concentrations used in the enzyme reactions were 0.3 mg/mL, including the studies of Km and inhibition.
Km
The incubation mixture (135 µL) contained microsomal protein (0.3 mg/mL), substrates (0–200 µM for β-estradiol, 0–180 µM for midazolam, 0–750 µM for diclofenac, and 1-naphthol, respectively), MgCl2 (5 mM), alamethicin (0.025 mg/mL), 1,4-lactone (5 mM), and phosphate buffer (0.1 M, pH 7.4, contained 0.1 mM ethylenediaminetetraacetic acid). Reactions were initiated by adding UDPGA (final concentration: 5 mM) to the mixture after pre-incubation at 37°C for 5 min. The reaction mixtures were incubated at 37°C for 30 min, and then stopped with the addition of equal volume acetonitrile (containing 0.2% formic acid and 50 nM flufenac acid as internal standard). The reactions were performed with Tecan Freedom EVOlyzer.Citation17 All the samples were triplicate, and each reaction was repeated. After being centrifugated at 1000g for 5 min on the filter plate, 20 µL filtrated samples were injected into the mass specrometry for liquid chromatography-mass spectrometry (LC-MS/MS) analysis.
IC50
Each expressed human UGT enzymes were prepared as one lot for all the inhibitions studies and enzyme kinetic experiments. The substrate concentration was prepared approximately around the Km. Inhibitor was prepared in the final concentrations of 0, 0.5, 2, 10, 20, 50, 150, and 300 µM, respectively. The stock solution of inhibitor was prepared in dimethyl sulfoxide, which concentration was controlled at 0.5%. The structures of 20 inhibitors for human UGTs are shown in .
Figure 1. The structures of the 20 chemicals studied as the inhibitors of human UGTs.
Inhibition type analysis
For inhibition type estimation, the substrate concentrations of UGT1A1 were prepared at 5, 10, 20, and 40 µM, while the inhibitor concentrations were in a series of 0, 10, 20, 50, and 100 µM for midazolam, testosterone, oleandomycin, paxlitaxel, and α-naphthoflavone, 0, 15, 30, 75, and 150 µM for omeprazole, and 0, 1.5, 3, 7.5, and 15 µM for ketoconazole, respectively.
Preparation of glucuronide metabolites
Glucuronide metabolites were prepared to perform the LC-MS/MS determination.Citation18 The compositions of incubation mixtures were similar with the assays of Km value determination, except that the concentrations of substrate and microsome were 1 mM and 1 mg/mL. Two-fold of the incubation volume of ice-cold acetonitrile, containing 0.2% formic acid (v/v) was added to the incubation mixture to stop the reactions. Then the reaction mixtures were centrifuged at 16,000 rpm for 30 min. The supernatants were collected and subjected to the solid phase extraction system, with Strata C18-E cartridge from Phenomenex (100 mg/mL; Torrance, CA). The column was conditioned with 3 mL of methanol followed by 1 mL of water. After loading the samples, polar impurities were removed with 1 mL of water and the samples were eluted with 1 mL of methanol.
LC-MS/MS conditions
Determinations of all analytes were carried out with an LC-MS/MS system, which consists of an HPLC apparatus (Shimadzu, Japan, including a SCL-10ADvp controller, two LC-10ADvp pumps and a DGU-14A solvent degasser), and a PE/SCIEX Applied Biosystems API 3000 triple-quadrupole mass spectrometer (Foster City, CA) equipped with electrospray ionization source. A MPS3C auto sampler (GERSTEL, Mülheim an der Ruhr, Germany) was used for sample injection. The HPLC column was a Gemini C18 110A column (2.0 × 100 mm, 5.0 µm) equipped with an ODS guard column (4 mm × 2.0 mm i.d.) from Phenomenex.
Mobile phases of A: acetonitrile/H2O (5/95) and B: acetonitrile/H2O (95/5) were employed and 0.1% formic acid were used in all chromatography runs. A gradient program started at 10% of B and increased to 30% until 0.8 min. It was followed by linear gradient to 100% at 2.4 min and this was remained. The flow rate was 0.2 mL/min, and the column temperature was ambient.
The multiple reaction monitoring acquisitions and positive ionization mode were used to monitor the metabolites except midazolam-N-glucuronide metabolized by UGT1A4, which was monitored with the negative ionization mode. Additionally, IS could be analyzed with both negative and positive modes. The glucoronide metabolites were monitored as m/z: 447.1/271.1 for β-estradiol-3-glucuronide (UGT1A1), 319.3/143.2 for 1-naphthol-glucuronide (UGT1A6), 469.9/193.2 for diclofenac-glucuroinde (UGT2B7 and 2B10) and the positive ion m/z: 502.3/326.2 for midazolam-N-glucuronide, respectively. Both m/z: 280/236.1 (negative) and 282/264 (positive) were used for IS. Mass spectrometric conditions were optimized to obtain maximum sensitivity. Data was acquired using the Analyst 1.4 software.
Data analysis
The Km and Vmax values were determined by Michaelis–Menten kinetics (Equation 1) and Hill model (Equation 2) with Grafit, version 5.0 and Origin 7.5.
The IC50 value (half-maximal inhibitory concentration) of each inhibitor from the inhibitions towards multiple UGTs’ glucuronidation was estimated by fitting the data to the equation 3 using Grafit, version 5.0.
Where I represents the inhibitor concentration and s stands for the slope factor. The Eadie–Hofstee plots were generated to analysis the inhibition for UGT1A1.
Results and discussion
The substrates selection, Km and Vmax values of five UGTs
It has been well known that β-estradiol-3-glucuronidation is selectively catalyzed by UGT1A1,Citation19 midazolam-N-glucuronidation by UGT1A4,Citation20 1-naphthol-glucuronidation by UGT1A6,Citation21 and diclofenac-glucuronidation non-selectively by UGT2B7Citation22 and 2B10, respectively. These four substrates were thus used for these five UGTs. The enzyme kinetic behaviours of UGT-mediated glucuronidations would follow Michaelis–Menten model and/or Hill model.Citation19,Citation21,Citation22 In the present study, both equations were applied to calculate the Km and Vmax values. The results are shown in . The plots of velocity versus substrate concentration and plots of Eadie–Hofstee are also shown in .
Table 1. Km and Vmax values for five expressed human UGTs*
Figure 2. The enzyme kinetics of expressed human UGT1A1, 2B7, 1A4, and 1A6. UGT1A1-mediated β-estradiol-3-glucuronidation (A), UGT2B7-mediated diclofenac-glucuronidation (B), UGT1A4-mediated midazolam-N-glucuronidation (C), and UGT1A6-mediated 1-naphthol-glucuronidation (D) were fitted with Michaelis–Menten model and Eadie–Hofstee plots, respectively.
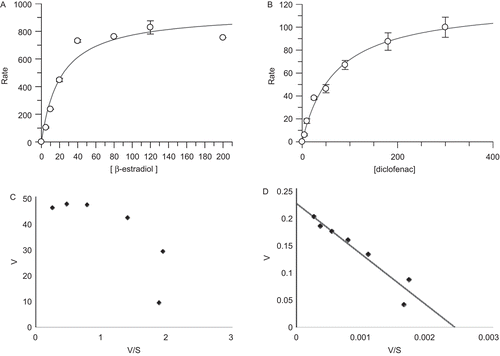
Monitoring the metabolite formation, the Km and Vmax were determined to be 21 ± 6 µM and 937 ± 77 pmol/min/mg for UGT1A1-mediated β-estradiol-3-glucuronidation, respectively. The reaction was fitted with Michaelis–Menten kinetic model (). The Km values of these UGT-mediated reactions were determined to be 13 ± 4, 84 ± 7, 69 ± 9, and 56 ± 12 µM for UGT1A4, 1A6, 2B7 (), and 2B10, respectively. Since their metabolite standards of 1-naphthol-glucuronide, midazolam-N-glucuronide, and diclofenac-glucuronide were not commercially available, the regression Vmax values were not determined. Additionally, the β-estradiol-17-glucuronidation was also studied for UGT2B7, and the Km and Vmax values were measured to be 28 ± 5 µM and 15 ± 1 pmol/min/mg, respectively (data not shown). The Km value of UGT2B7-mediated diclofenac-glucuronidation was determined to be 69 ± 9 µM, approximately 2-fold of the Km for β-estradiol-17-glucuronidation. The S50 values of the Hill equation for UGT1A1 and UGT1A4 were determined to be 16 ± 2 and 11 ± 0.6 µM, respectively. The S50 values of the Hill equation for UGT1A6 and UGT2B7 were similar to their Km values fitted with Michaelis–Menten equation. UGT2B10 was not convergent when fitted with Hill equation (data not shown).
The data from these UGT-mediated glucuronidations were used to calculate their Km values according to Michaelis–Menten model. However, their Eadie–Hofstee plots were displayed differently as shown in . Eadie–Hofstee plots for UGT1A1 and 1A4 () exhibited hooked-like curves. This was consistent with the previous studies of UGT1A1-mediated β-estradiol-3-glucuronidation which was fitted with Hill equation.Citation19,Citation23 In contrast, UGT1A4-mediated midazolam-N-glucuronidation was reported to fit with Michaelis–Menten kinetics and the substrate inhibition model.Citation20,Citation24 Additionally, almost linear relationships in Eadie–Hofstee plots, except for the lowest concentration points of the substrates, were fitted for UGT2B7- and 1A6-mediated glucuronidations (), where the substrates depletion with more than 15% and/or the protein-bondings may be taken into account.
The IC50 values of five UGTs
The inhibitory potentials of 20 compounds which are the selective substrates and inhibitors of CYPsCitation25 were investigated with five recombinant human UGTs: UGT1A1, UGT1A4, UGT1A6, UGT2B7, and UGT2B10. Some of these compounds displayed weak inhibitions to UGT-mediated glucuronidations, like coumarin, which was reported to show strong inhibition towards carbonic anhydrase recently,Citation26,Citation27 exhibited weak inhibition towards all the five UGTs, with IC50 values exceed 300 µM. But some of these tested chemicals exhibited strong inhibitory potentials. As shown in and , the IC50 value of 23 ± 6 µM was measured for α-naphthoflavone inhibiting UGT1A4-mediated metabolism (); the IC50 values of 4.9 ± 0.3 and 20 ± 3 µM were for paclitaxel inhibiting UGT1A4 () and UGT1A1, respectively; the IC50 values of 23 ± 1 and 26 ± 5 µM were for midazolam inhibiting UGT1A6 () and UGT1A1, respectively; the IC50 value of 23 ± 7 µM was for cyclosporine A inhibiting UGT1A4 (); the IC50 values of 4.1 ± 0.8, 19 ± 2, and 22 ± 5 µM were for ketoconazole inhibiting UGT1A1 (), UGT1A6, and UGT2B10, respectively.
Table 2. Determination of the IC50 values*.
Figure 3. The inhibition plots of expressed human UGT1A4-mediated midazolam-N-glucuronidation by α-naphthoflavone (A), and paclitaxel (B); UGT1A6-mediated 1-naphthol-glucuronidation by midazolam (C); UGT1A4-mediated midazolam-N-glucuronidation by cyclosporine A (D); UGT1A1-mediated β-estradiol-3-glucuronidation by ketoconazole (E). Their inhibition IC50 values were calculated to be 23 ± 6, 4.9 ± 0.3, 23 ± 1, 23 ± 7, and 4.1 ± 0.8 µM, respectively.
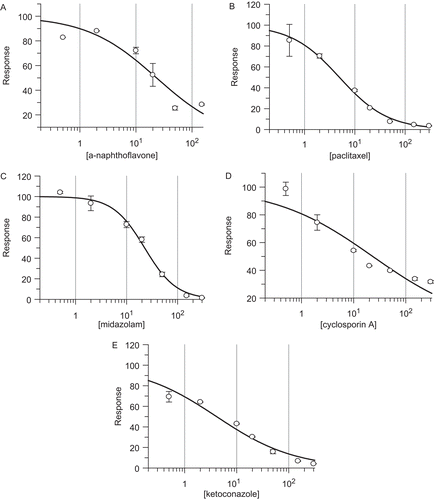
Especially, it is known that the maximal serum concentration of ketoconazole in human is in a range of 4.2–6.2 µg/mL (7.9–11.7 µM)Citation28 under 200 mg single dose treatment, and approximately 4.43 µg/mL (5.2 µM) for paclitaxel following i.v. administration of Genetaxyl,Citation29 comparing to their IC50 values of 4.1 ± 0.8 µM for ketoconazole inhibiting UGT1A1 and 4.9 ± 0.3 µM for paclitaxel inhibiting UGT1A4, respectively. The significant risk of clinical drug interactions caused by these UGTs should be investigated more deeply in the future. Furthermore, the IC50 values of some CYP-substrates inhibiting UGT activities are in the similar range of their Km values of CYP-mediated drug metabolism. Bupropion exhibited a Km value of 82 µM for CYP2B6Citation30 and an IC50 value of 88 ± 28 µM in UGT2B10-mediated diclofenac metabolism, while tolbutamide displayed a Km of 181 µM for CYP2C9Citation31 and an IC50 of 281 ± 69 µM in UGT1A6-mediated 1-naphthol glucorunidation. Additionally, testosterone with a Km of 80 µM for CYP3A4/5Citation17 displayed an IC50 value of 43 ± 14 µM in UGT1A1-mediated β-estradiol-3-glucorunidation. Therefore, the significance of their UGT inhibition potency in clinics, while they are metabolized through CYP-mediated pathways, would be interested for the future study. Regarding the selective inhibitors of CYPs, their IC50 values in UGTs determined in this study were much higher than those in CYP inhibition (a range of 0.001–10 µM).Citation30
The inhibitions kinetics of UGT1A1
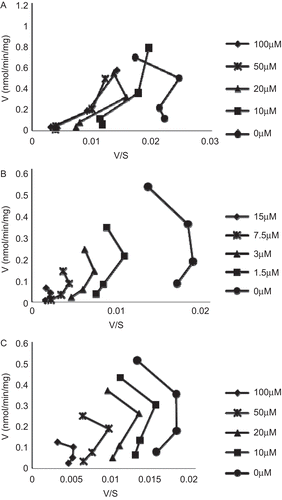
The inhibition kinetics of the following seven chemicals, paclitaxel, ketocazole, midazolam, omeprazole, α-naphthoflavone, oleandomycin, and testosterone, were studied towards UGT1A1-mediated β-estradiol-3-glucuronidation. Eadie–Hofstee plots are shown in , indicating mixing type inhibitions. The increasing concentrations of paclitaxel shifted the shape of the line from a curve to a more linear relationship in the Eadie–Hofstee plots (), similar to previously reported the effect of 17α-ethynylestradiol on UGT1A1-mediated β-estradiol-3-glucuronidation.Citation23 On the other hand, the inhibition connecting lines exhibited a similar shape patterns in the Eadie–Hofstee plots for ketoconazole and midazolam when the inhibitor concentrations were increased ().
Figure 4. Eadie–Hofstee plots for inhibitions kinetics of expressed human UGT1A1-mediated β-estradiol-3-glucuronidation by paclitaxel (A), ketoconazole (B), and midazolam (C). The concentrations of inhibitors were as shown in the figures, and the substrate concentrations were set at 5, 10, 20, and 40 µM for β-estradiol.
Conclusion
The β-estradiol-3-glucuronidation, midazolam-N-glucuronidation, 1-naphthol-glucuronidation, and diclofenac-glucuronidation were used to evaluate the Km values of the recombinant human UGT1A1, 1A4, 1A6, and 2B7/2B10, respectively. The Vmax values of β-estradiol-3-glucorunidation and β-estradiol-17-glucorunidation were also determined for UGT1A1 and UGT2B7, respectively. Twenty selective substrates and inhibitors for CYP metabolism were used to estimate their potentials on inhibiting UGT-mediated glucorunidation activities. Interestingly, some IC50 values determined in this study are similar to their Km range of CYP activities, such as bupropion associated with UGT2B10 and CYP2B6 (IC50: 88 ± 28 µM vs Km: 82 µM), tolbutamide associated with UGT1A6 and CYP2C9 (IC50: 281 ± 69 µM vs Km: 181 µM); and testosterone associated with UGT1A1 and CYP3A4/5 (IC50: 43 ± 14 µM vs Km: 80 µM). The results of inhibitions also indicated that ketoconazole and paclitaxel had strong inhibitory potentials on UGTs, especially UGT1A1 (IC50: 4.1 ± 0.8 µM) and UGT1A4 (IC50: 4.9 ± 0.3 µM), respectively.
Acknowledgements
The authors are very grateful for the help and cooperation from Yan Chen and Haitao Wang.
Declaration of Interest
This work was supported in part by the National High-tech R&D Program [Grant 2006AA02Z339] and [Grant 2008AA02Z314]; the Guangzhou Science and Technology Bureau [Grant 2006Z1-E4031, 2006P067]; and the Guangzhou Development District [Grant 2006Ss-P067].
References
- Hasler JA. Pharmacogenetics of cytochromes P450. Mol Aspects Med 1999;20:12–24, 5–137.
- Radominska-Pandya A, Czernik PJ, Little JM, Battaglia E, Mackenzie PI. Structural and functional studies of UDP-glucuronosyltransferases. Drug Metab Rev 1999;31:817–899.
- Pasternak GW, Bodnar RJ, Clark JA, Inturrisi CE. Morphine-6-glucuronide, a potent mu agonist. Life Sci 1987;41:2845–2849.
- Radominska-Pandya A, Bratton S, Little JM. A historical overview of the heterologous expression of mammalian UDP-glucuronosyltransferase isoforms over the past twenty years. Curr Drug Metab 2005;6:141–160.
- Terrier N, Benoit E, Senay C, Lapicque F, Radominska-Pandya A, Magdalou J, Fournel-Gigleux S. Human and rat liver UDP-glucuronosyltransferases are targets of ketoprofen acylglucuronide. Mol Pharmacol 1999;56:226–234.
- Shipkova M, Armstrong VW, Oellerich M, Wieland E. Acyl glucuronide drug metabolites: toxicological and analytical implications. Ther Drug Monit 2003;25:1–16.
- Kadakol A, Ghosh SS, Sappal BS, Sharma G, Chowdhury JR, Chowdhury NR. Genetic lesions of bilirubin uridine-diphosphoglucuronate glucuronosyltransferase (UGT1A1) causing Crigler–Najjar and Gilbert syndromes: correlation of genotype to phenotype. Hum Mutat 2000;16:297–306.
- Guillemette C. Pharmacogenomics of human UDP-glucuronosyltransferase enzymes. Pharmacogenomics J 2003;3:136–158.
- Ogilvie BW, Zhang D, Li W, Rodrigues AD, Gipson AE, Holsapple J, Toren P, Parkinson A. Glucuronidation converts gemfibrozil to a potent, metabolism-dependent inhibitor of CYP2C8: implications for drug-drug interactions. Drug Metab Dispos 2006;34:191–197.
- Backman JT, Kyrklund C, Neuvonen M, Neuvonen PJ. Gemfibrozil greatly increases plasma concentrations of cerivastatin. Clin Pharmacol Ther 2002;72:685–691.
- Benbrik E, Chariot P, Bonavaud S, Ammi-Saïd M, Frisdal E, Rey C, Gherardi R, Barlovatz-Meimon G. Cellular and mitochondrial toxicity of zidovudine (AZT), didanosine (ddI) and zalcitabine (ddC) on cultured human muscle cells. J Neurol Sci 1997;149:19–25.
- Trapnell CB, Klecker RW, Jamis-Dow C, Collins JM. Glucuronidation of 3′-azido-3′-deoxythymidine (zidovudine) by human liver microsomes: relevance to clinical pharmacokinetic interactions with atovaquone, fluconazole, methadone, and valproic acid. Antimicrob Agents Chemother 1998;42:1592–1596.
- Barbier O, Turgeon D, Girard C, Green MD, Tephly TR, Hum DW, Bélanger A. 3′-azido-3′-deoxythimidine (AZT) is glucuronidated by human UDP-glucuronosyltransferase 2B7 (UGT2B7). Drug Metab Dispos 2000;28:497–502.
- Nguyen N, Tukey RH. Baculovirus-directed expression of rabbit UDP-glucuronosyltransferases in Spodoptera frugiperda cells. Drug Metab Dispos 1997;25:745–749.
- Yueh MF, Nguyen N, Famourzadeh M, Strassburg CP, Oda Y, Guengerich FP, Tukey RH. The contribution of UDP-glucuronosyltransferase 1A9 on CYP1A2-mediated genotoxicity by aromatic and heterocyclic amines. Carcinogenesis 2001;22:943–950.
- Lowry OH, Rosebrough NJ, Farr AL, Randall RJ. Protein measurement with the Folin phenol reagent. J Biol Chem 1951;193:265–275.
- Yao M, Zhu M, Sinz MW, Zhang H, Humphreys WG, Rodrigues AD, Dai R. Development and full validation of six inhibition assays for five major cytochrome P450 enzymes in human liver microsomes using an automated 96-well microplate incubation format and LC-MS/MS analysis. J Pharm Biomed Anal 2007;44:211–223.
- Soars MG, Mattiuz EL, Jackson DA, Kulanthaivel P, Ehlhardt WJ, Wrighton SA. Biosynthesis of drug glucuronides for use as authentic standards. J Pharmacol Toxicol Methods 2002;47:161–168.
- Fisher MB, Campanale K, Ackermann BL, VandenBranden M, Wrighton SA. In vitro glucuronidation using human liver microsomes and the pore-forming peptide alamethicin. Drug Metab Dispos 2000;28:560–566.
- Klieber S, Hugla S, Ngo R, Arabeyre-Fabre C, Meunier V, Sadoun F, Fedeli O, Rival M, Bourrie M, Guillou F, Maurel P, Fabre G. Contribution of the N-glucuronidation pathway to the overall in vitro metabolic clearance of midazolam in humans. Drug Metab Dispos 2008;36:851–862.
- Uchaipichat V, Mackenzie PI, Guo XH, Gardner-Stephen D, Galetin A, Houston JB, Miners JO. Human udp-glucuronosyltransferases: isoform selectivity and kinetics of 4-methylumbelliferone and 1-naphthol glucuronidation, effects of organic solvents, and inhibition by diclofenac and probenecid. Drug Metab Dispos 2004;32:413–423.
- King C, Tang W, Ngui J, Tephly T, Braun M. Characterization of rat and human UDP-glucuronosyltransferases responsible for the in vitro glucuronidation of diclofenac. Toxicol Sci 2001;61:49–53.
- Williams JA, Ring BJ, Cantrell VE, Campanale K, Jones DR, Hall SD, Wrighton SA. Differential modulation of UDP-glucuronosyltransferase 1A1 (UGT1A1)-catalyzed estradiol-3-glucuronidation by the addition of UGT1A1 substrates and other compounds to human liver microsomes. Drug Metab Dispos 2002;30:1266–1273.
- Hyland R, Osborne T, Payne A, Kempshall S, Logan YR, Ezzeddine K, Jones B. In vitro and in vivo glucuronidation of midazolam in humans. Br J Clin Pharmacol 2009;67:445–454.
- Bjornsson TD, Callaghan JT, Einolf HJ, Fischer V, Gan L, Grimm S, Kao J, King SP, Miwa G, Ni L, Kumar G, McLeod J, Obach RS, Roberts S, Roe A, Shah A, Snikeris F, Sullivan JT, Tweedie D, Vega JM, Walsh J, Wrighton SA. The conduct of in vitro and in vivo drug-drug interaction studies: a Pharmaceutical Research and Manufacturers of America (PhRMA) perspective. Drug Metab Dispos 2003;31:815–832.
- Maresca A, Temperini C, Vu H, Pham NB, Poulsen SA, Scozzafava A, Quinn RJ, Supuran CT. Non-zinc mediated inhibition of carbonic anhydrases: coumarins are a new class of suicide inhibitors. J Am Chem Soc 2009;131:3057–3062.
- Maresca A, Temperini C, Pochet L, Masereel B, Scozzafava A, Supuran CT. Deciphering the mechanism of carbonic anhydrase inhibition with coumarins and thiocoumarins. J Med Chem 2010;53:335–344.
- Huang YC, Colaizzi JL, Bierman RH, Woestenborghs R, Heykants J. Pharmacokinetics and dose proportionality of ketoconazole in normal volunteers. Antimicrob Agents Chemother 1986;30:206–210.
- Chu Z, Chen JS, Liau CT, Wang HM, Lin YC, Yang MH, Chen PM, Gardner ER, Figg WD, Sparreboom A. Oral bioavailability of a novel paclitaxel formulation (Genetaxyl) administered with cyclosporin A in cancer patients. Anticancer Drugs 2008;19:275–281.
- Walsky RL, Obach RS. Validated assays for human cytochrome P450 activities. Drug Metab Dispos 2004;32:647–660.
- Tang C, Lin Y, Rodrigues AD, Lin JH. Effect of albumin on phenytoin and tolbutamide metabolism in human liver microsomes: an impact more than protein binding. Drug Metab Dispos 2002;30:648–654.