Abstract
Two series of pyridine derivatives were synthesised and evaluated for their in vivo anti-malarial activity against Plasmodium berghei. The anti-malarial activity was determined in vivo by applying 4-day standard suppressive test using chloroquine (CQ)-sensitive P. berghei ANKA strain–infected mice. Compounds 2a, 2g and 2h showed inhibition of the parasite multiplication by 90, 91 and 80%, respectively, at a dose level of 50 µmol/kg. Moreover, The most active compounds (2a, 2g and 2h) were tested in vitro against CQ-resistant Plasmodium falciparum RKL9 strains where compound 2g showed promising activity with IC50 = 0.0402 µM. The compounds were non-toxic at 300 and 100 mg/kg through the oral and parenteral routes, respectively. The docking pose of the most active compounds (2a, 2g and 2h) in the active site of dihydrofolate reductase enzyme revealed several hydrogen and hydrophobic interactions that contribute to the observed anti-malarial activities.
Introduction
Malaria has infected humans and may have been a human pathogen for the entire history of the mankindCitation1. There are a limited number of drugs that can be used to treat or prevent malaria, but increased resistance to these drugs is of concern to health practitioners. This resistance appears to occur through spontaneous mutations that confer reduced sensitivity to a given drug or class of drugs. Newer anti-malarials were discovered in an effort to tackle this problem, but all these drugs are either expensive or have undesirable side effects.
Even the new generation of anti-malaria drugs appears to be less effective after a variable length of time due to the development of resistance in the parasites, especially the Plasmodium falciparum species. Recently, scientists reported the first evidence of resistance to the world’s most effective drug coartem in western Cambodian subjectsCitation2. With the continuous resistance to commonly used anti-malarial drugs is spreading, the search for new effective anti-malarial drugs is an urgent and a necessity in large parts of the world to contain and control the disease.
A number of scaffolds, including the pyridine nucleus, showed anti-malarial activities. Thus, in an effort to discover novel anti-malarial agents, new compounds were designed in the present work by incorporating some pharmacophoric assemblies to the pyridine nucleus.
The pyridine derivative 4-isonicotinic hydrazide, which is a leading drug in the treatment of tuberculosis, is an inhibitor of enoyl-ACP reductase, an important enzyme in the fatty acid biosynthesis. Thus, pyridine analogues may inhibit the biosynthesis of fatty acids that are fundamental for the survival of P. falciparum in the hostCitation3. Compounds that act on more than one target site are more liable to be active.
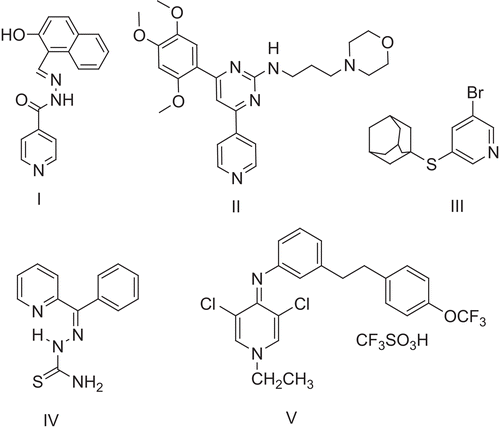
Several lead compounds containing a pyridine moiety attached to various cyclic and/or acyclic moieties have been reported (). Pyridine derivatives I, II4,5, adamantylthiopyridine III6, benzoylpyridinethiosemicarbazones IV7 and dichloropyridine derivative V8 showed pronounced anti-malarial activity ().
Figure 1. Chemical structures of pyridine derivatives having anti-malarial activity.
Many of the clinically relevant anti-malarial agents that have a known mechanism of action directly or indirectly affect pyrimidine metabolism. Drugs targeting dihydrofolate reductase (DHFR) or dihydropteroate synthetase (e.g., pyrimethamine, cycloguanil, sulphonamides and sulphones) disrupt folate metabolism, which is essential for the formation of thymidineCitation9. Also the lactate dehydrogenase enzyme of P. falciparum has the ability to rapidly use 3-acetyl pyridine NAD as a coenzyme in the reaction leading to the formation of pyruvate from lactateCitation10.
In the present study, various substituents at different positions on the pyridine nucleus were introduced to study the effect of such molecular variation on the anti-malarial activity. In addition to the targeted anti-malarial activity, docking pose of the most active compounds (2a, 2g and 2h) in the active site of DHFR enzyme and their acute toxicity profile were obtained.
Material and methods
Melting points were determined in open glass capillaries using an electro thermal BUCHI (B-540) melting point apparatus and are uncorrected. Infra red (IR) spectra were recorded on Perkin-Elmer (Postfach, Switzerland) 1430 IR spectrophotometer (Waltham, MA) using the KBr disc technique. 1H NMR spectra was recorded on a Bruker Avance DMX400 FT-NMR spectrometer (Rheinstetten, Germany) and the chemical shifts are given in δ (ppm) downfield from tetramethylsilane, which served as an internal standard. Splitting patterns were designated as follows: s: singlet; d: doublet; m: multiplet. Elemental analyses were performed on Perkin-Elmer 2400 elemental analyzer and were found within ±0.4% of the theoretical values. Follow up of the reactions and checking the purity of the compounds was made by thin layer chromatography on silica gel–pre-coated aluminium sheets (Type 60 GF254; Merck, Darmstadt, Germany) and the spots were detected by exposure to UV-lamp at λ = 254 nm and/or iodine chamber. Parasites were counted using BIO-PLUS microscope. All the chemicals were from Sigma Chemical Co. (St. Louis, MO) and the solvents were from AnalaR BDH (UK) unless otherwise stated.
Synthesis
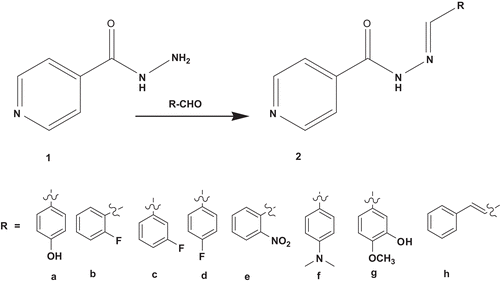
N′-arylideneisonicotinohydrazide (2a–h)
A mixture of selected aldehyde (10 mM), isonicotinic hydrazide 1 (1.37 g, 10 mM) and one drop HCl in ethanol (50 mL) was heated under reflux for 6–8 h. The reaction mixture was allowed to cool and the precipitate formed was filtered, washed with ethanol, dried and crystallized from ethanol–water mixture (7:3) (, and ).
Table 1. Physical and analytical data of compounds 2a–h, 4a–e and 5.
Table 2. Spectral data of compounds 2a–h, 4a–e and 5.
4-Aryl-1,2-dihydro-2-oxo-6-phenylpyridine-3-carbonitrile (4a–e)
A mixture of acetophenone 3 (1.20 g, 10 mM), ethyl cyanoacetate (1.13 g, 10 mM), the appropriate aldehyde (10 mM) and ammonium acetate (6.16 g, 80 mM) in ethanol (50 mL) was heated under reflux for 6 h. The reaction mixture was cooled and the formed precipitate was filtered, washed with ethanol, then washed successively with water, dried and crystallized from ethanol–water mixture (7:4) ( and ).
Ethyl 2-cyano-3-(4-(dimethylamino)phenyl)acrylate (5)
This compound was obtained as an unexpected product applying method used to prepare compounds 4a–e using 4-(dimethylamino)benzaldehyde A. To ensure the structure, this compound was synthesized by heating under reflux a mixture of 4-(dimethylamino)benzaldehyde A (1.49 g, 10 mM), ethyl cyanoacetate (1.13 g, 10 mM) and ammonium acetate (1.54 g, 20 mL) in ethanol (50 mL) for 5 h. The reaction mixture was concentrated, cooled and the formed precipitate was filtered, washed with ethanol, then washed successively with water, dried and crystallized from ethanol–water mixture (4:1) ( and ).
In vivo anti-malarial activity
In vivo anti-malarial activity test of the synthesized compounds was performed using a 4-day standard suppressive testCitation11 with some modifications. This is the most widely used preliminary test, in which the efficacy of a compound is assessed by the comparison of blood parasitaemia and mouse survival times in treated and untreated miceCitation12.
On day 0, the test mice were injected with 0.2 ml of 2 × 107 parasitised erythrocytes, (P. berghei ANKA strain) intra venously. After 2 h, the infected mice were weighed and randomly divided into 15 groups of six mice each per cage. Groups 1–13 received the synthesized compounds at 50 µmol/kg dose levels and served as treatment groupCitation13. Group 14 received the vehicle (7% Tween, 3% ethanol in water) and served as a negative control. Group 15 received the standard drug Choloroquine diphosphate (CQ, Sigma-Aldrich, Deisenhofen, Germany) at the same dose level (50 µmol/kg) and served as a positive controlCitation14.
On days 1–3, animals in the experimental groups were treated again (with the same dose of the synthesized compounds and same route daily) as in day 0. On day 4 (i.e. 24 hr after the last dose or 96 hr post-infection), blood smear from all test animals was prepared using Giemsa stain. Level of parasitaemia was determined microscopically by counting 4 fields of approximately 100 erythrocytes per field. The difference between the mean value for the control group (taken as 100%) and those of the experimental groups was calculated and expressed as percent reduction or activity.
Untreated control mice typically die approximately 1 week after infectionCitation15. For treated mice the survival time (in days) was recorded and the mean survival time was calculated in comparison with that of the negative control groupCitation16.
Percentage parasitaemia and suppression were calculated using the following formulae:
In vitro anti-malarial assay
P. falciparum strains RKL9 (CQ resistant) was maintained in a continuous culture using the standard method described by Trager and JensenCitation12. Parasites were cultured in human AB (+) erythrocytes in RPMI-1640 media (GIBCOBRL, Paisely, Scotland) supplemented with 25 mM HEPES buffer, 10% human AB (+) serum and 0.2% sodium bicarbonate (Sigma, St. Louis, MO) and maintained at 5% CO2. Cultures containing predominantly early ring stages were synchronized by addition of 5% d-sorbitol (Sigma) lysisCitation17 and used for testing. Initial culture was maintained in small vials with 10% haematocrit, i.e., 10 mL erythrocytes containing 1.0% ring stage parasite in 100 µl complete media. The culture volume per well for the assay was 100 mL. Number of parasites for the assay was adjusted at 1–1.5% by diluting with fresh human AB (+) RBC. Assay was done in 96-well microtitre flat-bottomed tissue culture plates incubated at 37°C for 24 h in presence of three-fold serial dilutions of compounds and CQ diphosphate for their effect on schizont maturation. Test compounds were dissolved in ethanol and further diluted with RPMI-1640 medium (the final ethanol concentration did not exceed 0.5%, which did not affect parasite growth). CQ diphosphate was dissolved in aqueous medium. Test was done in duplicate wells for each dose of the drugs. Solvent control culture containing the same concentrations of the solvent as present in the test wells was done with RPMI-1640 containing 10% AB (+) serum.
Parasite growth was found to be unaffected by the solvent concentrations used in the test. Growth of the parasites from duplicate wells of each concentration was monitored in Giemsa-stained blood smears by counting number of schizont per 100 asexual parasites. Percent schizont maturation inhibition was calculated by the formula: (1 − Nt/Nc) × 100 where, Nt and Nc represent the number of schizont in the test and control wells, respectively.
Acute toxicity
The oral acute toxicity of the most active compounds 2a, 2g and 2h was investigated using male mice (20 g each, Medical Research Institute, Alexandria University) according to previously reported methods. The animals were divided into groups of six mice each. The compounds were given orally, suspended in 1% gum acacia, in doses of 1, 10, 100, 200, 250, 300 mg/kg. The mortality percentage in each group was recorded after 24 h18. Additionally the test compounds were investigated for their parenteral acute toxicity in groups of mice of six animals each. The compounds or their vehicle, propylene glycol (control), were given by intra-peritoneal injection in doses of 10, 25, 50, 75, 100 mg/kg. The percentage survival was followed up to 7 daysCitation19.
Docking
The co-ordinate from the X-ray crystal structure of dehydrofolate reductase (DHFR) enzyme used in this simulation was obtained from the Protein Data Bank (PDB ID: 1J3I), where the selective DHFR inhibitor WR99210 is bound to the active site. The ligand molecules were constructed using the builder module and were energy minimized. The active site of DHFR was generated using the MOE-Alpha Site Finder, Molecular Operating Environment’s (MOE-Dock 2005) module to rationalize the observed biological results in the in vivo studyCitation20. Then ligands were docked within this active site using the MOE-Dock. The lowest energy conformation was selected and the ligand interactions (hydrogen bonding and hydrophobic interaction) with DHFR were determined.
Results and discussion
Chemistry
The target compounds were synthesized according to the steps outlined in and . Condensation of the appropriate aldehyde derivatives a–h with isonicotinic hydrazide 1 in ethanol afforded the corresponding hydrazones 2a–h. The structures of these compounds were confirmed by IR spectra that showed lack of the absorption bands characteristic for NH2 group. The absence of this primary amine in the IR spectrum and aldehydic proton in the 1H NMR spectrum confirmed that the target compounds were formed.
Scheme 1. Synthesis of hydrazone derivatives.
Scheme 2. Synthesis of pyridone derivatives.
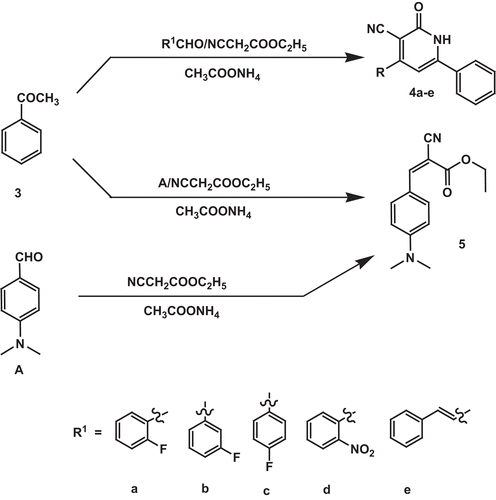
One of the synthetic procedures applied in the present work (Scheme 2) is regarded to be a multi-component reactionCitation21,Citation22. Applying one pot reaction, the selected aldehyde, ethyl cyanoacetate, acetophenone 3 and ammonium acetate in ethanol were heated under reflux to obtain compound 4a–e. The presence of amidic carbonyl group in the IR spectrum and the absence of aldehydic proton in the 1H NMR spectrum showed that the product obtained is in agreement with the proposed structure. In contrast, one pot reaction with p-dimethylaminobenzaldehyde, ethyl cyanoacetate, ammonium acetate and acetophenone did not produce the target compound. This may be attributed to the weak electrophilic property of p-dimethylbenzaldehyde, due to the electron donating effect of the p-dimethylamino group. Trials to obtain the target compound 1,2-dihydro-4-(4-dimethylaminophenyl)-2-oxo-6-phenylpyridine-3-carbonitrile by preparing the α,β-unsaturated ketone via condensation of acetophenone and p-dimethylbenzaldehyde in the presence of KOH, then cyclisation by ethyl cyanoacetate and ammonium acetate went in vain. The results obtained from elemental and spectral analyses clearly demonstrated that the compound obtained was not the targeted one, instead ethyl 2-cyano-3-(4-(dimethylamino)phenyl)acrylate 5 was obtained. To ensure the structure of compound 5, this compound was further synthesized by the reaction of 4-(dimethylamino)benzaldehyde A, ethyl cyanoacetate and ammonium acetate in ethanol. It is worth mentioning that the presence of ester functional group in the IR spectra and the number of protons from the 1H NMR spectral integration were exactly the same in the structure of the above unexpected compound and the targeted compound.
In vivo anti-malarial activity
shows the effect of the synthesised compounds against P. berghei infected mice. Compounds 2a, 2b, 2c, 2d, 2f, 2g, 2h and 4c showed more than 50% suppression of parasitaemia compared to the control and CQ treated groups. Compounds 2a, 2g and 2h are the most active compounds in this study with 90, 91 and 80% suppression, respectively. The only cyanopyridine derivative that showed promising activity was 4c. In general, isonicotinohydrazide derivatives were more active than cyanopyridine derivatives.
Table 3. In vivo anti-malarial activity of compounds 2a–h and 4a–e.
The in vivo biological activities of some of the synthesised isonicotinohydrazide derivatives showed encouraging results against P. berghei. Substitution of phenyl ring at the para-position with hydroxyl, flouro or methoxy group (2a, 2d and 2g) showed remarkable activity, which might be due the formation of hydrogen bonding with backbone of the receptor. Compound 2d showed promising activity. Moreover, compound having styryl moiety 2h showed good activity but still less than 2a and 2g. The presence of dimethylamino group at the para-position of the phenyl ring might decrease the activity of compound 2f. When the phenyl ring was substituted at para-position with hydroxyl group 2a, the activity was higher than that of compound 2e, which has nitro group at the para-position of the phenyl ring. This could be speculated to be due to the large nitro group that may not be able to interact with the backbone of the receptor.
In vitro anti-plasmodial activity
On testing the in vitro anti-plasmodial activity (), compounds 2a, 2g and 2h showed better activity than CQ diphosphate (IC50 = 0.188 µM) against CQ-resistant (RKL9) strain of parasite. Compound 2g was found to be the most potent against RKL9 (IC50 = 0.0402 µM). This may be attributed to the OH group at the para-position, which is capable of forming hydrogen bonding with the backbone of the receptor.
Table 4. In vitro anti-plasmodial activity against chloroquine-resistant (RKL9) strain of Plasmodium falciparum.
In vivo acute toxicity test
Compounds 2a, 2g and 2h were further evaluated for their oral acute toxicity in male mice using a previously reported methodCitation18,Citation19. The results indicated that the test compounds proved to be non-toxic and well tolerated by experimental animals up to 300 mg/kg. Moreover, these compounds were tested for their toxicity through the parenteral route. The results revealed that all test compounds were non-toxic up to 100 mg/kg.
Docking
Molecular docking studies further helps in understanding the various interactions between the ligands and enzyme active sites in detail. The determination of the 3D co-crystal structure of DHFR (PDB ID: 1J3I), complexed with a selective inhibitor, WR99210 (), has led to the development of a model for the topography of the anti-malarial drugs binding site in DHFR enzyme. This compound displayed hydrogen bond interactions with Ile 14, Asp 54 and Ile 164 in addition to hydrophobic interactions with Ile 14, Cys 15, Ala 16, Asp 54, Met 55, Phe 58 and Pro 113.
Figure 2. 3D view from a molecular modelling study, of the minimum-energy structure of the complex of WR99210 docked in DHFRE (PDB ID: 1J3I). Viewed using Molecular Operating Environment (MOE) module.
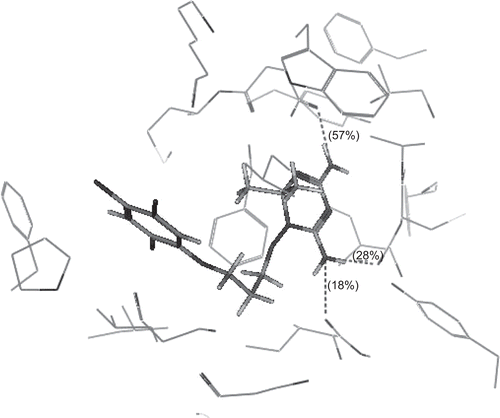
show the binding interactions of compounds 2a, 2g and 2h to the active site of DHFR, respectively, where they exhibited some similar interactions to WR99210. Compound 2a displayed hydrogen bond interactions with Phe 116, in addition to hydrophobic interactions with Ala 16, Leu 46, Asp 54, Met 55, Phe 58, Phe 116 and Arg 122. On the other hand, compound 2g showed hydrogen bond interactions with Phe 116 and Ser 120, in addition to hydrophobic interactions with Ser 111, Ile 112, Phe 116, Leu 119, Ser 120 and Arg 122. However, compound 2h displayed hydrogen bond interactions with Ser 111 in addition to hydrophobic interactions with Gly 44, Val 45, Leu 46, Met 55, Ser 108, Ser 111, Phe 116 and Leu 119. The higher activity of compounds 2a and 2g over compound 2h may be attributed to the arene–cation interaction between hydroxyphenyl group and Arg 122. The hydrogen bonds profile and their scores of the active compounds 2a, 2g and 2h and standard compound WR99210 are listed in . Although all compounds showed hydrogen bond interactions with the backbone of the enzyme, the residue of interaction may be different. Both compounds 2a, 2g interact with Phe 116 with hydrogen donor bond, but compound 2g had additional hydrogen acceptor bond with Ser 120 of excellent score. The standard compound WR99210 showed three hydrogen donor bonds with backbone of the enzyme, those with Ile 14 and Ile 164 are of weak score while that with Asp 54 of medium score.
Table 5. Calculated docking hydrogen bonding results for compounds WR99210, 2a, 2g and 2h.
Figure 3. 3D view from a molecular modelling study, of the minimum-energy structure of the complex of 2a docked in DHFRE (PDB ID: 1J3I). Viewed using Molecular Operating Environment (MOE) module.
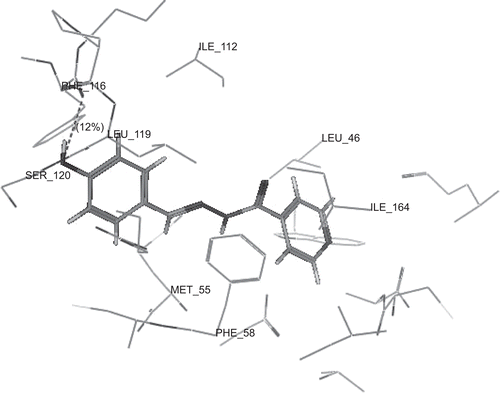
Figure 4. 3D view from a molecular modelling study, of the minimum-energy structure of the complex of 2g docked in DHFRE (PDB ID: 1J3I). Viewed using Molecular Operating Environment (MOE) module.
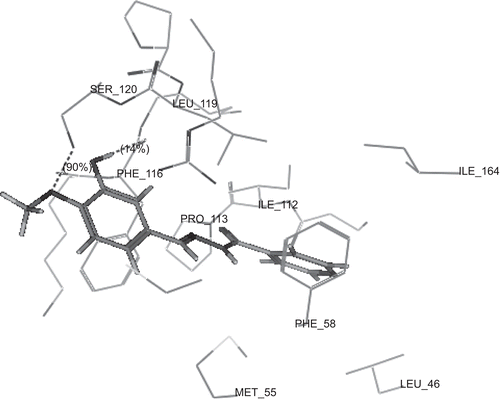
Figure 5. 3D view from a molecular modelling study, of the minimum-energy structure of the complex of 2h docked in DHFRE (PDB ID: 1J3I). Viewed using Molecular Operating Environment (MOE) module.
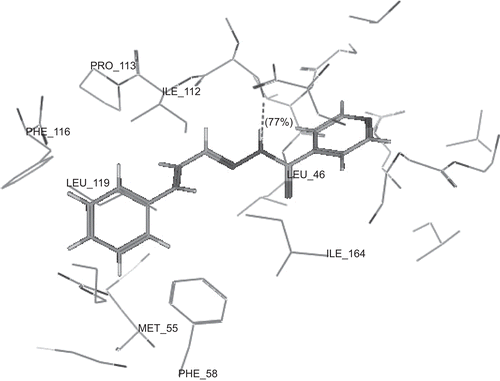
Conclusions
In this study, several pyridine derivatives were synthesised and tested for their anti-malarial activity both in vivo and in vitro against CQ-resistant and CQ-sensitive plasmodia. Compounds 2a, 2g and 2h exhibited promising anti-malarial activities against both malaria strains. Compound 2g was the most active in the present work. Acute toxicity studies showed that compounds 2a, 2g and 2h have good safety margin. Docking studies for 2a, 2g and 2h with the active site of DHFR (PDB ID: 1J3I) showed good binding profile. Therefore, compounds 2a, 2g and 2h would represent a fruitful matrix for the development of a new class of anti-malarial agents that would deserve further investigation and derivatisation.
It is worth to mention that the newly synthesised pyridine derivatives 2a, 2g and 2h (IC50 = 0.0402–0.0660 µM) exhibited higher anti-malarial activity against P. falciparum than the thiopyridine II (IC50 = 1–10 µM)Citation5. The current compounds also displayed pronounced activity compared to benzoylpyridinethiosemicarbazones IV (IC50 = 0.1 µM)Citation7. Furthermore, 2a, 2g and 2h showed superior activity over the dichloropyridine derivative VII (IC50 = 0.94 µM)Citation8, although the postulated anti-malarial mechanism in both groups of compounds is different.
Acknowledgements
The authors wish to thank the Egyptian Fund for Technical Cooperation with Africa, Ministry of Foreign Affairs, Egypt, for the financial support that enabled Prof. Adnan Bekhit and Prof. Abdel Maaboud Mohamed to be available in School of Pharmacy, Addis Ababa University.
Declaration of interest
The authors report no conflicts of interest. The authors alone are responsible for the content and writing of the paper.
References
- Joy DA, Feng X, Mu J, Furuya T, Chotivanich K, Krettli AU, Ho M, Wang A, White NJ, Suh E, Beerli P, Su XZ. Early origin and recent expansion of Plasmodium falciparum. Science 2003;300:318–321.
- Dondorp AM, Nosten F, Yi P, Das D, Phyo AP, Tarning J, Lwin KM, Ariey F, Hanpithakpong W, Lee SJ, Ringwald P, Silamut K, Imwong M, Chotivanich K, Lim P, Herdman T, An SS, Yeung S, Singhasivanon P, Day NP, Lindegardh N, Socheat D, White NJ. Artemisinin resistance in Plasmodium falciparum malaria. N Engl J Med 2009;361:455–467.
- Surolia A, Ramya TN, Ramya V, Surolia N. ‘FAS’t inhibition of malaria. Biochem J 2004;383:401–412.
- Agarwal A, Kumkum Srivastava, Puri SK, Prem MSC. Synthesis of 4-pyrido-6-aryl-2-substituted amino pyrimidines as a new class of antimalarial agents. Bioorg Med Chem 2005;13:6226–6232.
- Rollas S, Küçükgüzel SG. Biological activities of hydrazone derivatives. Molecules 2007; 12:1910–1939.
- Prachayasittikul S, Treeratanapiboon L, Ruchirawat S, Prachayasittikul V. Novel activities of 1-adamantylthiopyridines as antibacterials, antimalarials and anticancers. EXCLI J 2009;8:121–129.
- Pingaew R, Prachayasittikul S, Ruchirawat S. Synthesis, cytotoxic and antimalarial activities of benzoyl thiosemicarbazone analogs of isoquinoline and related compounds. Molecules 2010; 15:988–996.
- Rodrigues T, Guedes RC, dos Santos DJ, Carrasco M, Gut J, Rosenthal PJ, Moreira R, Lopes F. Design, synthesis and structure-activity relationships of (1H-pyridin-4-ylidene)amines as potential antimalarials. Bioorg Med Chem Lett 2009; 19:3476–3480.
- Phillips MA, Rathod PK. Plasmodium dihydroorotate dehydrogenase: a promising target for novel anti-malarial chemotherapy. Infect Disord Drug Targets 2010; 10:226–239.
- Makler MT, Hinrichs DJ. Measurement of the lactate dehydrogenase activity of Plasmodium falciparum as an assessment of parasitemia. Am J Trop Med Hyg 1993; 48:205–210.
- Peters W, Robinson BL. (1999) Parasitic infection models: Handbook of animal models of infection. London: Academic Press, 757–773.
- Trager W, Jensen JB. Human malaria parasites in continuous culture. Science 1976;193:673–675.
- Loiseaul PM, Xuong ND. Plasmodium berghei mouse model: Antimalarial activity of new alkaloid salts and of thiosemicarbazone and acridine derivatives. Trop Med and Inter Health 1996;1:379–384.
- Kiseko K, Hiroyuki M, Syun-ichi F, Ryuiichi F, Tomotaka K, Seiji M. Anti-malarial activity of leaf-extract of Hydrangea macrophyla, a common Japanese plant. Acta Med Okayama 2000;54:227–232.
- Kalra BS, Chawla S, Gupta P, Valecha N. Screening of antimalarial drug. Ind J Pharmacol 2006;38:1–12.
- Cravo P, Culleton R, Hunt P, Walliker D, Mackinnon MJ. Antimalarial drugs clear resistant parasites from partially immune hosts. Antimicrob Agents Chemother 2001; 45:2897–2901.
- Lambros C, Vanderberg JP. Synchronization of Plasmodium falciparum erythrocytic stages in culture. J Parasitol 1979; 65:418–420.
- Verma M, Tripathi M, Saxena AK, Shanker K. Antiinflammatory activity of novel indole dervatives. Eur J Med Chem 1994;29:941–946.
- Bekhit AA, Fahmy HTY. Design and synthesis of some substituted 1H-pyrazolyl-oxazolidines or 1H-pyrazolyl-thiazolidines as anti-inflammatory-antimicrobial agents. Arch Pharm Pharm Med Chem 2003; 336:111–118.
- Molecular Operating Environment (MOE). Version 2005–06. Chemical Computing Group, Inc: Montreal, Canada. Available at: http://www.chemcomp.com
- Bekhit AA, Baraka AM. Novel milrinone analogs of pyridine-3-carbonitrile derivatives as promising cardiotonic agents. Eur J Med Chem 2005;40:1405–1413.
- Domling A. Isocyanide based multi component reactions in combinatorial chemistry. High Throughput Screen 1998;1:1–22.