Abstract
Carbonic anhydrase inhibitors (CAI) are valuable molecules as they have several therapeutic applications, including anti-glaucoma activity. In this study, inhibition of three human carbonic anhydrase (hCA, EC 4.2.1.1) isozymes I, II and VI with a series of bisphenol and bromophenol derivatives was investigated. Molecular docking studies of a set of such inhibitors within CA I and II were also performed. KI values of the molecules 2–9 were in the range of 10.025–892.109 μM for hCA I, 1.437–59.107 μM for hCA II and 11.143–919.182 μM for hCA VI, respectively. Reported inhibitory activities of molecules 2–9 will assist in better understanding of structure-activity relationship studies of CAI.
Introduction
Carbonic anhydrase (CA, EC 4.2.1.1) is a zinc(II)-dependent enzyme that catalyses the reversible hydration of carbon dioxide to hydrogencarbonate and a proton. This enzyme plays an important role in physiological anion exchange processesCitation1,Citation2. Thus far, at least 16 CA isozymes have been described in mammalsCitation2–6. CA II is primarily found in red blood cells and also in many other secretory tissues of the gastrointestinal tract, kidney, lung, eye and so onCitation2–4. CA VI is a secretory enzyme that was initially described in the ovine parotid gland, saliva and normal human serumCitation4,Citation5. Other CA isoforms are found in a variety of tissues where they participate in several important biological processes such as acid–base balance, respiration, carbon dioxide and ion transport, bone resorption, ureagenesis, gluconeogenesis, lipogenesis and electrolyte secretionCitation1–13. CA isozymes involved in these processes are important therapeutic targets with the potential to be inhibited/activated for the treatment of a range of disorders such as oedema, glaucoma, obesity, cancer, epilepsy and osteoporosisCitation8–10,Citation13.
Recently, Our and Supuran’s groups investigated the interaction of CA I and II isozymes with several types of phenols such as the simple phenol, hydroxy-/methoxy-substituted benzoic acids as well as di-/tri-methoxy benzenes, anti-oxidant bisphenols and several of its substituted derivatives, for example, salicyclates and some of their derivativesCitation11–13. In this study, we extended these earlier investigations to bromophenols, some of which are widely used as anti-oxidant food additives or drugs. Bromophenols, frequently isolated from red algae of the family Rhodomelaceae, have some important biological activitiesCitation14–22. The bisphenols or related methoxy derivatives 3–6 were previously demonstrated to possess strong anti-oxidant propertiesCitation16.
In this study, we have purified human CA I, II and VI (hCA I, hCA II and hCA VI) from human blood and examined the in vitro inhibition effects of some bisphenol, metoxy and bromophenol compounds on these enzymes.
Materials and methods
CNBr-activated sepharose 4B, protein assay reagents, p-aminobenzene sulfonamide L-tyrosine, 4-nitrophenylacetate (NPA) and chemicals for electrophoresis were purchased from Sigma-Aldrich Co, Munich, Germany. All other chemicals were of analytical grade and obtained from either Sigma or Merck.
CA purification assay
Purification of hCA I and hCA II were performed as previously describedCitation13. The methods proposed by Murakami and Sly and Kivela et al. were modified and used for purification of CA VI from human serumCitation4,Citation7. Serum was obtained from fresh human blood at the Blood Center of the Research Hospital at Atatürk University. The blood samples were centrifuged at 5000 rpm for 15 min, the precipitant was removed and serum was isolated. The pH was adjusted to 8.7 with solid Tris. Sepharose-4B-aniline-sulfanylamide affinity column was equilibrated with 25-mM Tris-HCl/0.1-M Na2SO4 (pH 8.7). The affinity gel was washed with 25-mM Tris-HCl/22-mM Na2SO4 (pH 8.7). The human carbonic anhydrase (hCA-VI) isozyme was eluted with 0.25 M H2NSO3H/25cmM Na2HPO4 (pH = 6.7). All procedures were performed at 4°CCitation4,Citation5.
CA activity assay and kinetic studies
CA activity was assayed by following the change in absorbance at 348 nm of 4-NPA to 4-nitrophenylate ion over a period of 3 min at 25°C using a spectrophotometer (Shimadzu UV-VIS) according to the method described by Verpoorte et al.Citation23 The enzymatic reaction, in a total volume of 3.0 mL, contained 1.4-mL 0.05-M Tris–SO4 buffer (pH 7.4), 1-mL 3-mM 4-NPA, 0.5-mL H2O and 0.1-mL enzyme solution. A reference measurement was obtained by preparing the same cuvette without enzyme solution. The inhibitory effects of the phenolic compounds were examined. All compounds were tested in triplicate at each concentration used. Different concentrations of the compounds were used. Control cuvette activity in the absence of inhibitor was taken as 100%. For the compounds, an activity (%)−[inhibitor] graphs were drawn. In these experiments, 4-NPA was used as substrate at five different concentrations (0.15–0.75 mM). KI-s were obtained from IC50 by the Cheng–Prussoff equation, and the Lineweaver–Burk curves were drawnCitation24.
Protein determination
Protein quantity was determined spectrophotometrically at 595 nm according to the Bradford method during the purification steps, using bovine serum albumin as the standardCitation25.
SDS polyacrylamide gel electrophoresis
SDS polyacrylamide gel electrophoresis was performed after purification of the enzymes. It was carried out in 10% and 3% acrylamide for the running and the stacking gel, respectively, containing 0.1% SDS according to Laemmli procedure. A 20 μg sample was applied to the electrophoresis medium. Gels were stained for 1.5 h in 0.1% Coommassie Brilliant Blue R-250 in 50% methanol and 10% acetic acid, then destained with several changes of the same solvent without the dyeCitation26.
Molecular docking
Glide-Induced Fit Docking (IFD) program of Schrodinger molecular modelling platform has been used for docking studies (). Extra precision (XP) option is usedCitation26–28.
Figure 1. SDS polyacrylamide gel electrophoresis bands of carbonic anhydrase (CA) I, II, VI and standard proteins (Lane 1, CA I; Lane 2, CA II; Lane 3, CA VI; Lane 4, standards: rabbit phosphorylase B [97,400 Da], bovine albumin [66,000 Da], chicken ovalbumin [45,000 Da], bovine carbonic anhydrase [29,000 Da] and chicken egg white lysozyme [16,500 Da]).
![Figure 1. SDS polyacrylamide gel electrophoresis bands of carbonic anhydrase (CA) I, II, VI and standard proteins (Lane 1, CA I; Lane 2, CA II; Lane 3, CA VI; Lane 4, standards: rabbit phosphorylase B [97,400 Da], bovine albumin [66,000 Da], chicken ovalbumin [45,000 Da], bovine carbonic anhydrase [29,000 Da] and chicken egg white lysozyme [16,500 Da]).](/cms/asset/96aaa30d-5191-4cb4-b566-022dad3e5f4d/ienz_a_596836_f0001_b.gif)
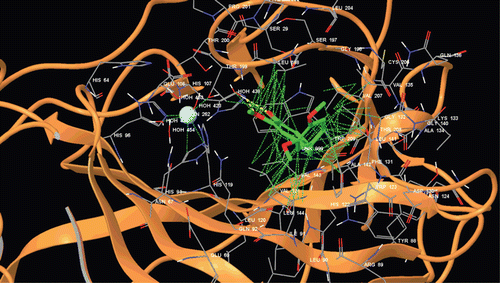
Figure 2. Docking of compound 2 within the hCA II active site.
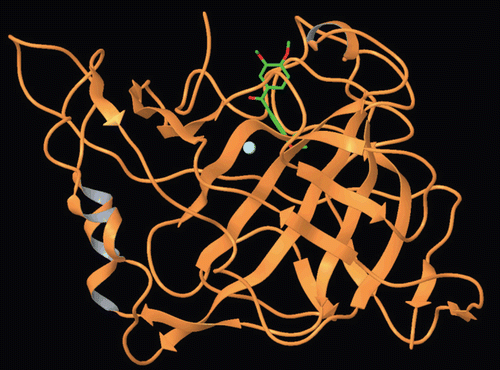
Figure 3. Binding mode of adduct and compound 2 at hCA-I.
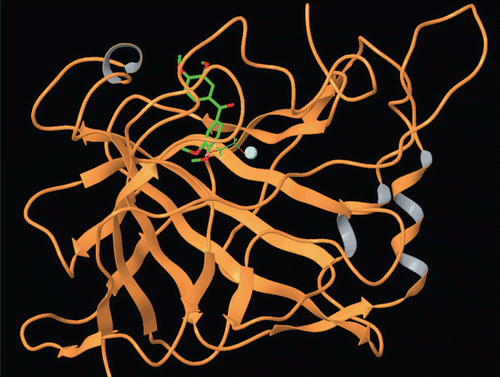
Figure 4. Docking of compound 2 within the hCA I active site.
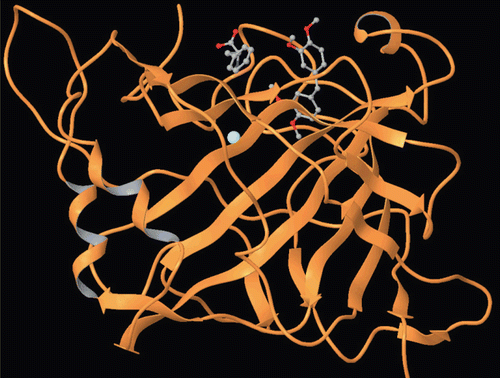
Figure 5. Docking of compound 4 within the hCA II active site.
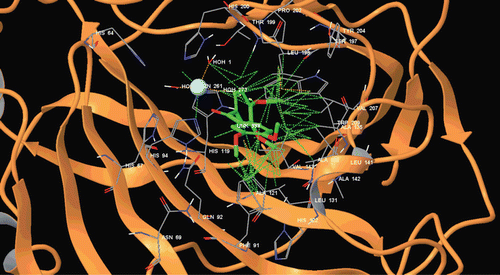
Figure 6. Binding mode of adduct and compound 4 at hCA-II.
Results and discussion
Since simple phenol (PhOH) has been shown to be the only competitive inhibitor with CO2 as substrate for the main isoform of CA, that is, human CA II (hCA II)Citation29, we investigated these phenolic derivatives. The X-ray crystal structure for the adduct of hCA II with phenolCitation29 showed that this compound binds to CA by anchoring its OH moiety to the zinc-bound water/hydroxide ion of the enzyme active site, through a hydrogen bond as well as through a second hydrogen bond to the NH amide of Thr199, an amino acid conserved in all α-CAs and critically important for the catalytic cycle of these enzymesCitation3–13. The hydrophobic part of the hCA II active site contains the phenyl moiety of phenol, where CO2, binds in the precatalytic complex, explaining the behaviour of phenol as a unique CO2 competitive inhibitor. Supuran et al. has recently investigated the interactions of phenol and some of its substituted derivatives with all mammalian CA enzymesCitation10–13. Their results demonstrated low micromolar/submicromolar inhibitory of molecules as well as the possibility to design isozyme-selective CA inhibitors (CAIs). The inhibition profile of various CA isozymes with this class of agents is very diverse, with inhibition constants ranging from the millimolar to the submicromolar range for many simple phenolsCitation10–13. Thus, it seemed reasonable to us to extend the previous studiesCitation10–13, including in this investigation phenols with clinical and anti-oxidant applications as food additives, such as compounds 3–6Citation16. Other structurally related derivatives such as 2, 7 and 8 were also included in this study.
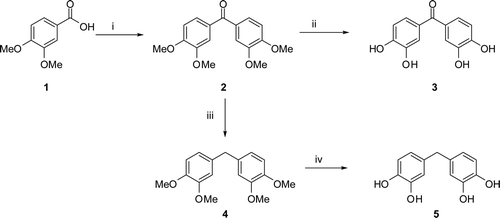
Bis(3,4-dimethoxyphenyl)methanone (6) was obtained from the reaction of 3,4-dimethoxybenzoic acid (6) with veratrole in polyphosphoric acid according to the literature methodCitation20,Citation22. The Wolff−Kishner reduction of 3 with hydrazine hydrate and KOH at 190°C gave bis(3,4-dimethoxyphenyl)methane (4). The literature procedureCitation14–20 described for o-demethylation of aryl methyl ethers with BBr3 was applied to compounds 1 and 3 to give corresponding phenols bis(2,3-dihydroxyphenyl)methanone (3) and 4,4′-methylenedibenzen-1,2-diol (5) (). Compounds 2–9 were synthesised according to our previously published proceduresCitation14–22.
The purification of the two CA isozymes was performed with a simple one-step method by a Sepharose-6B-aniline-sulfanilamide affinity column chromatograpyCitation30–32. hCA I was purified, 102-fold with a specific activity of 865.7 EU/mg and overall yield of 48.4%, hCA II was purified, 625-fold with a specific activity of 5306 EU/mg and overall yield of 52.2% and hCA VI was purified, 75.3-fold with a specific activity of 213 EU/mg and overall yield of 17.8%Citation22–25,Citation29–35. SDS polyacrylamide gel electrophoresis was performed after the purification of these enzymes, and the electrophoretic pattern was photographed (). Inhibitory effects of bisphenol, metoxy and bromophenol compounds on enzyme activities were tested under in vitro conditions; KI values were calculated from Lineweaver–Burk graphs and are given in Citation26.
Table 1. hCA I, II and VI inhibition data with compounds 1–11 and acetazolamide (12), by an esterase assay with 4-nitrophenylacetate as substrateCitation23.
We report here the first study on the inhibitory effects of bisphenol, metoxy and bromophenol compounds of type 2–9 on the esterase activity of hCA I, II and VI. Acetazolamide (AZA) 12 has been used as a negative control in our experiments and for comparison reasonsCitation13,Citation30. The previous reports by Senturk et al.Citation30–32 investigated other anti-oxidant phenol derivatives (including salicylic acid and propofol) by using esterase assay. Data of show the following regarding inhibition of hCA I, II and VI with phenols 2–9, by an esterase assayCitation23, with 4-NPA as substrate:
(i) Against the slow cytosolic isozyme hCA I, compounds 2–9 behave as weak inhibitors, with KI values in the range of 18.086–892.109 μM. 1,2,5-tribromo-3,4-dimethoxybenzene 9 was an ineffective hCA I inhibitor (KI of 892.109 μM), similarly to the structurally related compound 4 (KI 537.311 μM). A second group of derivatives, including 6 and 7, showed better inhibitory activity as compared with the previously mentioned methoxybenzenes, with KI values of 25.341–32.145 μM, (). Therefore, the nature of the groups in ortho- and ortho′- to the phenolic OH moiety strongly influences hCA I inhibitory activity. It is also interesting to note that the biphenyl derivatives 3 and 5 were much better hCA I inhibitors as compared with the corresponding metoxy-bisphenols 2 and 4 from which they were prepared. AZA 12 is also a medium CA I with this assay and substrate against hCA I (KI of 26.2 μM)Citation30. Kinetic investigations (Lineweaver–Burk plots, data not shown) indicate that similarly to sulfonamides and inorganic anionsCitation3–13,Citation36–47, all the investigated compounds act as non-competitive inhibitors with 4-NPA as substrate, that is, they bind in different regions of the active site cavity as compared with the substrate. However, the binding site of 4-NPA itself is unknown, but it is presumed to be in the same region as that of CO2, the physiological substrate of this enzymeCitation29.
(ii) A better inhibitory activity has been observed with compounds 2–9 investigated in this study for the inhibition of the rapid cytosolic isozyme hCA II (). Thus, two derivatives, that is, 2 and 4, showed moderate hCA II inhibitory activity with KI-s in the range of 47.832–59.107 μM (), whereas the remaining six derivatives were quite effective hCA II inhibitors, with KI-s in the range of 1.437–29.82 μM (). Structure-activity relationship is thus quite sharp for this small series of bisphenol, methoxy and bromophenol compounds: the 4,4′-dimethoxy-bis derivatives 2 and 4 are ineffective leads. This trend is maintained when different groups are present in the para or meta position to the phenol OH moiety, such as in compounds 6 and 7. Without methoxy-group, same compounds 3 and 5 are also effective hCA II inhibitors. The best hCA II inhibitor in this series of derivatives was the bulky, bisphenol derivative 3, which has a KI value of 1.437 μM. Derivative 3 showed better inhibitory profile compared with compounds 2, 4–7. It must be stressed that KI-s measured with the esterase method are always in the micromolar range because hCA I and II are weak esterasesCitation48–56.
(iii) 1,2,5-tribromo-3,4-dimethoxybenzene 9 and some of its congeners such as 2, 4 and 8 are also weak inhibitors of the secreted isozyme hCA VI, with KI-s of 158.592–919.182 μM. However, again the 3,4,5,6-tetrabromobenzene-1,2-diol 7 is medium potency inhibitors (KI of 29.138 μM), and derivatives 3, 5 and 6 show a higher affinity for this isozyme, with inhibition constants in the range of 11.143–13.692 μM ().
In a recent study, it was reported that derivatives of salicylic acidCitation13, a simple compound lacking the sulfonamide, sulfamate or related functional groups that are typically found in all known CA inhibitors, acts as a CA I inhibitor and could represent the starting point for a new class of inhibitors that may have advantages for patients with sulfonamide allergies (acetylsalicylic acid acts as a prodrug, being hydrolysed in situ with formation of a sodium salicylete derivative, which is the real enzyme inhibitor)Citation51. However, Innocenti et al.Citation52 showed that compared with sulphonamides, thioxolone was inefficient for generating isozyme-selective inhibitors. Because it is found that except for hCA I, which was inhibited in the nanomolar range (KI of 91 nM), the remaining 12 mammalian CA isoforms (CA II–CA XV) were inhibited with a very flat profile by this compound (KI-s in the range of only 4.93–9.04 μM). In contrast to thioxolone, 3,5-dichloro-4-hydroxybenzenesulfonamide as well as the clinically used heterocyclic sulfonamide AZA showed KI-s in the range of 58 nM–8.6 μM and 2.5 nM–200 μM, respectively, against the 13 investigated mammalian CAs. The sulfonamide zinc-binding group is thus superior to the thiol group (from the thioxolone hydrolysis product) for generating CA inhibitors with a diverse and sometimes isozyme-selective inhibition profile against the mammalian enzymes. However, it is critically important to explore further classes of potent CAIs to detect compounds with a different inhibition profile as compared with the sulfonamides and their bioisosteres and to find novel applications for the inhibitors of these widespread enzymes.
Bisphenol, methoxy and bromophenol compounds 2–9 influence the activity of CA isozymes due to the presence of the different functional groups (OH, OCH3 and Br) present in their aromatic scaffold. Thus, our findings indicate another class of possible CAIs of interest, in addition to the well-known sulfonamides/sulfamates/sulfamides, the phenols/biphenyl diphenols bearing bulky ortho moieties in their molecules. Some bisphenol, methoxy and bromophenol compounds investigated in this study showed effective hCA I and II inhibitory activity, in the low micromolar range, by the esterase method, which usually gives KI-s an order of magnitude higher as compared with the CO2 hydrase assayCitation51–58. These findings point out that substituted phenolic compounds may be used as leads for generating potent CAIs eventually targeting other isoforms, which have not been assayed yet for their interactions with such agents.
In silico studies
In this study, to better understand the binding mechanisms of studied molecules, fully flexible docking methodology for both receptor residues and docked ligands was used. Docking studies are performed using Glide XP–IFD algorithm, which was implemented with the Prime module under Schrodinger molecular modelling packageCitation26–28,Citation56,Citation57. The compounds 2–9 were docked at the binding site of the targets (hCA I and hCA II). Glide/IFD docking scores of docked inhibitors at hCA I and II targets and corresponding binding interactions were tabulated in (supporting information).
Table 2. Docking binding scores of the molecules.
Conclusions
Together with in vitro studies, we also performed molecular docking studies of compounds 2–9 within the hCA I and II active sites. The hCA I and II adducts for which the X-ray crystal structures have been reported in complexes with activators or inhibitors were used in the docking. AZA 12 together with compounds 2–9 have been docked to the active sites of hCA I and II. The results of the docking experiments are presented in (supporting information) for hCA I and II and are shown in some details in –. Such data may be important for designing compounds with enhanced affinity and eventually more selectivity for various CA isoforms.
Acknowledgments
We are grateful to Prof. Dr. Claudiu T. Supuran, University of Florence, for various valuable studies on CAIs.
Declaration of interest
This study was financed by Turkish Republic Prime Ministry State Planning Organization (DPT), (project no: 2010K120440) and Agri Ibrahim Cecen University Scientific Research Council, (project no: Agri BAP-2010/K-10) for (MS) and by the Scientific and Technological Research Council of Turkey (TUBITAK, Grant No: TBAG-107T/348) for (SG, HTB and AM).
References
- Maren TH. The kinetics of HCO3- synthesis related to fluid secretion, pH control, and CO2 elimination. Annu Rev Physiol 1988;50:695–717.
- Supuran CT. Carbonic anhydrases: novel therapeutic applications for inhibitors and activators. Nat Rev Drug Discov 2008;7:168–181.
- Hilvo M, Baranauskiene L, Salzano AM, Scaloni A, Matulis D, Innocenti A et al. Biochemical characterization of CA IX, one of the most active carbonic anhydrase isozymes. J Biol Chem 2008;283:27799–27809.
- Kivelä J, Parkkila S, Waheed A, Parkkila AK, Sly WS, Rajaniemi H. Secretory carbonic anhydrase isoenzyme (CA VI) in human serum. Clin Chem 1997;43:2318–2322.
- Karhumaa P, Leinonen J, Parkkila S, Kaunisto K, Tapanainen J, Rajaniemi H. The identification of secreted carbonic anhydrase VI as a constitutive glycoprotein of human and rat milk. Proc Natl Acad Sci USA 2001;98:11604–11608.
- Supuran CT, Scozzafava A. Carbonic anhydrases as targets for medicinal chemistry. Bioorg Med Chem 2007;15:4336–4350.
- Murakami H, Sly WS. Purification and characterization of human salivary carbonic anhydrase. J Biol Chem 1987;262:1382–1388.
- Sly WS, Hu PY. Human carbonic anhydrases and carbonic anhydrase deficiencies. Annu Rev Biochem 1995;64:375–401.
- Pastorekova S, Parkkila S, Pastorek J, Supuran CT. Carbonic anhydrases: current state of the art, therapeutic applications and future prospects. J Enzyme Inhib Med Chem 2004;19:199–229.
- Ekinci D, Ceyhun SB, Sentürk M, Erdem D, Küfrevioglu OI, Supuran CT. Characterization and anions inhibition studies of an a-carbonic anhydrase from the teleost fish Dicentrarchus labrax. Bioorg Med Chem 2011;19:744–748.
- Innocenti A, Vullo D, Scozzafava A, Supuran CT. Carbonic anhydrase inhibitors: interactions of phenols with the 12 catalytically active mammalian isoforms (CA I-XIV). Bioorg Med Chem Lett 2008;18:1583–1587.
- Innocenti A, Beyza Oztürk Sarikaya S, Gülçin I, Supuran CT. Carbonic anhydrase inhibitors. Inhibition of mammalian isoforms I-XIV with a series of natural product polyphenols and phenolic acids. Bioorg Med Chem 2010;18:2159–2164.
- Bayram E, Senturk M, Kufrevioglu OI, Supuran CT. In vitro inhibition of salicylic acid derivatives on human cytosolic carbonic anhydrase isozymes I and II. Bioorg Med Chem 2008;16:9101–9105.
- Gribble GW. Naturally occurring organohalogen compounds-a survey. J Nat Prod 1992;55:1353–1395.
- Gribble GW. The diversity of naturally occuring organobromine compounds. Chem Soc Rev 1999;28:335–346.
- Balaydin HT, Gülçin Í, Menzek A, Göksu S, Sahin E. Synthesis and antioxidant properties of diphenylmethane derivative bromophenols including a natural product. J Enzyme Inhib Med Chem 2010;25:685–695.
- Sentürk M, Ekinci D, Göksu S, Supuran CT. Effects of dopaminergic compounds on carbonic anhydrase isozymes I, II, and VI. J Enzyme Inhib Med Chem (In Press).
- Talaz O, Gülçin I, Göksu S, Saracoglu N. Antioxidant activity of 5,10-dihydroindeno[1,2-b]indoles containing substituents on dihydroindeno part. Bioorg Med Chem 2009;17:6583–6589.
- Akbaba Y, Balaydin HT, Goksu S, Sahin E, Menzek A. Total synthesis of the biologically active, naturally occurring 3,4-dibromo-5-[2-bromo-3,4-dihydroxy-6-(methoxymethyl) benzyl]benzene-1,2-diol and regioselective o-demethylation of aryl methyl ethers. Helv Chim Acta 2010;93:1127–1135.
- Balaydin HT, Akbaba Y, Menzek A, Sahin E, Goksu S. First and short syntheses of biologically active, naturally occurring brominated mono- and dibenzyl phenols. Arkivoc 2009;XIV:75–87.
- Pearl IA. Reactions of vanillin and its derived compounds. XXIII. The synthesis of 4,4′-dihydroxy-3,3′-dimethoxybenzophenone. J Am Chem Soc 1954;76:3635–3637.
- Cabaton N, Zalko D, Rathahao E, Canlet C, Delous G, Chagnon MC et al. Biotransformation of bisphenol F by human and rat liver subcellular fractions. Toxicol in Vitro 2008;22:1697–1704.
- Verpoorte JA, Mehta S, Edsall JT. Esterase activities of human carbonic anhydrases B and C. J Biol Chem 1967;242:4221–4229.
- Lineweaver H, Burk D. The determination of enzyme dissocation constants. J Am Chem Soc 1934;56:658–666.
- Bradford MM. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal Biochem 1976;72:248–254.
- Schrodinger Suite, Schrodinger, LLC, New York, USA 2007 (web page: www.schrodinger.com).
- Sherman W, Day T, Jacobson MP, Friesner RA, Farid R. Novel procedure for modeling ligand/receptor induced fit effects. J Med Chem 2006;49:534–553.
- Friesner RA, Murphy RB, Repasky MP, Frye LL, Greenwood JR, Halgren TA et al. Extra precision glide: docking and scoring incorporating a model of hydrophobic enclosure for protein-ligand complexes. J Med Chem 2006;49:6177–6196.
- Nair SK, Ludwig PA, Christianson DW. Two-site binding of phenol in the active site of human carbonic anhydrase II: structural implications for substrate association. J Am Chem Soc 1994;116:3659–3660.
- Sentürk M, Gülçin I, Dastan A, Küfrevioglu OI, Supuran CT. Carbonic anhydrase inhibitors. Inhibition of human erythrocyte isozymes I and II with a series of antioxidant phenols. Bioorg Med Chem 2009;17:3207–3211.
- Sentürk M, Gülçin I, Beydemir S, Küfrevioglu OI, Supuran CT. In Vitro Inhibition of Human Carbonic Anhydrase I and II Isozymes with Natural Phenolic Compounds. Chem Biol Drug Des 2011;77:494–499.
- Ceyhun SB, Senturk M, Yerlikaya E, Erdogan O, Kufrevioglu OI, Ekinci D. Purification and characterization of carbonic anhydrase from the teleost fish Dicentrarchus labrax (European Seabass) liver and toxicological effects of metals on enzyme activity. Environ Toxicol Phar 2010;32:69–74.
- Laemmli UK. Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature 1970;227:680–685.
- Parkkila S, Kaunisto K, Rajaniemi L, Kumpulainen T, Jokinen K, Rajaniemi H. Immunohistochemical localization of carbonic anhydrase isoenzymes VI, II, and I in human parotid and submandibular glands. J Histochem Cytochem 1990;38:941–947.
- Wilbur KM, Anderson NG. Electrometric and colorimetric determination of carbonic anhydrase. J Biol Chem 1948;176:147–154.
- Supuran CT, Scozzafava A, Casini A. Carbonic anhydrase inhibitors. Med Res Rev 2003;23:146–189.
- Scozzafava A, Mastrolorenzo A, Supuran CT. Carbonic anhydrase inhibitors and activators and their use in therapy. Expert Opin Ther Pat 2006;16:1627–1664.
- Thiry A, Dogné JM, Supuran CT, Masereel B. Carbonic anhydrase inhibitors as anticonvulsant agents. Curr Top Med Chem 2007;7:855–864.
- Ekinci D, Sentürk M, Beydemir S, Küfrevioglu OI, Supuran CT. An alternative purification method for human serum paraoxonase 1 and its interactions with sulfonamides. Chem Biol Drug Des 2010;76:552–558.
- Ekinci D, Beydemir S, Küfrevioglu OI. In vitro inhibitory effects of some heavy metals on human erythrocyte carbonic anhydrases. J Enzyme Inhib Med Chem 2007;22:745–750.
- Sentürk M, Talaz O, Ekinci D, Cavdar H, Küfrevioglu OI. In vitro inhibition of human erythrocyte glutathione reductase by some new organic nitrates. Bioorg Med Chem Lett 2009;19:3661–3663.
- Abdülkadir Coban T, Beydemir S, Gülcin I, Gücin I, Ekinci D, Innocenti A et al. Sildenafil is a strong activator of mammalian carbonic anhydrase isoforms I-XIV. Bioorg Med Chem 2009;17:5791–5795.
- Supuran CT. Carbonic anhydrases–an overview. Curr Pharm Des 2008;14:603–614.
- Ekinci D, Cavdar H, Talaz O, Sentürk M, Supuran CT. NO-releasing esters show carbonic anhydrase inhibitory action against human isoforms I and II. Bioorg Med Chem 2010;18:3559–3563.
- Alp C, Ekinci D, Gültekin MS, Sentürk M, Sahin E, Küfrevioglu OI. A novel and one-pot synthesis of new 1-tosyl pyrrol-2-one derivatives and analysis of carbonic anhydrase inhibitory potencies. Bioorg Med Chem 2010;18:4468–4474.
- Ceyhun SB, Senturk M, Erdogan O, Kufrevioglu OI. In vitro and in vivo effects of some pesticides on carbonic anhydrase enzyme from rainbow trout (Oncorhynchus mykiss) gills. Pestic Biochem Physiol 2010;97:177–181.
- Nishimori I, Minakuchi T, Onishi S, Vullo D, Scozzafava A, Supuran CT. Carbonic anhydrase inhibitors. DNA cloning, characterization, and inhibition studies of the human secretory isoform VI, a new target for sulfonamide and sulfamate inhibitors. J Med Chem 2007;50:381–388.
- Supuran CT, Mincione F, Scozzafava A, Briganti F, Mincione G, Ilies MA. Carbonic Anhydrase Inhibitors. Part 52. Metal Complexes of Heterocyclic Sulfonamides: A New Class of Strong Topical Intraocular Pressure-lowering Agents With Potential Use as Antiglaucoma Drugs. Eur J Med Chem 1998;33:247–254.
- Supuran CT, Scozzafava A. Novel aromatic/heterocyclic sulfonamides and their metal complexes as inhibitors of carbonic anhydrase isozymes I, II and IV. J Enzym Inhib 1997;12:37–51.
- Abbate F, Winum JY, Potter BV, Casini A, Montero JL, Scozzafava A et al. Carbonic anhydrase inhibitors: X-ray crystallographic structure of the adduct of human isozyme II with EMATE, a dual inhibitor of carbonic anhydrases and steroid sulfatase. Bioorg Med Chem Lett 2004;14:231–234.
- Barrese AA 3rd, Genis C, Fisher SZ, Orwenyo JN, Kumara MT, Dutta SK et al. Inhibition of carbonic anhydrase II by thioxolone: a mechanistic and structural study. Biochemistry 2008;47:3174–3184.
- Innocenti A, Maresca A, Scozzafava A, Supuran CT. Carbonic anhydrase inhibitors: thioxolone versus sulfonamides for obtaining isozyme-selective inhibitors? Bioorg Med Chem Lett 2008;18:3938–3941.
- Senturk M, Irfan Kufrevioglu O, Ciftci M. Effects of some analgesic anaesthetic drugs on human erythrocyte glutathione reductase: an in vitro study. J Enzyme Inhib Med Chem 2009;24:420–424.
- Sahin A, Senturk M, Ciftci M, Varoglu E, Kufrevioglu OI. The effects of chemical and radioactive properties of Tl-201 on human erythrocyte glucose 6-phosphate dehydrogenase activity. Nucl Med Biol 2010;37:389–394.
- Ceyhun SB, Sentürk M, Ekinci D, Erdogan O, Ciltas A, Kocaman EM. Deltamethrin attenuates antioxidant defense system and induces the expression of heat shock protein 70 in rainbow trout. Comp Biochem Physiol C 2010;152:215–223.
- Akkemik E, Senturk M, Ozgeris FB, Taser P, Ciftci M. In vitro eff ects of some drugs on human erythrocyte glutathione reductase. Turk J Med Sci 2011;41:235–241.
- Durdagi S, Sentürk M, Ekinci D, Balaydin HT, Göksu S, Küfrevioglu ÖI, et al. Kinetic and docking studies of phenol-based inhibitors of carbonic anhydrase isoforms I, II, IX and XII evidence a new binding mode within the enzyme active site. Bioorg Med Chem 2011;19:1381–1389.
- Oztürk Sarikaya SB, Topal F, Sentürk M, Gülçin I, Supuran CT. In vitro inhibition of a-carbonic anhydrase isozymes by some phenolic compounds. Bioorg Med Chem Lett (In Press).