Abstract
A novel class of inhibitors of the enzyme γ-glutamyl transpeptidase (GGT) were evaluated. The analog OU749 was shown previously to be an uncompetitive inhibitor of the GGT transpeptidation reaction. The data in this study show that it is an equally potent uncompetitive inhibitor of the hydrolysis reaction, the primary reaction catalyzed by GGT in vivo. A series of structural analogs of OU749 were evaluated. For many of the analogs, the potency of the inhibition differed between the hydrolysis and transpeptidation reactions, providing insight into the malleability of the active site of the enzyme. Analogs with electron withdrawing groups on the benzosulfonamide ring, accelerated the hydrolysis reaction, but inhibited the transpeptidation reaction by competing with a dipeptide acceptor. Several of the OU749 analogs inhibited the transpeptidation reaction by slow onset kinetics, similar to acivicin. Further development of inhibitors of the GGT hydrolysis reaction is necessary to provide new therapeutic compounds.
| Abbrevations | ||
| GGT, | = | γ-glutamyl transpeptidase |
| GpNA, | = | D- or L-glutamic acid γ-4-nitroanalide HCl |
| glygly, | = | glycyl-glycine |
| DMSO, | = | dimethyl sulfoxide |
Introduction
γ-Glutamyl transpeptidase (GGT) is a cell surface enzyme that catalyzes the cleavage of the γ-glutamyl bond of glutathione (GSH) and other γ-glutamyl compoundsCitation1. In humans, the expression of GGT is restricted predominantly to the apical surface of ducts and glands where fluids leave the bodyCitation2. The highest concentration of GGT is on the apical surface of the proximal tubule cells in the kidney where it prevents excretion of GSH into the urine by cleaving GSH present in the glomerular filtrateCitation2,Citation3. Aberrant expression and localization of GGT is observed in many disease states including cancerCitation4. Inhibiting GGT would sensitize tumours to chemotherapy and may be therapeutic in other diseasesCitation5–8. However, GGT inhibitors that have been evaluated clinically are glutamine analogs and are neurotoxicCitation9–13. We have previously reported the discovery of a novel inhibitor of GGT, OU749, which is not a glutamine analogCitation14.
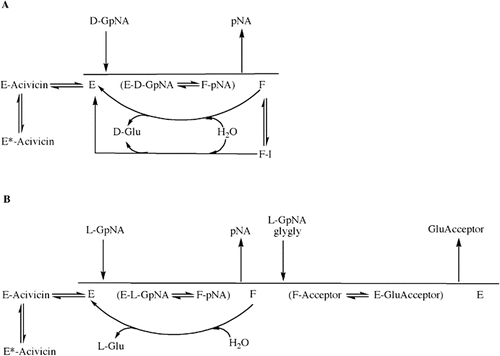
GGT can catalyze a hydrolysis reaction or a transpeptidation reaction. The first steps in both reactions are the cleavage of the γ-glutamyl bond of the substrate in the enzyme-substrate complex (ES) and the formation of a transient enzyme-glutamyl substrate complex (F-form of the enzyme). As the substrate is cleaved, the γ-glutamyl group forms a transient acyl bond with the enzyme, and the remainder of the substrate is released ()Citation15–17. In human GGT, the acyl bond forms between the γ-carbon of the γ-glutamyl substrate and the hydroxyl (β-oxygen) on the side chain of Thr-381Citation18. In the hydrolysis reaction, water hydrolyzes the acyl bond between the γ-glutamyl group and the nucleophilic residue, releasing both glutamate and the enzymeCitation19,Citation20. In the transpeptidation reaction, the γ-glutamyl group is transferred to the amine of an acceptor, thereby forming a new γ-glutamyl compoundCitation21. The transpeptidation reaction occurs by a modified ping-pong mechanismCitation16,Citation22. The pH and amino acid concentrations in extracellular fluids where GGT is localized favor the hydrolysis reaction, and previous studies have indicated that the hydrolysis reaction is the predominant reaction catalyzed by GGT in vivoCitation22,Citation23.
Figure 1. GGT reactions with γ-Glutamyl Substrates. Cleavage of glutathione, a physiologic substrate of GGT, is shown. The γ-glutamyl bond between the γ-carbon of glutamate and the amine of cysteine is cleaved by GGT. The γ-carbon of glutamate forms an acyl covalent bond with the hydroxyl group on the side chain of Thr-381 of human GGT. The acyl bond can either be hydrolyzed releasing glutamate (Glu), or an acceptor nucleophile can attack forming a new γ-glutamyl compound (transpeptidation reaction). The reactions proceeds via a Ping-Pong mechanism.
We previously identified OU749, a novel inhibitor of the GGT transpeptidation reactionCitation14. Studies of its inhibition revealed that it is uncompetitive against l-γ-glutamyl paranitroanilide (l-GpNA) in the transpeptidation reaction, binding the enzyme-glutamyl substrate complex (F-form) and competing with the acceptorCitation14. Our hypothesis is that OU749 and its structural analogs inhibit both the hydrolysis and transpeptidation reactions catalyzed by GGT. In this study, we performed in-depth kinetic analyses of the inhibition of both reactions by OU749 and a series of new structural analogs. We evaluated the potency with which the compounds inhibited the reactions and the mechanisms of inhibition. These data were compared to the inhibition by the glutamine analog, acivicin, a slow binding inhibitor with a slow rate of release. Our studies of both the hydrolysis and transpeptidation reactions have been conducted at physiologic pH. The standard GGT assay used by other investigators is conducted at pH 8.0 or higher, which may alter the physiologic cleavage mechanism due to decreased protonation of the amino acid side chains within the active siteCitation24. In addition, in the transpeptidation reaction, the presence of high concentrations of acceptor may induce conformational changes in the enzyme similar to the effects of hippurateCitation24. The data from this study provide insights into the essential features of both the acceptors and inhibitors of the GGT reaction.
Methods
Inhibitors
OU749 was purchased from ChemBridge Corp (San Diego, CA). Sodium benzosulfonamide was purchased from Sigma (St. Louis, MO).
Synthesis of compounds 2–20 (refer to and for structures)
Synthesis of synthetic intermediates TDA1-10 (). Two mmol of the appropriate phenyl acetic acid and 2 mmol of thiosemicarbazide were dissolved in 1 mL of POCl3 and refluxed for 45 minutes. The reaction was then cooled to room temperature, and 3 mL of water were added carefully. The solution was then refluxed for 4 hours. The reaction mixture was filtered hot, and the solid was washed with warm water. The filtrate was basified with saturated KOH, and the solid was isolated by filtration. The solid was recrystallized from ethanol. The characteristics of each intermediate are as follows: TDA-1: 283 mg (64%), off-white crystals, mp 195–197°C; 1H NMR (300 MHz, DMSO) δ 3.81 (s, 3H), 4.19 (s, 2H), 5.01 (br s, 2H), 6.88 (d, 2H, 8.4), 7.22 (d, 2H, 8.4). MS (m/z) = 244.05 [M+Na]+; TDA-2: 203 mg (53%), off-white crystals, mp 182–184°C; 1H NMR (300 MHz, DMSO) δ 4.14 (s, 2H), 7.04 (s, 2H), 7.31 (m, 5H). MS (m/z) = 214.04 [M+Na]+; TDA-3: 317 mg (61%), off-white crystals, mp 182°C; 1H NMR (300 MHz, DMSO) δ 4.19 (s, 2H), 7.10 (s, 2H), 7.28 (d, 1H, 8.1), 7.58 (s, 1H), 7.60 (d, 1H, 8.1). MS (m/z) = 281.96 [M+Na]+; TDA-4: 307 mg (68%), off-white crystals, mp 196–198°C; 1H NMR (300 MHz, DMSO) δ 4.16 (s, 2H), 7.06 (s, 2H), 7.30 (d, 2H, 8.1), 7.39 (d, 2H, 8.1). MS (m/z) = 248.00 [M+Na]+; TDA-5: 205 mg (50%), off-white crystals, mp 204–206°C; 1H NMR (300 MHz, DMSO) δ 2.27 (s, 3H), 4.09 (s, 2H), 7.02 (s, 2H), 7.14 (s, 4H). MS (m/z) = 228.06 [M+Na]+; TDA-6: 217 mg (52%), off-white crystals, mp 206–208°C; 1H NMR (300 MHz, DMSO) δ 4.15 (s, 2H), 7.04 (s, 2H), 7.16 (t, 2H, 8.4) 7.32 (t, 2H, 8.4). MS (m/z) = 232.03 [M+Na]+; TDA-7: 368 mg (78%), off-white crystals, mp 187°C; 1H NMR (300 MHz, DMSO) δ 4.34 (s, 2H), 7.12 (s, 2H), 7.57 (d, 2H, 8.7), 8.20 (d, 2H, 8.7). MS (m/z) = 259.03 [M+Na]+; TDA-8: 216 mg (48%), off-white crystals, mp 193–195°C; 1H NMR (300 MHz, DMSO) δ 4.18 (s, 2H), 7.07 (s, 2H), 7.25 (d, 1H, 7) 7.34 (m, 3H). MS (m/z) = 248.00 [M+Na]+; TDA-9: 194 mg (41%), off-white crystals, mp 185–187°C; 1H NMR (300 MHz, DMSO) δ 2.86 (s, 6H), 3.99 (s, 2H), 6.68 (d, 2H, 8.4), 6.97 (s, 2H), 7.07 (2, 2H, 8.4). MS (m/z) = 257.08 [M+Na]+; TDA-10: 207 mg (50%), off-white crystals, mp 194–196°C; 1H NMR (300 MHz, DMSO) δ 2.26 (s, 3H), 4.10 (s, 2H), 7.02 (s, 2H), 7.06 (m, 3H), 7.21 (t, 1H, 7.5). MS (m/z) = 228.05 [M+Na]+.
Figure 2. Synthesis scheme for the OU749 analogs. Synthesis and purification of the synthetic intermediates TDA1-10 (A) was required before synthesis of the OU749 analogs Compounds 2–4 and Compounds 6–20 (B). Synthesis of Compound 5 is derived from Compound 11 (C).
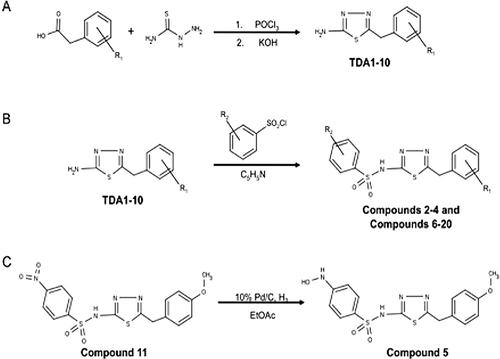
Synthesis of compounds 2–4 and compounds 6–20 ()
One mmol of the appropriate thiodiazole compound was dissolved in 1 mL of pyridine and cooled to 0°C. The appropriate benzenesulfonyl chloride (1.1 mmol) was then dissolved in 0.5 mL of pyridine and added to the reaction mixture. The reaction was allowed to warm to room temperature and stirred overnight. The reaction was diluted with 10 mL of water and acidified with dilute HCl. The solid was then filtered and washed with water. The product was recrystallized from ethanol. The characteristics of each OU749 analogs is as follows: OU749: 153 mg (42%), off-white crystals, mp 122–124°C; 1H NMR (300 MHz, DMSO) δ 3.75 (s, 3H), 4.12 (s, 2H), 6.93 (d, J = 8.6 Hz, 2H), 7.25 (d, J = 8.5 Hz, 2H), 7.64 – 7.45 (m, 3H), 7.76 (dd, J = 6.9, 1.5 Hz, 2H), 14.06 (s, 1H). MS (m/z) = 384.04 [M+Na]+; Compound 2: 259 mg (69%), off-white crystals, mp 143–145°C; 1H NMR (300 MHz, CDCl3) δ 2.41 (s, 3H), 3.84 (s, 3H), 4.07 (s, 2H), 6.91 (d, J = 8.8 Hz, 2H), 7.19 (d, J = 8.8 Hz, 2H), 7.25 (d, J = 7.9 Hz, 2H), 7.79 (d, J = 8.3 Hz, 2H), 11.57 (s, 1H). MS (m/z) = 398.06 [M+Na]+; Compound 10: 155 mg (39%), off-white crystals, mp 124–126°C; 1H NMR (300 MHz, CDCl3) δ 3.82 (s, 3H), 4.05 (s, 2H), 6.90 (d, J = 8.6 Hz, 2H), 7.17 (d, J = 8.4 Hz, 2H), 7.41 (d, J = 8.4 Hz, 2H), 7.81 (d, J = 8.4 Hz, 2H), 11.46 (s, 1H). MS (m/z) = 418.01 [M+Na]+; Compound 8: 208 mg (48%), off-white crystals, mp 135–137°C; 1H NMR (300 MHz, DMSO) δ 3.75 (s, 3H), 4.14 (s, 2H), 6.92 (d, J = 8.5 Hz, 2H), 7.25 (d, J = 8.5 Hz, 2H), 7.71 (dd, J = 8.4, 2.1 Hz, 1H), 7.82 (d, J = 8.4 Hz, 1H), 7.91 (d, J = 2.1 Hz, 1H), 14.25 (s, 1H). MS (m/z) = 451.97 [M+Na]+; Compound 9: 178 mg (45%), off-white crystals, mp 168–170°C; 1H NMR (300 MHz, CDCl3) δ 3.82 (s, 3H), 3.84 (s, 3H), 4.04 (s, 2H), 6.89 (d, J = 6.9 Hz, 2H), 6.92 (d, J = 7.2 Hz, 2H), 7.17 (d, J = 8.5 Hz, 2H), 7.82 (d, J = 8.8 Hz, 2H), 11.00 (s, 1H). MS (m/z) = 414.06 [M+Na]+; Compound 6: 27 mg (7%), off-white crystals, mp 170°C; 1H NMR (300 MHz, CDCl3) δ 3.81 (s, 3H), 4.01 (s, 2H), 6.88 (d, J = 8.6 Hz, 2H), 7.15 (d, J = 8.6 Hz, 2H), 7.48 (t, J = 7.8 Hz, 1H), 7.51 - 7.60 (m, 2H), 7.93 – 7.80 (m, 1H), 8.01 (d, J = 8.2 Hz, 1H), 8.24 (d, J = 7.3 Hz, 1H), 8.80 – 8.60 (m, 1H), 10.60 (s, 1H). MS (m/z) = 434.06 [M+Na]+; Compound 3: 185mg (47%), off-white crystals, mp 134–136°C; 1H NMR (300 MHz, CDCl3) δ 3.83 (s, 3H), 4.09 (s, 2H), 6.91 (d, J = 8.5 Hz, 2H), 7.19 (d, J = 8.5 Hz, 2H), 7.39 (t, J = 7.8 Hz, 1H), 7.51 (d, J = 7.8 Hz, 1H), 7.78 (d, J = 7.8 Hz, 1H), 7.91 (s, 1H). MS (m/z) = 418.01 [M+Na]+; Compound 4: 113 mg (26%), off-white crystals, mp 104–106; 1H NMR (300 MHz, DMSO) δ 3.74 (s, 3H), 4.13 (s, 2H), 6.92 (d, J = 8.4 Hz, 2H), 7.24 (d, J = 8.5 Hz, 2H), 7.60 (dd, J = 8.3, 2.3 Hz, 1H), 7.82 (d, J = 2.1 Hz, 1H), 7.99 (d, J = 8.4 Hz, 1H), 14.27 (s, 1H). MS (m/z) = 451.97 [M+Na]+; Compound 11: 214 mg (53%), off-white crystals, mp 187–190°C; 1H NMR (300 MHz, DMSO) δ 3.74 (s, 3H), 4.14 (s, 2H), 6.92 (d, J = 8.7 Hz, 2H), 7.25 (d, J = 8.7 Hz, 2H), 8.00 (d, J = 9.0 Hz, 2H), 8.35 (d, J = 9.0 Hz, 2H), 14.34 (s, 1H). MS (m/z) = 429.03 [M+Na]+; Compound 7: 110mg (26%), off-white crystals, mp 178–179°C; 1H NMR (CDCl3) (δ, ppm; J, hertz) 1.27 (s, 9H), 3.75 (s, 3H), 4.12 (s, 2H), 6.93 (d, 2H, 8.7), 7.25 (d, 2H, 8.7), 7.56 (d, 2H, 8.4), 7.67 (d, 2H, 8.4). MS (m/z) = 440.15 [M+Na]+; Compound 12: 254mg (59%), off-white crystals, mp 169–170°C; 1H NMR (CDCl3) (δ, ppm; J, hertz) 3.75 (s, 3H), 4.14 (s, 2H), 6.93 (d, 2H, 8.7), 7.25 (d, 2H, 8.7), 7.93 (d, 2H, 8.4), 7.97 (d, 2H, 8.4). MS (m/z) = 452.03 [M+Na]+; Compound 13: 223 mg (59%), off-white crystals, mp 198–200°C; 1H NMR (300 MHz, DMSO) δ 4.23 (s, 2H), 7.52 – 7.24 (m, 5H), 8.01 (d, J = 8.7 Hz, 2H), 8.36 (d, J = 8.7 Hz, 2H), 14.36 (s, 1H). MS (m/z) = 399.02 [M+Na]+; Compound 20: 219 mg (49%), off-white crystals, mp 199–201°C; 1H NMR (300 MHz, DMSO) δ 4.26 (s, 2H), 7.35 (dd, J = 8.1, 1.7 Hz, 1H), 7.63 (d, J = 8.2 Hz, 1H), 7.66 (d, J = 1.5 Hz, 1H), 8.03 (d, J = 8.9 Hz, 2H), 8.36 (d, J = 8.8 Hz, 2H), 14.41 (s, 1H). MS (m/z) = 466.94 [M+Na]+; Compound 16: 201 mg (49%), off-white crystals, mp 156–158°C; 1H NMR (300 MHz, DMSO) δ 4.24 (s, 2H), 7.37 (d, J = 8.4 Hz, 2H), 7.43 (d, J = 8.4 Hz, 2H), 8.02 (d, J = 8.9 Hz, 2H), 8.36 (d, J = 8.8 Hz, 2H), 13.86 (s, 1H). MS (m/z) = 432.98 [M+Na]+; Compound 17: 256 mg (66%), off-white crystals, mp 207–209°C; 1H NMR (300 MHz, DMSO) δ 2.29 (s, 3H), 4.16 (s, 2H), 7.17 (d, J = 8.4 Hz, 2H), 7.22 (d, J = 8.2 Hz, 2H), 8.00 (d, J = 8.6 Hz, 2H), 8.35 (d, J = 8.5 Hz, 2H), 14.34 (s, 1H). MS (m/z) = 413.04 [M+Na]+; Compound 19: 215mg (55%), off-white crystals, mp 189–192°C; 1H NMR (300 MHz, DMSO) δ 4.22 (s, 2H), 7.19 (t, J = 8.8 Hz, 2H), 7.47 – 7.33 (m, 2H), 8.01 (d, J = 8.8 Hz, 2H), 8.35 (d, J = 8.8 Hz, 2H), 14.37 (s, 1H). MS (m/z) = 417.01 [M+Na]+; Compound 14: 153 mg (36%), off-white crystals, mp 180–183°C; 1H NMR (300 MHz, DMSO) δ 4.42 (s, 2H), 7.63 (d, J = 8.7 Hz, 2H), 8.02 (d, J = 9.0 Hz, 2H), 8.23 (d, J = 8.8 Hz, 2H), 8.36 (d, J = 9.0 Hz, 2H), 14.55 (s, 1H). MS (m/z) = 422.02 [M+H]+; Compound 15: 194 mg (47%), off-white crystals, mp 185–188°C; 1H NMR (300 MHz, DMSO) δ 4.26 (s, 2H), 7.34 – 7.28 (m, 1H), 7.39 (dd, J = 5.4, 2.9 Hz, 2H), 7.45 (t, J = 1.9 Hz, 1H), 8.02 (d, J = 8.8 Hz, 2H), 8.36 (d, J = 8.8 Hz, 2H), 14.41 (s, 1H). MS (m/z) = 432.98 [M+Na]+; Compound 18: 211 mg (54%), off-white crystals, mp 178–180°C; 1H NMR (300 MHz, DMSO) δ 2.30 (s, 3H), 4.18 (s, 2H), 7.12 (d, J = 7.9 Hz, 3H), 7.26 (t, J = 7.6 Hz, 1H), 8.01 (d, J = 8.8 Hz, 2H), 8.35 (d, J = 8.8 Hz, 2H), 14.34 (s, 1H). MS (m/z) = 413.04 [M+Na]+.
Synthesis of compound 5 ()
Compound 11 (130 mg, 0.32 mmol) and 10%Pd/C (15.4 mg) were dissolved in EtOAc (7 mL). The reaction was degassed five times and stirred at RT for 24 hours under H2. The reaction was filtered through celite and concentrated in vacu. Product was recrystallized from EtOAc and Hexanes. The characteristics of Compound 5 are: 112 mg (89%), off-white crystals, mp 167–168°C; 1H NMR (300 MHz, DMSO) δ 3.74 (s, 3H), 4.10 (s, 2H), 6.83 (d, J = 8.8 Hz, 2H), 6.92 (d, J = 8.6 Hz, 2H), 7.24 (d, J = 8.6 Hz, 2H), 7.52 (d, J = 8.7 Hz, 2H), 8.65 (s, 1H), 8.93 (s, 1H), 13.87 (s, 1H). MS (m/z) = 415.05 [M+Na]+.
Enzyme isolation
Human GGT1 (P19440), lacking the transmembrane domain, was expressed in Pichia pastoris and isolated as previously describedCitation14. The specific activity of the purified enzyme was 406.5 units/mg. One unit of GGT activity was defined as the amount of enzyme that released 1 µmol of paranitroaniline/min at 37°C at pH 7.4 in the transpeptidation reaction.
Hydrolysis reaction
The assay buffer contained: 100 mM Na2HPO4, 3.2 mM KCl, 1.8 mM KH2PO4, and 27.5 mM NaCl pH 7.4. The concentration of the d-GpNA (Bachem,Torrance, CA) substrate was varied from 0.25 mM to 3 mM d-GpNA. The reaction was initiated with the addition of 19 mU GGT. The reaction was incubated at 37°C and monitored continuously at 405 nm by a Bio-Rad model 680 microplate reader with Microplate Manager 5.2 software (Bio-Rad, Hercules, CA).
Transpeptidation reaction
The same assay buffer was used for both the hydrolysis reaction and the transpeptidation reaction. The transpeptidation reaction included 40 mM glycylglycine (glygly, Sigma, St. Louis, MO) as the acceptor. The concentration of the substrate for the transpeptidation reaction, l-GpNA (Sigma) was varied from 0.25 mM to 3 mM. The concentration of l-GpNA was 3 mM for experiments in which the concentration of glygly was varied. To initiate the transpeptidation reaction, 4 mU GGT were added. The reaction was incubated at 37°C and monitored continuously at 405 nm.
Data Analysis
Samples in each experiment were run in triplicate. Each inhibitor was evaluated in two or more independent experiments. Double reciprocal plots were generated to assess data quality and determine the correct rate equation for data fitting. Data were fitted using the proper rate equation and the Marquardt-Levenberg algorithm supplied with the Enzfitter program (BIOSOFT, Cambridge, UK). Kinetic parameters with standard errors were estimated using a simple weighting method.
Data adhering to Michaelis-Menten Kinetics were fitted to eq. 1. Data for initial rate patterns with d-, l-GpNA varied at different fixed levels of glygly were fitted to eq. 2, which describes a ping pong kinetic mechanism. Data for competitive and uncompetitive inhibition were fitted to eqs. 3 and 4. Data for the dependence of Vmax and Vmax/KD-GpNA on the concentration of activator were fitted using eq. 5.Citation25
In eqs 1–5, v and Vmax are the initial and maximum rates, respectively, Ka and Kb are Michaelis constants for substrates A and B, respectively, and Kis and Kii are slope and intercept inhibition constants. In eq. 5, a is the value of Vmax or Vmax/KD-GpNA at zero activator/inhibitor, KID is the activation or inhibition constant, and KIN is a constant that causes the parameter to go to a constant value at infinite concentrations of X, the activator/inhibitor. The product a(KID/KIN) is the value of Vmax or Vmax/KD-GpNA at infinite activator/inhibitorCitation25.
For irreversible inhibition studies, time courses were fit to eq. 6.
In eq. 6, A is the burst amplitude, t is time, and k is the first order rate constant for formation of the inactivated enzyme. Graphs, averages, and standard error between experiments were calculated using Prism GraphPad Software (San Diego, CA).
Results
Initial Rate Studies with D- and L-GpNA
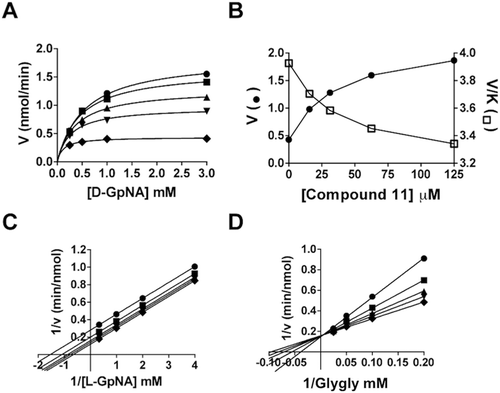
In this study, we examined the inhibition of both the GGT hydrolysis and transpeptidation reactions by OU749, and a new series of structural analogs. The hydrolysis and transpeptidation reactions were analyzed separately by using different stereo-isomers of the γ-glutamyl-p-nitroanilide (GpNA) substrate. We first characterized the kinetics of both reactions in the absence of inhibitors. Cleavage of the γ-glutamyl bond of either d- or l-GpNA releases p-nitroaniline, which has a ϵ405 of 7,680 M−1cm−1, allowing for the reactions to be monitored continuously with high sensitivity. The d-glutamyl isomer of GpNA was used as the substrate for the hydrolysis reaction, in which water serves as the acceptor. The l-glutamyl isomer of GpNA, with glycylglycine (glygly) as an acceptor, was used for the transpeptidation reaction. l-GpNA can serve as both a substrate and a low affinity acceptor, while d-GpNA is unable to serve as an acceptorCitation18,Citation21,Citation26. Therefore, in the absence of an added acceptor, GGT exclusively catalyzes a hydrolysis reaction with d-GpNA as the substrate. When l-GpNA is used as a substrate, the GGT reaction is a mixture of both the hydrolysis and transpeptidation reactions. Under these conditions, the rate of the reaction is the sum of hydrolysis and transpeptidation reactions, with the hydrolysis reaction contributing more at low l-GpNA concentrations and transpeptidation dominating at saturating substrate concentrations. For the hydrolysis reaction, the second order rate constants, V/KGpNA, were similar for d-GpNA and l-GpNA (13.2 ± 0.1 vs 7.8 ± 0.2 min−1nM−1), indicating that, at limiting substrate concentrations, the rate of the hydrolysis reaction is similar with d-GpNA and l-GpNA (). However, the maximum rate (Vmax) obtained with l-GpNA was approximately three times faster than that measured with d-GpNA, and the KGpNA for l-GpNA was five times higher than that measured for d-GpNA. This is at least partially due to the ability of l-GpNA to serve as an acceptor, resulting in a transpeptidation reaction, which is faster than the hydrolysis reaction. Therefore, to study the hydrolysis reaction in isolation, we used d-GpNA as the substrate without any added acceptor.
Table 1. Kinetic parameters for reaction with d- and l-GpNA.
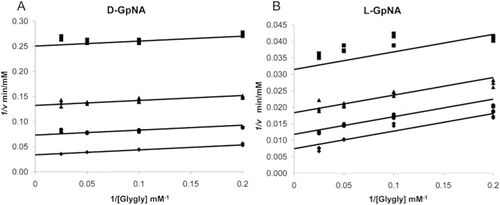
Characterization of the transpeptidation reaction with both d- and l-GpNA showed that addition of the acceptor, glygly, accelerates the reaction with both substrates (). In the presence of glygly, the kinetic mechanism is ping-pong with d-GpNA or l-GpNA as the donor and glygly acting as an acceptorCitation21,Citation27. In both cases, data fit well to the rate equation for a ping-pong mechanism, eq. 2, as demonstrated by the parallel lines in the double reciprocal plots ( and ). The measured Vmax for d- and l-GpNA reactions increased in the presence of glygly by 25- and 22-fold, respectively (). However, some notable differences were observed in the kinetics of the transpeptidation reaction with d- versus l-GpNA. The ratio of the maximum rates of the two reactions was almost 3:1 in favor of l-GpNA over d-GpNA in the presence of glygly (141 ± 11 vs 53 ± 11 mM/min·nM). While there was no change in the V/K for d-GpNA in the presence of an acceptor, the V/K for l-GpNA increased by about 15-fold in the presence of glygly. Insights into the mechanism of the reaction with l-GpNA provided by these data are included in the discussion. To evaluate the affect of the inhibitors on the transpeptidation reaction, we used the physiologic l-isomer as a substrate with glygly as the acceptor.
Figure 3. Initial velocity pattern with d-GpNA or l-GpNA as the substrate and glygly as an accepter. Initial rates were measured at pH 7.4 and 37 °C. Double recipricol plots of the initial velocity versus the glygly concentration with 0.25 mM (♦),0.5 mM (•),1 mM (▴), or 3 mM (▪) d-GpNA (A) or l-GpNA (B).
Inhibition of the GGT hydrolysis and transpeptidation reactions by OU749
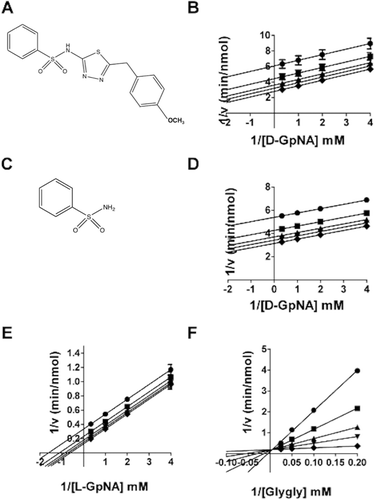
OU749 () is an uncompetitive inhibitor of GGT transpeptidationCitation14. To determine the mechanism by which OU749 inhibits the hydrolysis reaction, the rate of the hydrolysis reaction was determined as a function of d-GpNA concentration and different concentrations of OU749, including zero. Double reciprocal plots of the data gave rise to a series of parallel lines (). This pattern is indicative of uncompetitive inhibition and binding of OU749 to the covalent E-γ-glutamyl complex (the F-form of the enzyme). This is the same mode of inhibition observed previously for OU749 versus l-GpNA with glygly as an acceptorCitation14. A Kii of 73 µM was obtained for OU749 in the hydrolysis reaction, which is similar to the Kii of 68 µM that was obtained in the transpeptidation reaction with the l-GpNA substrate plus acceptor glygly (). These values are comparable to the Kii value of 73 µM for OU749 measured previously in the transpeptidation reactionCitation14. Maintaining the l-GpNA substrate concentration at 3 mM while varying the concentration of the acceptor, glygly, gave rise to a competitive inhibition profile with a Kis of 54 µM, confirming previously reported data with glygly as an acceptorCitation14. Thus, OU749 inhibits GGT by binding to the acceptor site.
Table 2. Inhibition of GGT by structural analogs of OU749.
Figure 4. Kinetic analysis of GGT inhibition by OU749 and SBS. The structure of OU749 (A). Double-reciprocal plot of the initial velocity of the hydrolysis of d-GpNA in the presence of 0 (♦), 15.2 µM (▾), 31.25 µM (▴), 62.5 µM (▪), 125 µM (•) OU749 (B). The structure of SBS (C). Double reciprocal plot of the initial velocity of the hydrolysis of d-GpNA in the presence of 0 (♦), 6.25 mM (▾), 12.5 mM (▴), 25 mM (▪), 50 mM (•) SBS (D). Double reciprocal plot of the initial velocity of the transpeptidation reaction with varying concentrations of l-GpNA and 40 mM glygly (E) or varying concentrations of glygly with 3 mM l-GpNA (F) in the presence of 0 (♦), 6.25 mM (▾), 12.5 mM (▴), 25 mM (▪), 50 mM (•) SBS. Data shown are average triplicate values ± S.D. For many data points the S.D. is smaller than the symbol.
OU749 substructure inhibition of GGT
Sodium benzosulfonamide (SBS, ) is a substructural component of OU749. The effect of SBS on both the hydrolysis and transpeptidation reactions was evaluated. The data show that SBS is an uncompetitive inhibitor verus d-GpNA in the hydrolysis reaction (). This is the same mechanism observed for OU749 (). However, in the hydrolysis reaction, SBS was more than 2,000-fold less effective than OU749 with a Kii of 160 ± 81 mM (). These data suggest that, although the SBS moiety provides some of the binding energy of OU749, it is a minor contributor. SBS also qualitatively mimics OU749 as an inhibitor of transpeptidation. It is uncompetitive versus l-GpNA () and competitive versus glygly (). SBS was several orders of magnitude less potent than OU749 at inhibiting the transpeptidation reaction. For SBS, the Kii and Kis values were, 28.8 ± 1.6 mM and 2.28 ± 0.07 mM, respectively, compared with 68 µM and 54 µM for OU749. The difference in the potency of SBS versus OU749 as an inhibitor of both the hydrolysis and transpeptidation reactions implies that the benzosulfonamide portion of OU749 contributes to the inhibition of GGT by blocking access of the acceptor to the acyl bond, although SBS is much less effective than OU749 in this regard. To determine the impact of the benzosulfonamide ring on GGT inhibition, we modified this substructure in the context of OU749 and assessed the effect of these modifications in the hydrolysis and transpeptidation reactions.
Modification of the benzosulfonamide ring of OU749
A series of structural analogs of OU749 with substitutions at the para (X), meta (W), or ortho (V) positions on the benzosulfonamide ring were evaluated as inhibitors of both the hydrolysis and transpeptidation reactions of GGT (). The compounds fall into a number of classes of inhibitory potency compared to OU749. Compounds 2–4, with p-methyl, m-chloro, or p, o-dichloro substitutions had no effect on inhibition of the hydrolysis reaction but were three to five times more effective inhibitors of the transpeptidation reaction. These data suggest that the aforementioned substitutions do not alter the stability of the ES complex. These compounds are equivalent to OU749 in their ability to block access of water to the acyl bond but are more effective than OU749 in blocking access of the dipeptide acceptors to the acyl bond in the transpeptidation reaction. Compounds 5–7 with the hydrophilic p-hydroxylamino, the bulky l-naphthylsulfonamide ring, or p-tBu substitutions are two to five times less effective than OU749 in inhibiting the hydrolysis reaction but have smaller effects as inhibitors of transpeptidation. The latter is also true of compounds 8–10 with p-, m-dichloro, p-methoxy or p-chloro substituents, which are 40 to 75 times poorer inhibitors of the hydrolysis reaction but, with the exception of the bulkier p-methoxy, are better inhibitors of the transpeptidation reaction. Comparison of the data obtained with compounds 3 and 4 with the data for compounds 8 and 10 reveals that the combination of Cl substituents alters the ability of the inhibitor to block hydrolysis of the acyl bond but has little effect on the transfer of the acyl bond to an acceptor. For the hydrolysis reaction, the detrimental effect of having a Cl substituent in the para position can be ameliorated by ortho, but not meta, substitution in the same molecule, while meta substitution alone generates no effect.
Introduction of a highly electron-withdrawing group, -NO2 or -CF3, in the para (X) position (Compounds 11 and 12, ) resulted in activation of the hydrolysis reaction. However, addition of these groups maintained inhibition of the transpeptidation reaction, with less than a 2-fold effect on the Kii relative to OU749. Data are shown for Compound 11 (). A detailed analysis of the kinetics of the hydrolysis reaction is shown in and . At each of the concentrations of Compound 11, including zero, data were first fitted to eq. 1 to obtain estimates of Vmax and Vmax/KD-GpNA. The limiting rate constants were fitted to eq. 5, which allows for an estimate of the values of the rate constant at zero and infinite concentration of the compound and a determination of its activation constant (Kact). Note that activation is observed on Vmax, while inhibition is observed for Vmax/KD-GpNA (). The compound is thus a V-type activator in the hydrolysis reaction. GGT inhibition by Compound 11 was also analyzed in the transpeptidation reaction. Compound 11 is an uncompetitive inhibitor versus l-GpNA in the transpeptidation reaction, indicating that it binds to the l-γ-glutamyl intermediate, presumably at the acceptor site (). The Kis was 93 µM. The Kii of Compound 11 while varying glygly in the transpeptidation reaction was 96 µM (). The affinity of Compound 11 for the d-γ-glutamyl enzyme intermediate and the l-γ-glutamyl enzyme intermediate differ by only 2.5-fold, suggesting that there may be a minor difference in the acceptor site or a slight difference in the orientation of the acyl bond depending on the isomer of glutamate that is bound.
Figure 5. Kinetic analysis of GGT inhibition by Compound 11. Substrate velocity curve of the hydrolysis reaction versus d-GpNA concentration in the presence of 0 (♦), 15.2 µM (▾), 31.25 µM (▴), 62.5 µM (▪), 125 µM (•) Compound 11 (A). Velocity vs Compound 11 concentration (closed symbols) and V/K vs Compound 11 concentration (open symbols) plots illustrate the activation of the hydrolysis reaction (B). Double-reciprocal plots of the initial velocities of the transpeptidation reaction varying l-GpNA with 40 mM glygly (C) or varying glygly with 3 mM l-GpNA (D) in the presence of 0 (♦), 15.2 µM (▾), 31.25 µM (▴), 62.5 µM (▪), 125 µM (•) Compound 11. Data shown are average triplicate values ± S.D.
Modification of the benzyl ring, structure activity studies
In order to determine whether modification of the phenyl ring remote to the benzosulfonamide ring has an effect on the behaviour of Compound 11, a series of structural analogs of Compound 11 were synthesized with modifications to this portion of the inhibitor. Kinetic parameters for modifications of the benzyl ring of nitro compounds are shown in . A 3- to 6-fold activation of the Vmax for hydrolysis (the ratio of V∞ to Vo in ) was observed with an activation constant, Kact, of 22–54 µM for Compounds 13 thru 19. These activators may alter the conformation of the enzyme or be oriented differently than OU749, allowing greater access of water to the acyl bond.
Table 3. Kinetic parameters for activators of the D-GpNA hydrolysis reaction and Inhibition of the L-GpNA transpeptidation reaction.
In the transpeptidation reaction, the nitro-containing derivatives of Compound 11 behave in a manner that is both qualitatively and quantitatively similar to the parent compound, OU749 (). Addition of a chlorine group at the Y position resulted in an inhibitor (, Compound 16) with a Kii of 61.9 µM in the transpeptidation reaction. An OCH3 at the Y position increased the l-GpNA Kii in the transpeptidation reaction less than 2-fold and increased the glygly Kis more than 2-fold (, Compound 11) when compared with Compound 16. No substitution on the benzyl ring (, Compound 13) increased the l-GpNA Kii by almost 2-fold compared to Compound 16, while only slightly increasing the glygly Kis. Addition of a methyl group at the Y position yielded results similar to having no substitution (, Compound 17). Addition of any of a series of chemical groups at the Z position reduced the strength of this family of inhibitors in the transpeptidation reaction by 2-fold, while slightly increasing the strength of the compound as a competitive inhibitor of the acceptor, glygly (, Compounds 15 and 18). Addition of an NO2 at the Y position (, Compound 14) more than doubled the Kii and Kis relative to Compound 16 in the transpeptidation reaction. There is very little difference in the behaviour of any of these compounds in the transpeptidation reaction with the exception of Compound 19 and Compound 20, which exhibited time dependent inhibition and, together with acivicin, will be discussed below.
Acivicin and acivicin-like inhibition
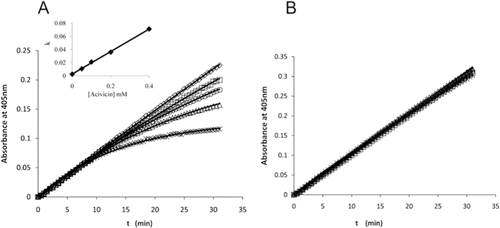
Acivicin is an analog of γ-glutamyl donor substrates [GpNA, GSH, etc.] and exhibits reversible competitive inhibition against these substrates with a very slow rate of releaseCitation27. The time course for production of pNA in the presence of increasing concentrations of acivicin is shown in . The time course for hydrolysis of 0.287 mM d-GpNA in the absence of acivicin is linear for at least 30 minutes (). In the presence of increasing concentrations of acivicin, the initial rate is unchanged, but the rate decreases with increasing time, with the most pronounced decrease seen at the highest acivicin concentration (). A fit of the time courses to the equation for a first-order process gives an observed rate constant, kobs, that is a linear function of acivicin concentration. This linear dependence is expected for an irreversible inhibitor, where kobs is the pseudo-first order net on-rate constant for modification of enzyme by acivicin times the concentration of acivicin. The slope of the line depicted in the inset of thus describes the second order rate constant, kon, as 0.171 mM−1min−1. Since acivicin is competitive versus d-GpNA, kobs is equal to the pseudo-first order rate constant divided by (1 + [d-GpNA]/KD-GpNA). Given a KD-GpNA of 0.16 mM (), the kon, corrected for the presence of d-GpNA, is 0.48 mM−1min−1.
Figure 6. Time courses for the hydrolysis of d-GpNA in the presence of acivicin. The release of pNA from 0.287 mM d-GpNA (A) or 2 mM d-GpNA (B) was monitored over time in the presence of acivicin (concentrations of acivicin noted in A). The reactions were conducted at pH 7.4. The increase in the Km of d-GpNA with increasing concentrations of acivicin is shown (insert 6A).
Kinetic analysis of acivicin was also carried out for the transpeptidation reaction using l-GpNA or l-GpNA with glygly, and the results were qualitatively the same (data not shown). In the transpeptidation reaction with l-GpNA, the time courses were curvilinear even in the absence of acivicin. Unlike d-GpNA, which can only act as a donor to form the γ-glutamyl enzyme intermediate (F-form), l-GpNA can also act as an acceptorCitation18,Citation21,Citation26. As a result, a complete analysis was only carried out for the hydrolysis reaction using d-GpNA.
Increasing the d-GpNA concentration to saturation eliminated the development of inhibition by acivicin over the same time course as would be expected for competitive inhibition against d-GpNA (). Time-dependent inhibition of the transpeptidation reaction was observed for the monofluoro- (Compound 19) and dichloro- (Compound 20) substituted compounds shown in , suggesting that they also exhibit either irreversible or slow-onset inhibition. A complete analysis of this phenomenon is outside the scope of the present studies.
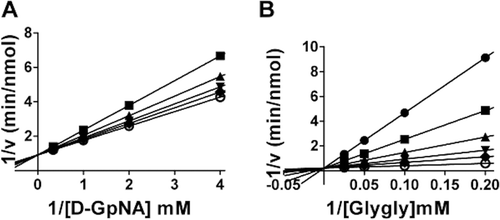
All of the nitro-containing OU749 analogs accelerated the hydrolysis reaction except Compound 20 (). Compound 20 has chlorines both para (Y) and meta (Z) on the benzyl ring and a NO2 at the para position on the benzosulfonamide ring. The inhibition mechanism of Compound 20 is unique among the OU749 analogs, because it is competitive with the γ-glutamyl substrate in the hydrolysis reaction, yielding a Kii with d-GpNA of 78 µM (). In addition to being competitive with the γ-glutamyl substrate, Compound 20 is also competitive with the acceptor glygly in the transpeptidation reaction (), with a Kis of 10 µM (). However, we were unable to establish a Kii with l-GpNA in the transpeptidation reaction, because Compound 20 exhibits slow-onset binding similar to acivicin. Thus, the transpeptidation reaction is too complex for accurate analysis and establishment of inhibition constants for this compound.
Figure 7. Kinetic analysis of GGT inhibition by Compound 20. Double-reciprocal plot of the initial velocity of the hydrolysis of d-GpNA in the presence of 0 (♦), 15.2 µM (▾), 31.25 µM (▴), 62.5 µM (▪), 125 µM (•) Compound 20 (A). Double reciprocal plot of the initial velocity of the transpeptidation reaction with varying concentrations of glygly with 3 mM l-GpNA (B) in the presence of 0 (○), 15.2 µM (♦), 31.25 µM (▾), 62.5 µM (▴), 125 µM (▪), 250 µM (•) Compound 20. Data shown are average triplicate values ± S.D. For many data points the S.D. is smaller than the symbol.
Discussion
In this analysis, new structural analogs of OU749 were evaluated for their inhibitory activity in both the hydrolysis and transpeptidation reactions. All of the new analogs were weaker inhibitors of the hydrolysis reaction relative to OU749, but some were more potent inhibitors than OU749 in the transpeptidation reaction. In addition to structure-activity information, the divergent results provide insights into the reaction catalyzed by GGT. In both the hydrolysis and transpeptidation reactions, OU749 is an uncompetitive inhibitor of the donor γ-glutamyl substrate. The data provided herein are consistent with OU749 binding to the F-form of the enzyme. Despite the fact that l-GpNA has the capacity to act as an acceptor in the transpeptidation reaction, its maximum turnover rate was only about three-times higher than that measured for d-GpNA in the absence of glygly (). Thus, although l-GpNA can act as an acceptor, it is not very effective. The second order rate constant measured with l-GpNA is similar to the rate constant with d-GpNA (8 min−1nM−1 and 13 min−1 nM−1, respectively), indicating that formation of the γ-glutamyl-enzyme intermediate (F-form) is independent of the stereochemistry of the γ-glutamyl moiety on the nitroanilide substrate. To develop effective inhibitors for therapeutic use, each compound must be evaluated in the physiologic hydrolysis reaction.
We began our comparative analyses by characterizing the substructural elements of OU749 that contribute to its efficacy as an inhibitor. The terminal benzosulfonamide moiety of OU749, SBS, was evaluated for activity as an inhibitor ( and ). SBS is an uncompetitive inhibitor versus d- and l-GpNA and a competitive inhibitor versus glygly in the l-GpNA reaction. We expanded our analysis to a series of OU749 analogs with modifications on the benzosulfonamide ring. As with SBS, Compounds 2–10 were more potent inhibitors of the transpeptidation reaction than the hydrolysis reaction (). However, compared to OU749, all of these structural analogs bound with either equal (Compounds 2–4) or lower (Compounds 5–10) affinity to the d-γ-glutamyl enzyme. As observed for glygly, analogs of OU749 with a strong electron-withdrawing group (-NO2 or – CF3) para to the sulfonamide group activate the hydrolysis reaction with d-GpNA (Compounds 11 and 12, ). Both analogs inhibit the reaction with l-GpNA and exhibit an affinity very similar to that of Compounds 2–10 in . Thus, as was observed for glygly in the absence of inhibitors, Compounds 11 and 12 activate the hydrolysis reaction. This suggests that like glygly, they bind to the acceptor site of the E~γ-d-glutamyl intermediate (F-form), and in doing so, they may alter the conformation of the intermediate in a manner that allows a more facile attack by water on the ester carbonyl. The SO2 of the benzosulfonamide group may interact with residues in or near the active site.
All of the OU749 analogs containing a nitroxide group (Compounds 11, 13–20), except Compound 20, accelerated the hydrolysis reaction. However, these same analogs inhibited the transpeptidation reaction with an affinity (Kii) very similar to that of Compounds 2–10 (). A series of analogs in which the methoxyphenyl ring of Compound 11 was modified () elicited no major effect on the inhibition of the transpeptidation reaction, with the exception of p-F or p-,m-dichloro analogs (Compounds 19 and 20), which exhibited a distinct mode of inhibition similar to that observed for acivicin.
Acivicin is an analog of the γ-glutamyl donor substrateCitation27–30. It exhibits time-dependent inhibition at low, but not at high, concentrations of l-GpNA (). The second order rate constant for binding acivicin, kon, is 0.48 mM−1 min−1. This is the same type of inhibition of the transpeptidation reaction as was observed for Compounds 19 and 20, indicating that they also exhibit either irreversible or time-dependent inhibition. Of further interest is the fact that the p-, m-dichloro analog (Compound 20) is the only compound in this series of analogs that does not accelerate the hydrolysis reaction but, rather, serves as a competitive inhibitor of the d-GpNA substrate. This (p-, m-dichloro analog) is the only OU749 analog that inhibits a GGT reaction by competing with the γ-glutamyl substrate.
To fully understand the affect of the OU749 analogs in the hydrolysis and transpeptidation reactions, the reactions had to be independently analyzed with both donor substrates, d- and l-GpNA. In the absence of the acceptor glygly, the second order rate kinetics are similar for both d- and l-GpNA in the GGT hydrolysis reaction, indicating the stereochemistry of the γ-glutamyl group of the substrate does not affect the binding and cleavage of the substrate in the hydrolysis reaction. However, when the acceptor glygly is present in the transpeptidation reaction, with l-GpNA serving as the donor substrate, the second order rate kinetics increase by 15-fold. This dramatic increase suggests three possibilities (i) the binding of glygly increases the rate of hydrolysis, (ii) glygly could be binding to another site, perhaps an affector site, instead of to the acceptor site that increases the turnover rate, or (iii) glygly could be binding free enzyme, forming an E-glygly complex, prior to l-GpNA binding. The third possibility would imply a sequential mechanism instead of a ping-pong mechanism. In a ping-pong mechanism, the donor substrate’s second order rate constant (V/K) will not change in the absence or presence of an acceptor molecule. A sequential mechanism can also yield the classic ping-pong plots, if the Km for glygly is greater than the Ki of glygly for the E-glygly complex. This is only possible when the glygly concentration is varied around the Km of glygly, as was carried out in these studies and previous studies of the GGT transpeptidation reactionCitation14,Citation31,Citation32.
It is difficult at best to model the results obtained in this study on a molecular level. The crystal structure for human GGT has not yet been determined. Homology models based on the crystal structures of bacterial GGT lack the resolution necessary for molecular modelingCitation33–35. Mutational analysis have indicated that the active nucleophile Thr-381, Arg-107 and Asp-423 interact with the α-amino and the α-carboxylate of glutamateCitation29–30,Citation36. Two adjacent serines (Ser-451 and Ser-452) have also been proposed to stabilize the rate-limiting transition state of the catalytic reactionCitation29. Identification of amino acids involved in the binding of the acceptor has been more elusive, and there is little solid structural work available to delineate the “acceptor site” of GGT from any organism. All of the inhibitors tested, with the exception of acivicin, target the acceptor site of human GGT. Although one could speculate as to the residues or sites with which different inhibitors interact, no definitive statements can be made until a reasonable structure is obtained with the acceptor site occupied.
A schematic representation of the hydrolysis and transpeptidation reaction in the presence of a high affinity competitive inhibitor (acivicin) and an uncompetitive inhibitor (i.e. OU749 and analogs, I) is presented in . In the hydrolysis reaction (), d-GpNA binds to GGT, and the γ-glutamyl moiety is cleaved, releasing pNA. The d-γ-glutamyl group is retained by GGT establishing the F-form. Water can then hydrolyze the acyl bond of the F-form, releasing d-glutamate and free enzyme. Acivicin can inhibit GGT by binding the free enzyme, competing with the γ-glutamyl substrate, thus inhibiting both the hydrolysis and transpeptidation reactions equally. After establishment of the F-form of GGT in the transpeptidation reaction with l-GpNA (), the acyl bond can be hydrolyzed by water, or attacked by an acceptor (i.e. glygly or l-GpNA). l-glutamate, l-γ-glutamyl-glygly, or l-γ-glutamyl-l-GpNA are then released, and the enzyme returns to the free enzyme state. OU749 and its analogs are uncompetitive, in that they bind the F-form of the enzyme, competing with the acceptor. Therefore, OU749 and its analogs only inhibit the second half of the reaction of GGT: the attack of the acyl bond by either water or an acceptor.
Figure 8. Proposed overall kinetic mechanism of inhibition and activation of GGT. Mechanism of hydrolysis of d-GpNA (A). Enzyme (E) is γ-glutamylated by d-GpNA with release of pNA to give a form of the enzyme (F) that can transfer the γ-glutamyl moiety to water. d-GpNA is not capable of acting as an acceptor. The ester bond can be hydrolyzed with rate constant khyd, and this rate is decreased in the presence of an inhibitor such as OU749 (F-I), or increased in the presence of an activator such as Compound 11 giving rate k’hyd. Acivicin can bind to the free enzyme at the acceptor site, and is slowly trapped (tightly bound) on enzyme. Mechanism of transpeptidation of GpNA (B). Enzyme (E) is γ-glutamylated by GpNA with release of pNA to give a form of the enzyme (F) that can transfer the γ-glutamyl moiety to an acceptor (l-GpNA and/or glygly). The ester bond can be hydrolyzed with rate constant khyd, but competition between water, l-GpNA and/or glygly decreases the rate of hydrolysis. As in panel A, acivicin can bind to the free enzyme at the acceptor site, and is slowly trapped on enzyme.
Expression of GGT on the surface of the cell initiates the cleavage of extracellular GSH, thereby releasing cysteine and providing an additional source of cysteine for intracellular GSH synthesisCitation5,Citation37. GSH and free cysteine within the cell are potent reducing agents that protect the cell against oxidative stress and detoxify electrophilic metabolites. Conjugation of many chemotherapy drugs to GSH result in their inactivation, and the conjugates are then exported from the cellCitation38. GSH has been shown to be increased in various cancers including breastCitation39, lungCitation40, bone marrowCitation41, ovarianCitation42, head and neck, and laryngealCitation43. Increased intracellular GSH has also been shown to contribute to the inhibition of apoptosis by inducing Bcl-2 and inducing resistance to antihormonal therapyCitation44,Citation45. Studies have shown that, in mice, GGT-positive tumours are more resistant to treatment with cisplatin than GGT-negative tumoursCitation6. In Phase I clinical trials, acivicin was found to be neurotoxic and cannot be used clinically as an inhibitor of GGTCitation10–13. Therefore, less toxic inhibitors need to be developed. A complete understanding of the mechanism of inhibition of GGT is crucial to optimize GGT inhibitors for clinical use. In the current study, the stereochemistry of the donor substrates provided insight into the heterogeneity of the acceptor site, and the potential interaction of the OU749 analogs within the acceptor site. These studies also emphasize the need to study the mechanism of inhibition of GGT in both the hydrolysis and transpeptidation reactions. The hydrolysis reaction is the physiologic reaction, and inhibitors of GGT need to be thoroughly analyzed and optimized in this reaction.
Declaration of interest
This work was supported, in whole or in part, by National Institutes of Health Grant RO1CA57530 (M.H.H.) and the Grayce B. Kerr endowment to the University of Oklahoma to support the research (to P.F.C.).
References
- Hanigan MH. γ-Glutamyl transpeptidase, a glutathionase: its expression and function in carcinogenesis. Chem Biol Interact 1998;111–112:333–342.
- Hanigan MH, Frierson HF Jr. Immunohistochemical detection of γ-glutamyl transpeptidase in normal human tissue. J Histochem Cytochem 1996;44:1101–1108.
- Lieberman MW, Wiseman AL, Shi ZZ, Carter BZ, Barrios R, Ou CN et al. Growth retardation and cysteine deficiency in γ-glutamyl transpeptidase-deficient mice. Proc Natl Acad Sci USA 1996;93:7923–7926.
- Hanigan MH, Frierson HF Jr, Swanson PE, De Young BR. Altered expression of γ-glutamyl transpeptidase in human tumors. Hum Pathol 1999;30:300–305.
- Ruoso P, Hedley DW. Inhibition of γ-glutamyl transpeptidase activity decreases intracellular cysteine levels in cervical carcinoma. Cancer Chemother Pharmacol 2004;54:49–56.
- Hanigan MH, Gallagher BC, Townsend DM, Gabarra V. γ-glutamyl transpeptidase accelerates tumor growth and increases the resistance of tumors to cisplatin in vivo. Carcinogenesis 1999;20:553–559.
- Hanigan MH, Pitot HC. γ-glutamyl transpeptidase–its role in hepatocarcinogenesis. Carcinogenesis 1985;6:165–172.
- Lowry MH, McAllister BP, Jean JC, Brown LA, Hughey RP, Cruikshank WW et al. Lung lining fluid glutathione attenuates IL-13-induced asthma. Am J Respir Cell Mol Biol 2008;38:509–516.
- Ahluwalia GS, Grem JL, Hao Z, Cooney DA. Metabolism and action of amino acid analog anti-cancer agents. Pharmacol Ther 1990;46:243–271.
- Weiss GR, McGovren JP, Schade D, Kufe DW. Phase I and pharmacological study of acivicin by 24-hour continuous infusion. Cancer Res 1982;42:3892–3895.
- Earhart RH, Koeller JM, Davis TE, Borden EC, McGovren JP, Davis HL et al. Phase I trial and pharmacokinetics of acivicin administered by 72-hour infusion. Cancer Treat Rep 1983;67:683–692.
- Fleishman G, Yap HY, Murphy WK, Bodey G. Phase II trial of acivicin in advanced metastatic breast cancer. Cancer Treat Rep 1983;67:843–844.
- Taylor S, Belt RJ, Joseph U, Haas CD, Hoogstraten B. Phase I evaluation of AT-125 single dose every three weeks. Invest New Drugs 1984;2:311–314.
- King JB, West MB, Cook PF, Hanigan MH. A novel, species-specific class of uncompetitive inhibitors of γ-glutamyl transpeptidase. J Biol Chem 2009;284:9059–9065.
- Tate SS, Meister A. Interaction of γ-glutamyl transpeptidase with amino acids, dipeptides, and derivatives and analogs of glutathione. J Biol Chem 1974;249:7593–7602.
- Tate SS, Meister A. Stimulation of the hydrolytic activity and decrease of the transpeptidase activity of γ-glutamyl transpeptidase by maleate; identity of a rat kidney maleate-stimulated glutaminase and γ-glutamyl transpeptidase. Proc Natl Acad Sci USA 1974;71:3329–3333.
- Castonguay R, Lherbet C, Keillor JW. Kinetic studies of rat kidney γ-glutamyltranspeptidase deacylation reveal a general base-catalyzed mechanism. Biochemistry 2003;42:11504–11513.
- Keillor JW, Castonguay R, Lherbet C. γ-glutamyl transpeptidase substrate specificity and catalytic mechanism. Meth Enzymol 2005;401:449–467.
- Han L, Hiratake J, Kamiyama A, Sakata K. Design, synthesis, and evaluation of γ-phosphono diester analogues of glutamate as highly potent inhibitors and active site probes of γ-glutamyl transpeptidase. Biochemistry 2007;46:1432–1447.
- Lherbet C, Keillor JW. Probing the stereochemistry of the active site of γ-glutamyl transpeptidase using sulfur derivatives of l-glutamic acid. Org Biomol Chem 2004;2:238–245.
- Thompson GA, Meister A. Hydrolysis and transfer reactions catalyzed by γ-glutamyl transpeptidase; evidence for separate substrate sites and for high affinity of L-cystine. Biochem Biophys Res Commun 1976;71:32–36.
- Elce JS, Broxmeyer B. γ-Glutamyltransferase of rat kidney. Simultaneous assay of the hydrolysis and transfer reactions with (glutamate-14C)glutathione. Biochem J 1976;153:223–232.
- Curthoys NP, Hughey RP. Characterization and physiological function of rat renal γ-glutamyltranspeptidase. Enzyme 1979;24:383–403.
- Allison RD, Meister A. Evidence that transpeptidation is a significant function of γ-glutamyl transpeptidase. J Biol Chem 1981;256:2988–2992.
- Cook PF, Cleland WW. Enzyme Kinetics and Mechanism. Garland Science, London, New York, 2007.
- Allison RD. γ-Glutamyl transpeptidase: kinetics and mechanism. Meth Enzymol 1985;113:419–437.
- Stole E, Smith TK, Manning JM, Meister A. Interaction of γ-glutamyl transpeptidase with acivicin. J Biol Chem 1994;269:21435–21439.
- Smith TK, Ikeda Y, Fujii J, Taniguchi N, Meister A. Different sites of acivicin binding and inactivation of γ-glutamyl transpeptidases. Proc Natl Acad Sci USA 1995;92:2360–2364.
- Ikeda Y, Fujii J, Anderson ME, Taniguchi N, Meister A. Involvement of Ser-451 and Ser-452 in the catalysis of human γ-glutamyl transpeptidase. J Biol Chem 1995;270:22223–22228.
- Castonguay R, Halim D, Morin M, Furtos A, Lherbet C, Bonneil E et al. Kinetic characterization and identification of the acylation and glycosylation sites of recombinant human γ-glutamyltranspeptidase. Biochemistry 2007;46:12253–12262.
- Thompson GA, Meister A. Modulation of the hydrolysis, transfer, and glutaminase activities of γ-glutamyl transpeptidase by maleate bound at the cysteinylglycine binding site of the enzyme. J Biol Chem 1979;254:2956–2960.
- Tate SS, Meister A. Serine-borate complex as a transition-state inhibitor of γ-glutamyl transpeptidase. Proc Natl Acad Sci USA 1978;75:4806–4809.
- Okada T, Suzuki H, Wada K, Kumagai H, Fukuyama K. Crystal structures of γ-glutamyltranspeptidase from Escherichia coli, a key enzyme in glutathione metabolism, and its reaction intermediate. Proc Natl Acad Sci USA 2006;103:6471–6476.
- Wada K, Irie M, Suzuki H, Fukuyama K. Crystal structure of the halotolerant γ-glutamyltranspeptidase from Bacillus subtilis in complex with glutamate reveals a unique architecture of the solvent-exposed catalytic pocket. FEBS J 2010;277:1000–1009.
- Morrow AL, Williams K, Sand A, Boanca G, Barycki JJ. Characterization of Helicobacter pylori γ-glutamyltranspeptidase reveals the molecular basis for substrate specificity and a critical role for the tyrosine 433-containing loop in catalysis. Biochemistry 2007;46:13407–13414.
- Ikeda Y, Fujii J, Taniguchi N, Meister A. Human γ-glutamyl transpeptidase mutants involving conserved aspartate residues and the unique cysteine residue of the light subunit. J Biol Chem 1995;270:12471–12475.
- Hanigan MH, Ricketts WA. Extracellular glutathione is a source of cysteine for cells that express γ-glutamyl transpeptidase. Biochemistry 1993;32:6302–6306.
- Estrela JM, Ortega A, Obrador E. Glutathione in cancer biology and therapy. Crit Rev Clin Lab Sci 2006;43:143–181.
- Perry RR, Mazetta JA, Levin M, Barranco SC. Glutathione levels and variability in breast tumors and normal tissue. Cancer 1993;72:783–787.
- Oberli-Schrämmli AE, Joncourt F, Stadler M, Altermatt HJ, Buser K, Ris HB et al. Parallel assessment of glutathione-based detoxifying enzymes, O6-alkylguanine-DNA alkyltransferase and P-glycoprotein as indicators of drug resistance in tumor and normal lung of patients with lung cancer. Int J Cancer 1994;59:629–636.
- Joncourt F, Oberli-Schrämmli AE, Stadler M, Buser K, Franscini L, Fey MF et al. Patterns of drug resistance parameters in adult leukemia. Leuk Lymphoma 1995;17:101–109.
- Raderer M, Scheithauer W. Clinical trials of agents that reverse multidrug resistance. A literature review. Cancer 1993;72:3553–3563.
- Mulder TP, Manni JJ, Roelofs HM, Peters WH, Wiersma A. Glutathione S-transferases and glutathione in human head and neck cancer. Carcinogenesis 1995;16:619–624.
- Mena S, Benlloch M, Ortega A, Carretero J, Obrador E, Asensi M et al. Bcl-2 and glutathione depletion sensitizes B16 melanoma to combination therapy and eliminates metastatic disease. Clin Cancer Res 2007;13:2658–2666.
- Rudin CM, Yang Z, Schumaker LM, VanderWeele DJ, Newkirk K, Egorin MJ et al. Inhibition of glutathione synthesis reverses Bcl-2-mediated cisplatin resistance. Cancer Res 2003;63:312–318.