Abstract
Novel dioxoacridine sulfonamide compounds were synthesized from reaction of cyclic 1,3-diketones, sulfanilamide (4-amino benzene sulfonamide) and aromatic aldehydes. The structures of these compounds were confirmed by using spectral analysis (IR, H-NMR, 13C-NMR, and mass). Human carbonic anhydrase isoenzymes (hCA I and hCA II) were purified from erythrocyte cells by affinity chromatography. The inhibitory effects of sulfanilamide, acetazolamide (AAZ), and newly synthesized sulfonamides on hydratase and esterase activities of these isoenzymes have been studied in vitro. The IC50 values of compounds for esterase activity are 0.71–0.11 µM for hCA I and 0.45–0.12 µM for hCA II, respectively. The Ki values of these inhibitors were determined as 0,38–0,008 µM for hCA I and 0,19–0,001 µM for hCA II, respectively.
Introduction
Carbonic anhydrases (CAs) (E.C. 4.2.1.1) are catalytic reversible hydration of carbon dioxide in a two-step reaction to yield bicarbonate and protonCitation1–5. Sixteen different CA isoenzymes were described in higher vertebrates, including humansCitation6. The isoenzymes relevant to the human eye are hCA I, hCA II and hCA IV. The isoenzymes of hCA I and hCA II are cytosolic, whereas hCA IV is membrane-boundCitation7–9. The CA inhibitors, which reduce aqueous production with a corresponding decrease in intraocular pressure (IOP), have been used as ocular hypotensive agents for the treatment of glaucomaCitation10,Citation11. The disease is the second leading cause of blindness worldwideCitation12,Citation13. The risk factors for glaucoma disease include age, race, ocular hypertension, severe myopia and a family history of glaucoma. Here, the strongest risk factors are age, race and ocular hypertension. An elevated IOP was formerly synonymous with glaucomaCitation7.
Sulfonamides, which are powerful CA inhibitors, are now widely used as the drug for the treatment or prevention of a variety of diseasesCitation4,Citation5,Citation14–16. They are the best known inhibitors of CA enzyme in the treatment of glaucoma in clinical medicineCitation4,Citation5,Citation15. These inhibitors are very effective in the treatment by reducing elevated IOP. Acetazolamide (AAZ) was the first compound used as the inhibitor of this potent hCA II. Then, it was suggested that reducing aqueous humor secretion might provide an effective means of lowering IOP to treat the diseaseCitation17. Afterwards some systemic sulfonamide drugs were mainly used clinically for a long time as anti-glaucoma agentsCitation17. The orally administered drugs had affected various CA isozymes present in other tissues and led to an entire range of side effects. To decrease systemic side effects of oral CA inhibitors, dorzolamide (DZA) and brinzolamide (BRZ) ophthalmic suspension approved for the treatment of glaucoma have been used as the topical CA inhibitorsCitation7,Citation11. Each drug may be an effective anti-glaucoma agent, but these drugs tend to pose tolerability problems in many patients because of local side effectsCitation18. Thus, it is required to develop new compounds and new drugs.
In this study, novel dioxoacridine derivatives have been synthesized and characterized by IR, 1H and 13C-NMR data and satisfactory mass spectral analyses, respectively. The synthesis of new CA inhibitors by using sulfanilamide and their effects on human CA isoenzymes (hCA I, hCA II) purified from human erythrocytes are reported. Further, the potential use of these compounds as new inhibitors of hCA I and hCA II isoenzymes in the treatment of glaucoma are investigated.
Material and methods
Materials
The chemicals used in the synthesis of dioxoacridine sulfonamide derivatives were provided from Merck and Aldrich Chemical Company and Sepharose 4B for affinity column and electrophoresis reagents were obtained from Sigma Chem. Co. All chemicals and solvents used for the synthesis were spectroscopic reagent grade. Melting points were measured on a Bibby Stuart Scientific apparatus. Fourier Transform Infrared (FT-IR) spectra were recorded from Bruker Optics, Andrtex 70 FT-IR spectrometer using ATR diamond crystal. The 1H-NMR, and 13C-NMR spectra were obtained with a Bruker DPX-400 FT-NMR instrument in CDCl3 and DMSO-d6 as solvent with trimethylsilane as the internal reference, at 400 and 100 MHz, respectively. Chemical shifts are expressed in δ units (ppm). The mass analyses were performed on Waters 2695 Alliance Micromass ZQ instrument LC/MS.
General procedure for preparation of dioxoacridine sulfonamide derivatives (4–12).
A mixture of a 5,5-dimethylcyclohexane-1,3-dione 1 (280 mg, 2 mmol), sulfanilamide 2 (172 mg, 1 mmol), 4-cyanobenzaldehyde 3 (131 mg, 1 mmol), and DBSA (420 mg, 10 mol%) in H2O (40 mL) was stirred at refluxing for 4 h (6 h for cyclohexane-1,3-dione derivatives; 10 h for 4,4-dimethylcyclohexane-1,3-dione derivatives). The progress of the reaction was monitored by TLC. Once the reaction is completed, the mixture was cooled to room temperature and solid filtered off and washed with H2O. The acridine-1,3-dion sulfonamide products were purified and recrystallized from the following solvent mixture for each compound (76%–91%).
4-(9-(4-Cyanophenyl)-3,3,6,6-tetramethyl-1,8-dioxo-1,2,3,4,5,6,7,8-octahydroacridine-10(9H)-yl)benzenesulfonamide (4)
As yellow crystals, (471 mg, 87%), mp 215°C (ethanol–H2O). 1H NMR (400 MHz, CDCl3) δ (ppm): 0.84 (s, 6H, 2 × CH3), 0.96 (s, 6H, 2 × CH3), 1.76–1.81 (m, 2H, −CH2), 1.99–2.23 (m, 6H, −CH2), 3.75 (s, 3H, −OCH3), 5.05 (s, 2H, SO2NH2), 5.29 (s, 1H, −CH), 7.41 (d, 2H, J = 8.72 Hz Ar-H), 7.44 (m, 4H, Ar-H), 8.20 (d, 2H, J = 8.38 Hz Ar-H); 13C NMR (100 MHz, CDCl3) δ (ppm): 27.23, 28.96, 32.36, 33.96, 40.53, 50.04, 112.61, 113.89, 120.93, 122.58, 127.38, 130.40, 131.59, 134.18, 143.56, 150.72, 161.14, 197.03. IR (cm−1): 3298 and 3172 w (NH2), 3052 w (Ar-H), 2960 w (C–H), 2229 (CN), 1641 s (C = O), 1628 and 1575 m (C = C), 1361, 1220, 1170, 1145, 1019; MS(CI) m/z 530.90 (M + 1).
4-(9-(4-Methoxyphenyl)-3,3,6,6-tetramethyl-1,8-dioxo-1,2,3,4,5,6,7,8-octahydro acridine-10(9H)-yl)benzenesulfonamide (5)
As yellow crystals, (0.481 mg, 90%), mp 227°C (ethanol–H2O). 1H NMR (400 MHz, CDCl3) δ (ppm): 0.85 (s, 6H, 2 × CH3), 0.98 (s, 6H, 2 × CH3), 1.75–1.81 (m, 2H, −CH2), 1.99–2.23 (m, 6H, −CH2), 3.78 (s, 3H, −OCH3), 5.06 (s, 2H, SO2NH2), 5.24 (s, 1H, −CH), 6.82 (d, 2H, J = 8.65 Hz Ar-H), 7.34 (d, 2H, J = 8.65 Hz Ar-H), 7.44 (d, 2H, J = 8.40 Hz Ar-H), 8.16 (d, 2H, J = 8.40 Hz Ar-H); 13C NMR (100 MHz, CDCl3) δ (ppm): 26.87, 28.99, 31.68, 32.64, 42.05, 51.10, 55.13, 113.29, 115.23, 123.05, 128.47, 130.88, 131.35, 138.15, 146.57, 157.61, 157.89, 195.68. IR (cm−1): 3303 and 3222 w (NH2), 3093 and 3071 w (Ar-H), 2997 w (C-H), 1633 s (C = O), 1626 and 1570 m (C = C), 1386, 1221, 1166, 1136, 1032; MS(CI) m/z 535.10 (M + 1), 428.00 (M-PhOCH3).
4-(3,3,6,6-tetramethyl-1,8-dioxo-9-(pyridin-2-yl)-1,2,3,4,5,6,7,8-octahydroacridine-10(9H)-yl)benzenesulfonamide (6)
As yellow crystals, (0.429 mg, 85%), mp 170°C (ethanol–H2O). 1H NMR (400 MHz, CDCl3) δ (ppm): 1.01 (s, 6H, 2 × CH3), 1.11 (s, 6H, 2 × CH3), 2.14–2.28 (m, 4H, −CH2), 2.43–2.58 (m, 4H, −CH2), 4.94 (s, 1H, −CH), 5.35 (s, 2H, SO2NH2), 7. 19 (qxd, 2H, J = 2.29 Hz, J = 1.44 Hz Ar-H), 7.53–7.64 (m, 4H, Ar-H), 8.27 (dxd, 1H, J = 7.41 Hz, J = 1.39 Hz Ar-H), 8.39 (dxq, 1H, J = 4.80 Hz, J = 0.64 Hz Ar-H); 13C NMR (100 MHz, CDCl3) δ (ppm): 27.15, 29.30, 32.30, 34.45, 40.86, 50.75, 114.33, 121.40, 122.10, 124.96, 130.95, 131.77, 135.71, 145.02, 148.89, 161.72, 163.37, 196.98. IR (cm−1): 3300 w (NH2), 3065 w (Ar-H), 2960 w (C–H), 1652 s (C = O), 1621 and 1570 m (C = C), 1360, 1223, 1196, 1165, 1001; MS(CI) m/z 506.78 (M + 1).
4-(9-(2,4-dimethoxyphenyl)-1,8-dioxo-1,2,3,4,5,6,7,8-octahydroacridine-10(9H)-yl) benzenesulfonamide (7)
As yellow crystals, 396 mg, 78%, mp 243°C (chloroform). 1H NMR (400 MHz, DMSO-d6) δ (ppm): 1.60–1.91 (m, 6H, 3 × CH2), 2.19–2.46 (m, 6H, 3 × CH2), 3.72 (s, 3H, −OCH3), 3.80 (s, 3H, −OCH3), 5.18 (s, 1H, −CH), 6.44 (dxd, 1H, J = 8.53 Hz, J = 2.60 Hz Ar-H), 6.50 (d, 1H, J = 2.54 Hz Ar-H), 7.24 (d, 1H, J = 8.57 Hz Ar-H), 7.62 (s, 2H, Ar-H), 7.69 (s, 2H, SO2NH2), 8.06 (d, 2H, J = 8.50 Hz Ar-H); 13C NMR (100 MHz, DMSO-d6) δ (ppm): 21.40, 27.94, 31.96, 36.51, 55.73, 55.91, 99.03, 103.14, 116.90, 122.08, 124.54, 130.66, 131.13, 133.46, 144.80, 156.09, 159.48, 161.29, 196.08. IR (cm−1): 3310 w (NH2), 3171 and 3031 w (Ar-H), 2971 w (C–H), 1641 s (C=O), 1608 and 1568 m (C=C), 1362, 1234, 1089, 1042; MS(CI) m/z 509.65 (M + 1).
4-(9-(4-methoxyphenyl)-1,8-dioxo-1,2,3,4,5,6,7,8-octahydroacridine-10(9H)-yl)benzene sulfonamide (8)
As yellow crystals, 0.397 mg, 83%, mp 181°C (ethanol–H2O). 1H NMR (400 MHz, CDCl3) δ (ppm): 1.54–1.93 (m, 6H, 3 × CH2), 1.96-2.30 (m, 6H, 3 × CH2), 3.52 (s, 3H, −OCH3), 5.11 (s, 1H, −CH), 6.64 (d, 2H, J = 8.80 Hz Ar-H), 6.77 (s, 2H, SO2NH2), 7.13 (d, 2H, J = 8.51 Hz Ar-H), 7.30 (d, 2H, J = 8.51 Hz Ar-H), 7.96 (d, 2H, J = 8.80 Hz Ar-H); 13C NMR (100 MHz, CDCl3) δ (ppm): 21.36, 27.95, 31.23, 36.48, 55.48, 113.70, 116.54, 122.16, 126.53, 130.69, 131.02, 138.29, 144.53, 150.63, 156.52, 196.43. IR (cm−1): 3312 w (NH2), 3125 and 3061 w (Ar-H), 2940 w (C–H), 1638 s (C = O), 1595 and 1539 m (C = C), 1385, 1131, 1084, 1011; MS(CI) m/z 479.22 (M + 1).
4-(9-(4-Cyanophenyl)-2,2,7,7-tetramethyl–1,8-dioxo–1,2,3,4,5,6,7,8-octahydroacridine–10(9H)-yl)benzenesulfonamide (9).
As yellow crystals, 471 mg, 89%, mp 299°C (ethanol–H2O). 1H NMR (400 MHz, CDCl3) δ (ppm): 0.82 (s, 6H, 2 × CH3), 0.93 (s, 6H, 2 × CH3), 1.63–1.72 (m, 2H, −CH2), 1.78–1.92 (m, 2H, −CH2), 1.99–2.10 (m, 2H, −CH2), 2.28–2.47 (m, 2H, −CH2), 5.11 (s, 1H, −CH), 6.80 (s, 2H, SO2NH2), 7.33–7.46 (m, 6H, Ar-H), 8.01 (d, 2H, J = 8.30 Hz Ar-H); 13C NMR (100 MHz, CDCl3) δ (ppm): 24.08, 24.16, 24.35, 32.57, 33.84, 39.91, 112.08, 114.50, 120.54, 122.46, 127.45, 130.27, 131.65, 133.71, 144.10, 151.03, 160.23, 204.60. IR (cm−1): 3356 and 3280 w (NH2), 3076 w (Ar-H), 2957 w (C–H), 2222 (CN), 1643 s (C = O), 1619 and 1566 (C = C), 1349, 1220, 1198, 1160, 1010; MS(CI) m/z 530.93 (M + 1).
4-(9-(4-Hydroxyphenyl)-2,2,7,7-tetramethyl–1,8-dioxo–1,2,3,4,5,6,7,8-octahydro acridine–10(9H)-yl)benzenesulfonamide (10)
As yellow crystals, 442 mg, 85%, mp 280°C (decompoze) (ethanol–H2O). 1H NMR (400 MHz, DMSO-d6) δ (ppm): 0.91 (s, 6H, 2 × CH3), 1.00 (s, 6H, 2 × CH3), 1.60–1.71 (m, 4H, −CH2), 1.88–1.92 (m, 2H, −CH2), 2.28–2.33 (m, 2H, −CH2), 4.98 (s, 1H, −CH), 7.61 (d, 2H, J = 8.45 Hz Ar-H), 7.05 (d, 2H, J = 8.45 Hz Ar-H), 7.55 (d, 2H, J = 7.62 Hz Ar-H), 8.02 (d, 2H, J = 7.62 Hz Ar-H), 8.25 (s, 1H, Ar-OH), 9.02 (s, 2H, SO2NH2); 13C NMR (100 MHz, DMSO-d6) δ (ppm): 24.12, 24.23, 24.38, 32.69, 33.81, 39.93, 114.56, 115.38, 122.49, 129.06, 130.50, 131.23, 139.51, 144.17, 154. 91, 160.31, 204.56. IR (cm−1): 3353 and 3278 w (NH2), 3066 and 3029 w (Ar-H), 2971 w (C–H), 1631 s (C = O), 1610 and 1586 m (C = C), 1362, 1222, 1161, 1145, 1016; MS(CI) m/z 521.83 (M + 1).
4-(9-(3,4-Dimethoxyphenyl)-2,2,7,7-tetramethyl–1,8-dioxo–1,2,3,4,5,6,7,8-octahydro acridine–10(9H)-yl)benzenesulfonamide (11)
As yellow crystals, 429 mg, 76%, mp 190°C (ethanol–H2O). 1H NMR (400 MHz, CDCl3) δ (ppm): 1.04 (s, 6H, 2 × CH3), 1.11 (s, 6H, 2 × CH3), 1.67–1.72 (m, 4H, 2 × CH2), 1.85–2.03 (m, 2H, −CH2), 2.15–2.28 (m, 2H, −CH2), 3.86 (s, 3H, −OCH3), 3.92 (s, 3H, −OCH3), 5.01 (s, 2H, SO2NH2), 5.29 (s, 1H, −CH), 6.78 (m, 2H, Ar-H), 7.11 (s, 1H, Ar-H), 7.47 (d, 2H, J = 8.20 Hz, Ar-H), 8.14 (d, 2H, J = 7.50 Hz, Ar-H); 13C NMR (100 MHz, CDCl3) δ (ppm): 24.17, 24.20, 24.33, 32.98, 33.73, 39.88, 55.23, 55.29, 112.55, 114.30, 114.65, 122.41, 122.49, 130.53, 131.24, 137.62, 144.26, 156.49, 157.20, 160.72, 204.61. IR (cm−1): 3306 and 3239 w (NH2), 3096 w (Ar-H), 2963 w (C–H), 1631 s (C = O), 1579 and 1510 (C = C), 1362, 1263, 1227, 1159, 1027; MS(CI) m/z 565.88 (M + 1).
4-(9-(2,4-Dichlorophenyl)-2,2,7,7-tetramethyl–1,8-dioxo–1,2,3,4,5,6,7,8-octahydro acridine–10(9H)-yl)benzenesulfonamide (12)
As yellow crystals, 521 mg, 91%, mp 270°C (decompoze) (ethanol–H2O). 1H NMR (400 MHz, DMSO-d6) δ (ppm): 0.81 (s, 6H, 2 × CH3), 0.97 (s, 6H, 2 × CH3), 1.52–1.58 (m, 2H, −CH2), 1.60–1.70 (m, 2H, −CH2), 1.84–1.95 (m, 2H, −CH2), 2.19–2.23 (m, 2H, −CH2), 5.20 (s, 1H, −CH), 7.30 (dxd, 1H, J = 6.00 Hz, J = 2.31 Hz, Ar-H), 7.38 (d, 1H, J = 2.77 Hz, Ar-H), 7.51 (d, 1H, J = 8.50 Hz, Ar-H), 7.61 (s, 2H, SO2NH2), 7.71 (m, 1H, Ar-H), 7.86 (m, 1H, Ar-H), 8.02 (d, 2H, J = 7.83 Hz, Ar-H); 13C NMR (100 MHz, DMSO-d6) δ (ppm): 24.13, 24.20, 24.30, 33.01, 33.69, 39.90, 114.88, 122.45, 127.04, 130.32, 130.44, 131.18, 131.27, 134.56, 137.71, 140.83, 144.20, 161.04, 204.51. IR (cm−1): 3384 and 3319 w (NH2), 3088 w (Ar-H), 2963 w (C–H), 1642 s (C = O), 1620 and 1573 (C = C), 1332, 1221, 1198, 1160, 1013; MS(CI) m/z 573.75 (M + 1).
Purification of carbonic anhydrase I and II from human erythrocytes
Erythrocytes were purified from human blood. The blood samples were centrifuged at 1500 rpm for 20 min and plasma was removed. Later, red cells were washed with NaCl (0.9%), and the erythrocytes were hemolyzed with 1.5 volumes of ice-cold water. Cell membranes were removed by centrifugation at 4°C, 20,000 rpm for 30 min. The pH of hemolysate was adjusted to 8.7 with solid Tris. The hemolysate was applied to affinity column with a structure of Sepharose-4B-L-tyrosine-p-aminobenzenesulfonamide and equilibrated with 25 mM Tris–HCl/0.1 M Na2SO4 (pH 8.7). The affinity gel was washed with solution of 25 mM Tris–HCl/22 mM Na2SO4 (pH 8.7). The hCA-I and hCA-II isozymes were diluted with the solution of 1 M NaCl/25 mM Na2HPO4 (pH 6.3) and 0.1 M NaCH3COO/0.5 M NaClO4 (pH 5.6), respectivelyCitation19. For protein content estimation, Bradford method was used with bovine serum albumin as a standardCitation20,Citation21. SDS polyacrylamide gel electrophoresis was performed after the purification of the enzyme (see )Citation22.
Figure 1. SDS-PAGE analysis of CA isoenzymes: (a) Standard hCA I, (b) isolated hCA I, (c) standard hCA II, (d) isolated hCA II.

Determination of hydratase and esterase activities of hCA I and hCA II
The CO2-hydratase activity of the enzyme was determined at 0°C in a veronal buffer (pH = 8.15) with pH-state method as indicator and saturated carbon dioxide solution as substrate in a final volume of 4.2 mL. The time (in seconds) taken for the solution to change from pH 8.15 to pH 6.5 was measured by pH meter. The enzyme unit (EU) is the enzyme amount that reduces the non-enzymatic reaction time by 50%. Activity of an enzyme unit was calculated by using the equation (t0−tc/tc) where to and tc are times for pH change of the non-enzymatic and enzymatic reactions, respectivelyCitation23–25. Esterase activities of hCA I and hCA II isoenzymes, eluted from affinity column, were determined by hydrolysis of p-nitrophenylacetate. The change of absorbance was determined at 348 nm after 3 minCitation26.
Determination of IC50 and Ki values of compounds
The study consists of two parts. In the first part, IC50 values have been obtained as in vitro. The IC50 values of inhibitors (4–12) were assayed by the hydrolysis of p-nitrophenyl acetate on the esterase activities of CA isoenzymes in the presence of various inhibitor concentrations. The absorbance was determined at 348 nm after 3 minCitation27. Regression analysis graphs were drawn by plotting inhibitor concentrations versus enzyme activity.
To determine Ki value as well as the inhibition type, three different inhibitor concentrations giving 30%, 50% and 70% inhibition were selected. At each of these inhibitor concentrations, enzyme activity was measured in the presence of various substrate concentrations (0.3 mM, 0.4 mM, 0.5 mM, 0.6 mM and 0.7 mM) and the data were linearized with Lineweaver–Burke plot for Vmax and the Ki determination. Enzyme activity was also measured in the presence of the same substrate concentrations but in the absence of any inhibitor to determine the VmaxCitation26.
Result and discussion
The general synthetic method shown in is employed to prepare dioxoacridine sulfonamide derivatives (4–12). All spectral data are in agreement with the assigned structures.
Scheme 1. Synthesis of acridine-1,3-dion sulfonamides (4–12).
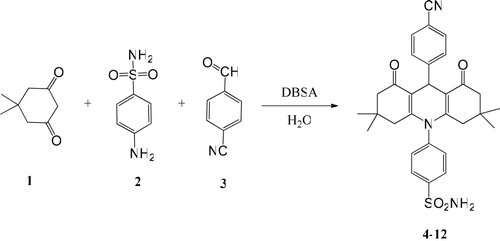
The synthesis of dioxoacridine sulfonamide compounds were realized in water in a single process through two successive reactions (Aldol condensation and Michael addition) and using a phase transfer catalyst-Bronsted acid as p-dodesilbenzensulphonic acid (DBSA). In recent years, using DBSA as a combine catalyst (phase transfer catalyst-Bronsted acid) has been a popular application in organic chemistryCitation28–30. Sulfonamide compounds were prepared by one pot reaction in processing high yields and simple work-up procedure.
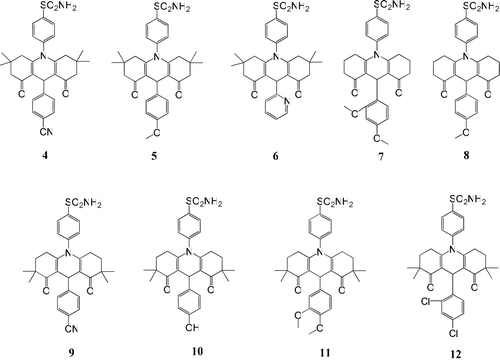
The infrared (IR) spectra of all the dioxoacridine sulfonamide compounds showed sharp peaks for the carbonyl groups in region between 1650 and 1631 cm−l Citation31. The compounds 4, 9 exhibited peaks that belong to CN group 2229 and 2222 cm−1, respectivelyCitation32. Besides, in the IR spectra of the compounds, aliphatic C–H stretching bands at 2997–2940 cm−1 and aromatic C–H stretching bands at 3171–3029 cm−1 were observed. The NH2 vibrations of dioxoacridine sulfonamide compounds were observed in the region between 3384 and 3172 cm−1 Citation33. The 1H-NMR spectra of compounds 4–6 showed singlet peaks that belong to protons of the methyl groups in position 3 and 6 between 0.84 and 1.11 ppm. The compounds 9–12 showed singlet peaks that belong to protons of the methyl groups in position 2 and 7 between 0.81 and 1.11 ppmCitation30. The CH2 group protons of the cyclohexene rings of the compounds 4–12 showed multiple peaks in the 1.54–2.58 ppm rangeCitation30. Signals for the methoxy group protons for compounds 5, 7, 8, 11 were shown in the range of 3.52–3.92 ppm. The signals for the CH protons at 4.98–5.35 ppm and signals for the aromatic protons in the range of 6.44–8.39 ppm were observed. Signals of pyridine ring protons for the compound 6 showed in the range of 7.01–8.39 ppm. Hydroxyl group proton of compound 10 was observed as broad signal at 8.25 ppm. The broad singlet peaks between 4.94 and 9.02 ppm were assigned to sulfonamide (−SO2NH2) groups protons of all the compounds 4–12 ().
Scheme 2. New CA inhibitors (4–12).
The concentration required inhibiting hCA I and hCA II activities of the purified proteins by 50% (IC50) and inhibition equilibrium constant (Ki) was determined for each compound. Sulfanilamide and AAZ were used as control compounds to compare inhibition potential of newly synthesized compounds 4–12.
According to in vitro studies, any inhibition effects of 4–12 compounds were not observed on hydratase activity of hCA I and hCA II isoenzymes. The IC50 and Ki values obtained from esterase activities of these compounds were shown in .
Table 1. The Ki values obtained from in vitro inhibition experiments.
All of the new inhibitor compounds are more effective than control compounds (sulfanilamide and AAZ) for hCA I and hCA II. Especially compounds 5, 6, 7, 8 and 10 have shown remarkable inhibition on hCA I and hCA II isoenzymes.
The Ki values of all the novel compounds are lower than control compounds for hCA I and hCA II.
Compounds 5, 7, 8 and 11 contain methoxy group, while compound 10 contain hydroxyl group. These compounds are the most effective inhibitors in the newly synthesized compounds. It is thought that these groups in abovementioned compounds increase interactions of inhibitor-zinc metal and inhibitor-histidine residues in the active site of hCA I and hCA II isoenzymesCitation34.
In summary, these new compounds have potential inhibitory effects; so these compounds can be candidates for in vivo studies of glaucoma treatment.
Conclusion
A series of dioxoacridine sulfonamide containing nitrile, methoxy, halogen, and hydroxyl groups were synthesized. Synthesized compounds have inhibitory potential on human erythrocyte hCA I and hCA II isoenzymes. So these compounds can be candidates for in vivo studies of glaucoma treatment.
Acknowledgments
The authors are grateful to Prof. Dr. Yılmaz Yıldırır, Gazi University, Ankara, Turkey.
Declaration of interest
The authors are very grateful to Dumlupinar University Research Fund for providing financial support for this project (Grant No. 2008-4).
References
- Supuran CT. Carbonic anhydrases: novel therapeutic applications for inhibitors and activators. Nat Rev Drug Discov 2008;7:168–181.
- Hewett-Emmett D, Tashian RE. Functional diversity, conservation, and convergence in the evolution of the alpha-, beta-, and gamma-carbonic anhydrase gene families. Mol Phylogenet Evol 1996;5:50–77.
- Briganti F, Mangani S, Orioli P, Scozzafava A, Vernaglione G, Supuran CT. Carbonic anhydrase activators: X-ray crystallographic and spectroscopic investigations for the interaction of isozymes I and II with histamine. Biochemistry 1997;36:10384–10392.
- Supuran CT, Puscas I. In carbonic anhydrase and modulation of physiologic and pathologic processes in the organism. Helicon: Timisoara 1994;29–111.
- Supuran CT, Conroy CW, Maren TH. Carbonic anhydrase inhibitors: Synthesis and inhibitory properties of 1,3,4-thiadiazole-2,5-bissulfonamide. Eur J Med Chem 1996;31:843–846.
- Supuran CT, Scozzafava A. Applications of carbonic anhydrase inhibitors and activators in therapy. Exp Opin Ther Pat 2002;12:217–242.
- Sugrue MF. Pharmacological and ocular hypotensive properties of topical carbonic anhydrase inhibitors. Prog Retin Eye Res 2000;19:87–112.
- Wistrand PJ, Schenholm M, Lönnerholm G. Carbonic anhydrase isoenzymes CA I and CA II in the human eye. Invest Ophthalmol Vis Sci 1986;27:419–428.
- Hageman GS, Zhu XL, Waheed A, Sly WS. Localization of carbonic anhydrase IV in a specific capillary bed of the human eye. Proc Natl Acad Sci USA 1991;88:2716–2720.
- Becher B. Decrease in intraocular pressure in man by a carbonic anhydrase inhibitor, Diamox: a preliminary report. Am J Ophthalmol 1954;37:13–15.
- Tamaki Y, Araie M, Muta K. Effect of topical dorzolamide on tissue circulation in the rabbit optic nerve head. Jpn J Ophthalmol 1999;43:386–391.
- Scozzafava A, Banciu MD, Popescu A, Supuran CT. Carbonic anhydrase inhibitors: inhibition of isozymes I, II and IV by sulfamide and sulfamic acid derivatives. J Enzym Inhib 2000;15:443–453.
- Schuman JS. Antiglaucoma medications: a review of safety and tolerability issues related to their use. Clin Ther 2000;22:167–208.
- Maren TH. The development of topical carbonic anhydrase inhibitors. J Glaucoma 1995;4:49–62.
- Reiss WG, Oles KS. Acetazolamide in the treatment of seizures. Ann Pharmacother 1996;30:514–519.
- Becker B. The mechanism of the fall in intraocular pressure induced by the carbonic anhydrase inhibitor, diamox. Am J Ophthalmol 1955;39:177–184.
- Mincione F, Starnotti M, Menabuoni L, Scozzafava A, Casini A, Supuran CT. Carbonic anhydrase inhibitors: 4-sulfamoyl-benzenecarboxamides and 4-chloro-3-sulfamoyl-benzenecarboxamides with strong topical antiglaucoma properties. Bioorg Med Chem Lett 2001;11:1787–1791.
- Scozzafava A, Mincione F, Menabuoni L, Supuran CT. Carbonic anhydrase inhibitors: topically acting antiglaucoma sulfonamides incorporating phthaloyl and phthalimido moieties. Drug Des Discov 2001;17:337–348.
- Rickli EE, Ghazanfar SA, Gibbons BH, Edsall JT. Carbonic anhydrases from human erythrocytes. preparation and properties of two enzymes. J Biol Chem 1964;239:1065–1078.
- Bradford MM. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal Biochem 1976;72:248–254.
- Bülbül M, Hisar O, Beydemir S, Ciftçi M, Küfrevioglu OI. The in vitro and in vivo inhibitory effects of some sulfonamide derivatives on rainbow trout (Oncorhynchus mykiss) erythrocyte carbonic anhydrase activity. J Enzyme Inhib Med Chem 2003;18:371–375.
- Laemmli UK. Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature 1970;227:680–685.
- Bülbül M, Erat M. Investigation of the effects of some sulfonamide derivatives on the activities of glucose-6-phosphate dehydrogenase, 6-phospho gluconate dehydrogenase and glutathione reductase from human erythrocytes. J Enzyme Inhib Med Chem 2008;23:418–423.
- Yenikaya C, Sari M, Bülbül M, Ilkimen H, Cinar B, Büyükgüngör O. Synthesis and characterisation of two novel proton transfer compounds and their inhibition studies on carbonic anhydrase isoenzymes. J Enzyme Inhib Med Chem 2011;26:104–114.
- Laemmli DK. Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature 1970;227:680–683.
- Wilbur KM, Anderson NG. Electrometric and colorimetric determination of carbonic anhydrase. J Biol Chem 1948;176:147–154.
- Landolfi C, Marchetti M, Ciocci G, Milanese C. Development and pharmacological characterization of a modified procedure for the measurement of carbonic anhydrase activity. J Pharmacol Toxicol Methods 1997;38:169–172.
- Jin TS, Zhang JS, Guo TT, Wang AQ, Li TS. One-pot clean synthesis of 1,8-dioxo-decahydroacridines catalyzed by p-dodecylbenezenesulfonic acid in aqueous media. Synlett 2004;12:1425–1427.
- Jin TS, Zhang JS, Xiao JC, Wang AQ, Li TS. Clean synthesis of 1,8-dioxo-octahydroxanthene derivatives catalyzed by p-dodecylbenezenesulfonic acid in aqueous media. Synlett 2004;5:866–870.
- Kaya M, Basar E, Colak F. Synthesis and antimicrobial activity of some bisoctahydroxanthene-1,8-dione derivatives. Med Chem Res 2010; DOI 10.1007/s00044-010–9459-2.
- Kaya M, Yıldırır Y, Türker L. Synthesis and laser activity of halo-acridinedione derivatives. J Heterocyclic Chem 2009;46:294–297.
- Kaya M, Yıldırır Y, Celik GY. Synthesis and antimicrobial activities of novel bisacridine-1,8-dione derivatives. Med Chem Res 2011;20:293–299.
- Yenikaya C, Sari M, Bülbül M, Ilkimen H, Celik H, Büyükgüngör O. Synthesis, characterization and antiglaucoma activity of a novel proton transfer compound and a mixed-ligand Zn(II) complex. Bioorg Med Chem 2010;18:930–938.
- Bayram E, Senturk M, Kufrevioglu OI, Supuran CT. In vitro inhibition of salicylic acid derivatives on human cytosolic carbonic anhydrase isozymes I and II. Bioorg Med Chem 2008;16:9101–9105.