Abstract
Twenty-two quinazoline derivatives have been synthesised and examined for their anti-tumour activity against three tumour cell lines, namely human breast cancer cell line (MCF-7), human cervical cancer cell line (HeLa) and human hepatoma cell line (HepG2). Twelve of the tested compounds have shown promising anti-tumour activity with an IC50 range of 5.0–9.7 µg/mL. Regarding the spectrum of activity, five compounds exhibited interesting anti-proliferative properties against the three tested cell lines comparable to the reference drug (dasatinib).
Keywords::
Introduction
With the aging of the world’s population, the westernisation of diet and the widespread environmental pollution, cancer has emerged as the top threat to human life worldwideCitation1. For the majority of cancers, chemotherapy, either alone or as an adjunct to radiotherapy or surgery, remains the treatment of choice. Most anti-cancer agents are broadly acting cytotoxic drugs. The heterogeneity of tumours, the lack of selectivity between normal and cancerous cells and the development of drug resistance are some of the major obstacles hindering the reach of an ultimate cure for such a deadly diseaseCitation2. In spite of the substantial progress in many aspects of cancer research, the current approaches have many disadvantages, including low efficacy and high degree of toxicity. Notably, many cancer chemotherapeutic agents are themselves carcinogenicCitation3. Therefore, there is an urgent need to rationally design novel, molecularly targeted anti-cancer therapies, which are more selective, ideally less toxic and ultimately more effective than traditional treatments.
Numerous quinazoline derivatives have been reported to have anti-cancer activityCitation4–6. In that context, these agents have shown inhibitory activity against thymidylate synthaseCitation5–7, dihydrofolate reductase and various receptor tyrosine kinasesCitation8–10. Dasatinib, the standard used in this study, is a multi-targeted kinase inhibitorCitation11.
Recently, quinazolines have shown inhibitory activity against NF-κB, a transcription factor that has been reported to be involved in cancer development and progression, as well as in resistance to chemotherapy and radiotherapyCitation12. Tobe et al.Citation13 have conducted a reporter gene-based screening, with the reporter DNA having the binding sequence for NF-κB and the luciferase gene, to find new structural class of NF-κB activation inhibitors. This effort led to the identification of quinazoline 1 as a lead compound (IC50 = 2.3 µM), .
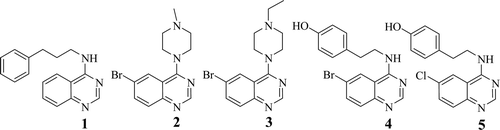
Huntington’s disease (HD), a member of the trinucleotide-repeat neurological disorders, is caused by a heritable, polyglutamine expansion mutation in the NH3-terminus of the Huntingtin protein (Htt). To search for small-molecule activators of Htt protein clearance, Rinderspacher et al.Citation14 used a unique two-tiered functional genetic screen and found that quinazoline derivatives 2–5 () suppressed tet-regulatable gene expression with IC50 of 1.3, 2.21, 1.1 and 0.71 µM, respectively.
In this work, a new series of quinazoline compounds of the general formula represented in were designed in such a way to accommodate phenyl group at position 2 to modify the electronic effects (increase lipophilicity) to study its effect on activity because most of the reported active compounds were devoid of this moiety. Some substituted amines and/or their oxo-isosteres at position 4, and/or piperidines were synthesised to impart structural similarity to compounds 1–5. We designed these compounds with one and two carbon spacer between the 4-quinazolin-amine backbone and the side chain at position 4 aiming to identify new candidates that may be of value as potent, selective and less toxic anti-tumour agents.
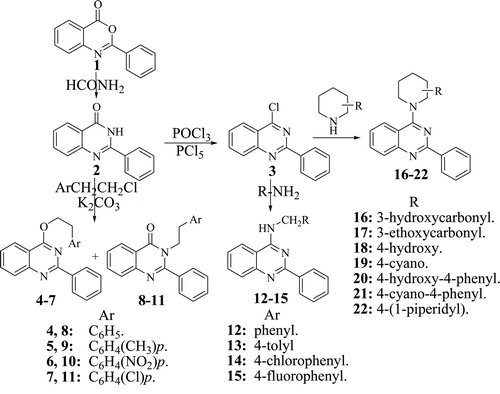
Scheme 1. Synthesis of the proposed anti-tumour quinazoline derivatives (4–22).
Results and discussion
Chemistry
The synthesis of the quinazoline derivatives 1–22 was carried out following Scheme 1. It outlines the synthetic pathway used to obtain compounds 4–11. The starting material 2-phenyl-4H-[3,1]benzoxazin-4-one 1, 2-phenyl-3H-quinazolin-4-one 2 and 2-phenyl-4-chloroquinazoline 3 were prepared in our laboratory following a reported procedureCitation15. Compound 2 was alkylated with some selected phenethyl chloride derivatives in dry dimethylformamide containing anhydrous potassium carbonate. It was reported that 3H-quinazolin-4-one derivatives react readily with alkyl halides at the 3-nitrogen and occasionally at the 4-oxygen atomCitation16. The variation of the product ratio has remained unclear until Bogentoft et al.Citation17 presumed that a steric hindrance effect of the 2-substituent of the quinazoline ring facilitated the 4-O alkylation. However, Hori et al.Citation18 reported that the ratio of O/N alkylation was not explicable by the steric hindrance alone, but other factors such as electronic effects should be considered.
In this work, a mixture of two products was obtained and separated by column chromatography. The first one had the alkyl group at the 3-nitrogen atom to afford 3-alkylquinazolin-4-one derivatives 4–7. The other one was identified to be the 4-alkoxy derivatives 8–11, where alkylation occurred at the 4-oxygen atom. The structures of these compounds were confirmed by infra red (IR) and nuclear magnetic resonance (NMR) spectra. Generally, compounds 4–7 showed the characteristic carbonyl absorption band around 1695–1680 cm−1 and no absorption band for the NH group. While the IR spectra of compounds 8–11 revealed the absence of both the NH and the endocyclic carbonyl C=O groups of the parent compounds, and no signal for the NH groups was seen in the 1H NMR spectra. On the other hand, the 1H NMR spectra of the latter compounds showed triplet at around δ 4.3, which corresponds to O-CH2, while the N-alkyl derivatives 8–11 showed their triplet at around δ 3.5, which corresponds to N-CH2. 13H NMR spectra of O-alkyl derivatives, 4–7 showed their CH2 carbons at around δ 65 ppm. However, the N-alkyl derivatives 8–11 showed their CH2 carbons at around δ 44 ppm.
The appropriate piperidine derivative was allowed to react with the 4-chloro derivative in acetone containing anhydrous potassium carbonate to afford the target 4-substituted derivatives 12–22. The structures of these compounds were confirmed by IR, NMR and mass spectra.
Pharmacology
All the new compounds were screened for their anti-tumour activity against HepG2 (human liver cancer cell line), MCF-7 (human breast cancer cell line) and HeLa (human cervical cancer cell line) using sulphorhodamine-B assay as described previouslyCitation19 and dasatinib as a reference drug. Each cell line was incubated with five concentrations (0–100 µg/mL) of each compound and the results were used to create compound concentration versus survival fraction curves. The concentration causing 50% cell growth inhibition (IC50) was calculated. Concerning sensitivity of cell lines to the synthesised compounds, MCF-7 cell line was shown to be the most sensitive toward the tested compounds followed by HeLa and HepG2 cell lines. As shown in , five compounds (14, 16–18, 22) showed broad spectrum inhibition of the three cell lines. In this respect, the potency of these compounds can be arranged in the following descending order: 22 > 14 > 18 > 16 > 17. Indeed, compounds 22, 14, 18 and 16 exhibited potent anti-tumour activity better than that of the standard drug dasatinib, whereas the potency of compound 17 was comparable to that of dasatinib. On the other hand, compounds 10 and 12 were moderately active against both human breast cancer cell line MCF-7 (IC50 = 9.7, 9.3 µg/mL, respectively) and human hepatoma cell line HepG2 (IC50 = 9.2, 7.8 µg/mL, respectively). With regard to selectivity of tested compounds against individual cell lines, most of the tested compounds showed a distinctive pattern of selectivity. Compounds 8, 9 and 12 showed high degree of selectivity against the human breast cancer cell line MCF-7 (IC50 = 9.3, 5.4, 9.3 µg/mL, respectively), while compounds 5, 13 and 15 selectively inhibited the human cervical cancer cell line HeLa with moderate activity (IC50 = 9.7, 9.7, 7.8 µg/mL). Except for the above-mentioned results, none of the compounds under test selectively inhibited HepG2. Regarding the structure-activity-relationship of the compounds presented in this study, it is clear that, generally, the 4-substituted quinazoline derivatives were generally more active than the 3-substituted ones. Within the substituted piperidine derivatives, the mono-substituted compounds proved to be far more promising than the di-substituted ones. It seems that electron releasing groups attached to the piperidine ring has better impact on activity than electron withdrawing ones; compounds 18 and 22 with 4-hydroxy and 4-piperidyl-piperidine moieties, respectively, showed the highest activity, followed by compounds 16 and 17 with 3-hydroxycarbonyl and 3-ethoxycarbonyl, respectively. While in compounds 20 and 21, the 4,4-di-substituted piperidine derivatives were inactive or very weakly active. Amongst the flexible 4-substituted benzylamine derivatives, compound 14 with 4-chloro group was the most active one followed by compound 12 (the unsubstituted), while the 4-fluorobenzyl derivative was the least active one.
Table 1. IC50 of synthesised compounds against cancer cell lines.
Conclusion
This work led to the development of novel anti-tumour molecules containing 4-substituted aminoquinazoline pharmacophore. Three cell lines, including MCF-7, HeLa and HepG2, were used to measure cytotoxic activity of the proposed quinazoline derivatives. Five compounds exhibited promising and more potent anti-tumour activity than the standard drug dasatinib and one compound showed activity similar to that of dasatinib. Most of the tested compounds showed varying degrees of activity against the three selected cell lines and few compounds were ineffective. Our preliminary results in this study where we considered three cancer cell lines will be followed by further studies to explore the exact mechanism of action of the most active compounds in addition to lead modifications.
Experimental protocol
Chemistry
General
All melting points (°C, uncorrected) were determined on a Stuart melting point apparatus (Stuart Scientific, Redhill, UK). Elemental analyses (C, H, N) were performed on Perkin-Elmer 2400 analyser (Perkin-Elmer, Norwalk, CT, USA) and were in full agreement with the proposed structures within ≥0.4% of the theoretical values. IR spectra (KBr) were recorded using Pye Unicam SP 1000 IR spectrometer (Thermoelectron, Egelbach, Germany) and expressed in wave number υ (cm−1). NMR spectra were obtained on a Bruker AC 300 Ultra Shield NMR spectrometer (Bruker, Munich, Germany) at 300 MHz for 1H and 75 MHz for 13C; the chemical shifts are expressed in δ (ppm) downfield from tetramethylsilane. Splitting patterns were designated as follows: s: singlet; d: doublet; t: triplet; m: multiplet. Electron impact mass spectra were recorded on a Varian Mat 311-A70eV instrument (Varian, Fort Collins, USA). Chemicals used were supplied from Sigma-Aldrich (Sigma-Aldrich Chemie GmbH, Steinheim, Germany).
Synthesis
Compounds 1–3
These compounds were prepared in our laboratory according to reported procedures.
General procedures for the synthesis of compounds 4–11
A mixture of 2-phenyl-4-chloroquinazoline 3 (0.666 g, 0.003 mol) and the appropriate alkyl halide (0.003 mol) in dry dimethylformamide (10 mL) in presence of anhydrous potassium carbonate (0.3 g) was stirred for 24 h at room temperature. The solvent was then diluted with water (30 mL) and the separated solid was filtered, washed with water, dried and crystallised from ethanol to afford compounds 4–1l.
4: Yield, 41%; m.p. 135–137°C; 1H NMR (CDCl3): δ 2.90–2.92 (t, 2H, C6H5CH2, J = 7.0 Hz), 4.34–4.36 (t, 2H, OCH2, J = 7.0 Hz), 7.60–8.22 (m, 14H, Ar-H). 13C NMR: δ 34.40 (C6H5CH2), 64.98 (OCH2), 118.11, 121.95, 126.50, 127.28, 128.16, 129.23, 130.05, 131.15, 133.80, 140.06, 151.34, 163.28, 181.02 (Ar-C). MS (EI): m/z 326 [M+, 32%]. Anal. (C22H18N2O) C, H, N.
General procedures for the synthesis of 2-phenyl 4-substituted amino quinazoline derivatives 12–22
A mixture of compound 3 (0.36 g, 0.0015 mol) and the appropriate piperidine derivative (0.0015 mol) in acetone (12 mL) in presence of anhydrous potassium carbonate (0.5 g) was heated under reflux for 5 h. The solvent was then evaporated under vacuum and the separated solid was filtered, washed with water, dried and crystallised from ethanol to afford compounds l2–22.
12: Yield, 87%; m.p. 159–161°C; 1H NMR (CDCl3): δ 4.33 (s, 2H, CH2), 6.70 (s, 1H, NH, exchangeable), 7.48–8.25 (m, 14H, Ar-H). 13C NMR: δ 45.84 (CH2), 117.0, 121.05, 126.42, 127.0, 127.41, 128.76, 129.83, 130.25, 130.64, 134.0, 142.16, 150.18, 157.33, 160.15. MS (EI): m/z 311 [M+, 65%]. Anal. (C21H17N3) C, H, N.
Pharmacology
Materials
Dasatinib was supplied by LC Laboratories® (Woburn, MA, USA). All other chemicals were obtained from Sigma Chemical Company (St. Louis, MO, USA). The cell lines MCF-7, HeLa and HepG2 were obtained from the American Type Culture Collection (Rockville, MD, USA). Tissue culture plates and flasks were purchased from Costar (Milan, Italy).
Evaluation of cellular cytotoxicity
The cytotoxic activity of compounds 4–22 against HepG2 cells, MCF-7 cells and HeLa cells was determined using sulphorhodamine-B assay as described by Skehan et al.Citation19. Briefly, stock solutions of the tested compounds were prepared in dimethyl sulphoxide (DMSO) and were used for serial dilutions in culture medium. The final concentration of DMSO in all wells was adjusted to 0.3%. The tested cell lines were grown in RPMI-1640 medium supplemented with 10% calf serum. Exponentially growing cells were suspended in the above-mentioned medium, seeded in sextuplicates onto 96-well plates at a density of 5 × 104 cells/well and incubated at 37°C in a humidified 5% CO2 atmosphere for 24 h. The cell medium in test wells was then changed to new culture medium containing the required concentrations (10, 25, 50, 100 µg/mL) of the tested compounds, while the cell medium in control wells was changed to new culture medium containing an equivalent volume of solvent (DMSO), and all cells were then incubated for 48 h. Following the 48-h exposure to the compounds, cells were fixed with 50% cold trichloroacetic acid for 1 h, stained for 30 min with 0.4% sulphorhodamine-B and then washed with 1% acetic acid. The plates were then air-dried and the optical density of each well was measured spectrophotometrically at 564 nm using the enzyme-linked immunosorbent assay microplate reader (Meter tech.*sigma;∑ 960, USA). Surviving fraction for each cell type was determined, and then IC50 was calculated for each compound under investigation. It is worth to mention that the cytotoxic activity of anti-cancer drug dasatinib, a multi-targeted kinase inhibitor of BCR-ABL and Src family kinasesCitation9, against the three cell lines was examined at the same concentrations of tested compounds and used as standard for comparative purposes.
Declaration of interest
This work was supported by a grant from King Abdulaziz city for science and technology (KACST; Grant No.: ARP-29–265) and the National Plan of Science, Technology and Innovation (Grant No. 10-MED1188-02), and a funding from the College of Pharmacy Research Center, King Saud University, Riyadh, Saudi Arabia.
References
- Wang Y, Ji P, Liu J, Broaddus RR, Xue F, Zhang W. Centrosome-associated regulators of the G(2)/M checkpoint as targets for cancer therapy. Mol Cancer 2009;8:1–13.
- MacDonald V. Chemotherapy: Managing side effects and safe handling. Can Vet j 2009;50:665–668.
- Sirisoma N, Pervin A, Zhang H, Jiang S, Willardsen JA, Anderson MB et al. Discovery of N-(4-methoxyphenyl)-N,2-dimethylquinazolin-4-amine, a potent apoptosis inducer and efficacious anticancer agent with high blood brain barrier penetration. J Med Chem 2009;52:2341–2351.
- Li J, Meng Y, Liu Y, Feng ZQ, Chen XG. F84, a quinazoline derivative, exhibits high potent antitumor activity against human gynecologic malignancies. Invest New Drugs 2010;28:132–138.
- Bavetsias V, Marriott JH, Melin C, Kimbell R, Matusiak ZS, Boyle FT et al. Design and synthesis of Cyclopenta[g]quinazoline-based antifolates as inhibitors of thymidylate synthase and potential antitumor agents(,). J Med Chem 2000;43:1910–1926.
- Gangjee A, Vidwans AP, Vasudevan A, Queener SF, Kisliuk RL, Cody V et al. Structure-based design and synthesis of lipophilic 2,4-diamino-6-substituted quinazolines and their evaluation as inhibitors of dihydrofolate reductases and potential antitumor agents. J Med Chem 1998;41:3426–3434.
- Conconi MT, Marzaro G, Guiotto A, Urbani L, Zanusso I, Tonus F et al. New Vandetanib analogs: Fused tricyclic quinazolines with antiangiogenic potential. Invest New Drugs 2010;.
- Yang SH, Khadka DB, Cho SH, Ju HK, Lee KY, Han HJ et al. Virtual screening and synthesis of quinazolines as novel JAK2 inhibitors. Bioorg Med Chem 2011;19:968–977.
- Gwanmesia PM, Romanski A, Schwarz K, Bacic B, Ruthardt M, Ottmann OG. The effect of the dual Src/Abl kinase inhibitor AZD0530 on Philadelphia positive leukaemia cell lines. bmc Cancer 2009;9:53.
- Schenone S, Bruno O, Radi M, Botta M. New insights into small-molecule inhibitors of Bcr-Abl. Med Res Rev 2011;31:1–41.
- Cortes J, Rousselot P, Kim DW, Ritchie E, Hamerschlak N, Coutre S et al. Dasatinib induces complete hematologic and cytogenetic responses in patients with imatinib-resistant or -intolerant chronic myeloid leukemia in blast crisis. Blood 2007;109:3207–3213.
- Baud V, Karin M. Is NF-kappaB a good target for cancer therapy? Hopes and pitfalls. Nat Rev Drug Discov 2009;8:33–40.
- Tobe M, Isobe Y, Tomizawa H, Nagasaki T, Takahashi H, Fukazawa T et al. Discovery of quinazolines as a novel structural class of potent inhibitors of NF-kappa B activation. Bioorg Med Chem 2003;11:383–391.
- Rinderspacher A, Cremona ML, Liu Y, Deng SX, Xie Y, Gong G et al. Potent inhibitors of Huntingtin protein aggregation in a cell-based assay. Bioorg Med Chem Lett 2009;19:1715–1717.
- Alafeefy AM, Kadi AA, Al-Deeb OA, El-Tahir KE, Al-Jaber NA. Synthesis, analgesic and anti-inflammatory evaluation of some novel quinazoline derivatives. Eur J Med Chem 2010;45:4947–4952.
- Burbuliene MM, Mazeikaite R, Vainilavicius P. Regioselective synthesis of N(3)- and O-acylmethyl derivatives of 2-methylthio-4(3H)-quinazolinone. J Heterocyclic Chem 2008;45:607–610
- Bogentoft C, Kronberg L, Danielsson B. Studies on the medicinal chemistry of oxoquinazolines. IV. N- and O-alkylation of some 2-substituted 3,4-dihydro-4-oxoquinazolines. Acta Pharm Suec 1969;6:489–500.
- Hori M, Ohtaka H. Effects of a 2-Substituent on the Ratio of N- and O-Alkylation of 4(3H)-Quinazolinones. Chem Pharm Bull 1993;41:1114–1117.
- Skehan P, Storeng R, Scudiero D, Monks A, McMahon J, Vistica D et al. New colorimetric cytotoxicity assay for anticancer-drug screening. J Natl Cancer Inst 1990;82:1107–1112.