Abstract
Plasmin plays important roles in various physiological systems. The identification of inhibitors controlling its regulation represents a promising drug-discovery challenge. To develop selective inhibitors of plasmin, structural information of the binding modes is crucial. Here, a computational docking study was conducted to provide structural insight into plasmin subsite interactions with substrates/inhibitors. Predicted binding modes of two peptide-substrates (D/L-Ile-Phe-Lys), and potent and weak inhibitors (YO-2 and PKSI-527) suggested non-prime and prime subsite interactions relevant to recognition by plasmin. Predicted binding modes also correlated well with the experimental structure–activity relationships for plasmin substrates/inhibitors, namely the differences of KM values between the D- and L-peptide-substrates and inhibitory potencies of YO-2 and PKSI-527. In particular, interaction observed at a hydrophobic pocket near S2 and at a tunnel-shaped hydrophobic S1′ was strongly suggested to be significantly involved in tight binding of inhibitors to plasmin. Our present findings may aid in the design of potent and selective plasmin inhibitors.
Introduction
Plasmin, which consists of a trypsin-type serine protease domain and five kringle domains, is activated from plasminogen by cleavage of the Arg561–Val562 peptide bond by plasminogen activators, including tissue plasminogen activator and urokinase plasminogen activatorCitation1,Citation2. Plasmin plays important roles in the fibrinolysis system, mainly by degrading fibrin clots, and also activates angiogenetic and metastatic proteases/factors, such as matrix metalloproteases. Thus, the ability to regulate plasmin activity is a clinical challenge to realize better therapeutics for various haematological diseases and thrombolytic therapy.
To date, a series of plasmin inhibitors with trans-4-aminomethylcyclohexane carboxylic acid (Tra) or cyclic ketone groups have been developedCitation3–5 and a few are in clinical useCitation6. Recently, it was revealed that plasmin contributes to the early stages of rheumatoid arthritis and facilitates influenza virus circulation, indicating the potential of applying plasmin inhibitors to emerging disease targetsCitation7,Citation8.
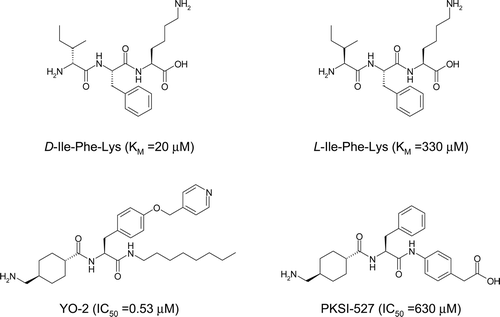
The selectivity of plasmin inhibitors towards non-target serine proteases is an important consideration for potential therapeutics since these proteases share highly homologous sequences. Although information related to protein structure and ligand interactions are relevant to designing selective inhibitors, to date, only one X-ray structure of the serine protease domain of plasmin (i.e. μ-plasmin) complexed with a covalently bound chloromethylketone inhibitor is availableCitation9. Here, to structurally elucidate the subsites of plasmin and better understand the subsite interactions with substrates and inhibitors, we conducted a computational docking study with the tripeptidic substrates D/L-Ile-Phe-Lys, YO-2Citation3 and PKSI-527Citation10 as representative plasmin inhibitors ().
Figure 1. Chemical structures of D/L-Ile-Phe-Lys, YO-2, and PKSI-527. KM and IC50 values for plasmin are in parenthesesCitation3,Citation4,Citation20.
Methods
Coordinates used for docking experiments
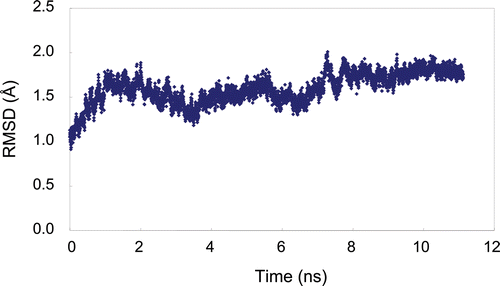
The protein data bank (PDB) IDs used in this study were 2FX6 for trypsin in complex with 2-aminobenzimidazoleCitation11, 1BML for plasmin in complex with streptokinase and no ligand moleculesCitation12, and 1FV9 for (micro)urokinase in complex with 2-amino-5-hydroxy-benzimidazoleCitation13. As described in the Introduction section, the X-ray structure of plasmin complexed with a covalently bound ligand is available with PDB ID 1BUICitation9. The 1BUI structure represents a productive complex that consists of the “enzyme” µ-plasmin, staphylokinase, and the “substrate” µ-plasmin. The substrate-binding site of the “enzyme” µ-plasmin is widely occupied by the bound proteins compared with the 1BML structure. To minimize the influence of the bound proteins on the conformations of plasmin subsites, the 1BML structure was used in this study. High resolution X-ray structures of urokinase in complex with different ligands are known. However, the 1FV9 structure with a moderate resolution of 3.0 Å was chosen for this study since the ligand of the 1FV9 structure possesses a P1 moiety that may not largely influence the conformation of prime and non-prime subsites in urokinase. The structures of the proteases were determined only using their catalytic domains. In the trypsin and urokinase structures, the ligand molecules complexed in the S1 site were removed for the docking experiments. In the plasmin structure, the X-ray structure was likely represented as a rigid status since the entire plasmin molecule was surrounded by streptokinase, and thus should be relaxed prior to the docking experiments. After Ala741 in the plasmin structure was mutated to Ser to handle the structure as wild-type and streptokinase was removed, the conformation relaxation of plasmin was performed by molecular dynamic (MD) calculations using AMBER 10 (University of California, San Francisco, USACitation14). MD simulation for 11 ns under periodic boundary conditions with constant pressure at 300 K and 1.0 atm with a 2 fs time step and 4897 water molecules was implemented after a 120 ps equilibration calculation. After the MD system reached conformational equilibrium, 50 structures sampled in 50 ps simulation starting from 10.5 ns were averaged and then minimized to obtain the coordinates of plasmin for the docking experiments (). The AMBER ff03 force field was appliedCitation15. The particle mesh Ewald method was employed for calculation of the full electrostatic energy of a periodic box with a cut-off distance for non-bonding interaction of 8.0 Å. All MD simulations were performed using the PMEMD module of the AMBER package. Manipulation of molecular structures in this study was done using the Maestro 9.0 molecular modelling package (Schrodinger Inc., Portland, USA) on a single Pentium IV central processing unit (3.0 GHz) computer with Linux/CentOS4.0.
Figure 2. Molecular dynamic simulation of plasmin over 11 ns. The root-mean square deviation (RMSD) of simulated structures from the initial structure is plotted versus time. The RMSD calculation is based on the protein main-chain atoms.
Docking protocol and experiments
For the docking experiments, Omega2.3, Fred2.2, and Syzbki1.3 (OpenEye Scientific Software Inc., Santa Fe, USA) were used as computational tools for the generation of multiple conformations of ligands, searching ligand poses in the binding pockets of proteins, and optimizing the conformations of poses and side-chains of proteins, respectivelyCitation16–18. Omega calculations for D- and L-Ile-Phe-Lys substrates, YO-2, and PKSI-527 produced 2289, 2543, 5713, and 931 conformers, respectively. The C-terminal structures of the substrates were considered to be an aldehyde form (CHO) to mimic reaction intermediates. In searching for docking poses using Fred2.2, the outer contour volumes in the receptor boxes of plasmin and urokinase were adjusted to 30586 and 32232 ÅCitation3, respectively. The top 1000 docking poses for each docking experiment were selected using consensus scores with PLP, Chemgauss3, and OEChemscore. The selected poses were optimized using Szybki1.3 with MMFF force-fields and Poisson–Boltzmann electrostatic potential. The cut-off distance of non-bonding interaction used was 18.0 Å, and the dielectric constant used for Poisson–Boltzmann calculation was 1.0. The optimization calculation was also carried out for the protein side-chain atoms which were located within 3.0 Å from the ligands. The optimization was terminated when the rms gradient reached 0.1 Å. The most probable docking pose selected from the 1000 poses was defined as the conformation with the lowest energy of ligand–protein interaction among conformations with energetically reasonable van der Waals and Coulombic interaction energies, which were within two standard deviations of the average energies of the 1000 poses. This energetic selection criterion was set to avoid selecting an artificial binding pose. For the selection of the substrates, the most probable docking pose was selected from the poses with lower and reasonable interaction energy, as defined above, and with the closest distance between the Oγ-atom of Ser741 and the C-terminal carbon of the substrates.
Results and discussion
Validation of computational protocol for serine protease system
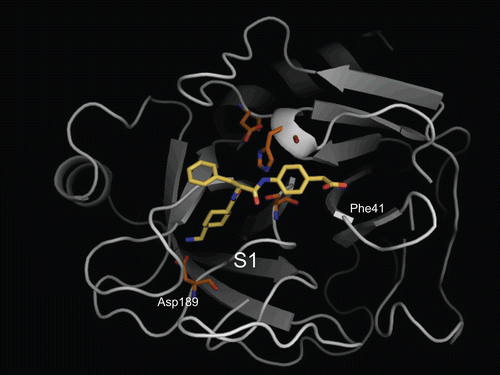
The validation of the computational docking protocol for the system of serine protease–ligand complexes was performed using trypsin and PKSI-527 as a test set. The x-ray structure of trypsin complexed with PKSI-527 is the only experimentally solved structure for a YO-2 series compoundCitation19. depicts the trypsin–PKSI-527 complex determined by the computational protocol, showing that the Tra moiety of PKSI-527 was bound to the S1 site of trypsin. The 4-carboxymethylanilide moiety of PKSI-527 was directed to Phe41 similarly as observed in the Form-I of the X-ray structure described in the literatureCitation19 whereas the position of the Phe moiety was slightly displaced from the Form-I structure. This observation corresponds well to that in the X-ray complex structure. The successful validation of trypsin complex suggests that the computational docking protocol used in this study can be applied for the serine protease system.
Figure 3. Predicted complex of trypsin with PKSI-527. The structure of trypsin is displayed as a cartoon model (grey) and the inhibitor PKSI-527 is shown as a stick model (yellow). The side-chains of His57, Asp102, Asp189, and Ser195 in the catalytic site of trypsin are shown as stick models (brown).
Prediction of D/L-Ile-Phe-Lys substrate-binding modes
To structurally estimate the subsites of plasmin, the binding poses of the tripeptidic substrates D-Ile-Phe-Lys and L-Ile-Phe-Lys were predicted using the computational docking protocol. The affinities of D-Ile-Phe-Lys-pNA and L-Ile-Phe-Lys-pNA (pNA = p-nitroanilide) to plasmin exhibit a 10-fold difference; the KM values of D- and L-forms are 20 and 330 μM, respectivelyCitation20. The overall binding poses of the two substrates were quite similar (). The side-chain of the Lys residue of both substrates interacted with Asp735 of plasmin as an S1/P1 interaction, consistent with that seen in the complexes of other trypsin-type proteases with various ligands. The numbers of residues in substrates and subsites in plasmin are labelled as follows: the scissile bond of substrate is defined as P1–P1′ bond. The N-terminal sides of the scissile bind in the substrate are labelled as P1–Pn. The corresponding subsites in the protease are labelled as S1–Sn. The C-terminal sides of the substrate and subsites from the scissile bond are labelled with prime numbers as P1′–Pn′ and S1′–Sn′, respectively. The carbonyl group of the Ile residue was hydrogen-bonded to the amino group of Gly762 in both complexes (3.8 Å distance), while this distance is slightly far from canonical hydrogen bonds as described below. The side-chains of the substrate Phe and Ile residues protruded in the same direction in the binding pocket and made some internal hydrophobic interactions/contacts (. The Phe side-chain was positioned on a wall composed of His603, Lys645, and Asp646, and the Ile side-chain faced towards the region of Arg719, Trp761, Gly762, and Leu763. These areas in plasmin most likely represent S2 and S3 sites, respectively.
Figure 4. Predicted complex of plasmin with D/L-Ile-Phe-Lys substrates. The structure of plasmin is displayed as a surface model (grey) and the ligands D- and L-Ile-Phe-Lys are shown as stick models (magenta for D-Ile-Phe-Lys and blue for L-Ile-Phe-Lys). The side-chains of residues in the binding and catalytic sites are shown as stick models (white). The underlined label is for the substrate. (a) Close-up view of S1/P1 interaction. (b) The overall binding mode of the substrates.
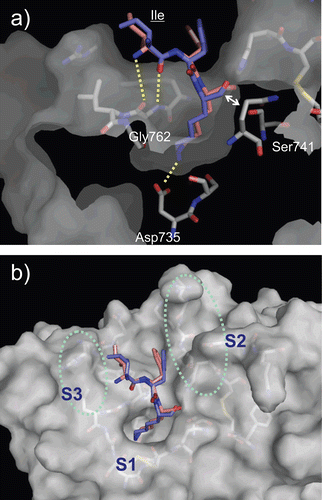
Our modelling also revealed that the amino group at the N-terminus of the substrates was differentially oriented between the D- and L-forms of the tripeptidic substrate (. The N-terminal amino group of the D-form was positioned towards the carbonyl group of the main-chain of Gly762, whereas that of the L-form was positioned away from Gly762. Although the atom–atom distance between the nitrogen of the D-form N-terminus and the oxygen of the Gly762 carbonyl was 4.3 Å, which is longer than canonical hydrogen-bonding distance, this distance may become shorter in a productive complex (i.e. a complex showing the state of the cleavage reaction). In this model, the C-terminus of the D-form substrate was positioned at 3.6 Å far from the oxygen of Ser741. In the productive complex, this distance should be much shorter approximately 1.4 Å. Therefore, the binding position of the substrate would be shifted to the direction of Ser741, and then the distance between the N-terminus of the substrate and Gly762 would be shortened into the range of hydrogen-bonding. In contrast, the L-form may not form a hydrogen bond between the N-terminal amino group and Gly762 even when the distance of the substrate is closer to Ser741 during the reaction, since the nitrogen atom was not directed towards Gly762 and the atom–atom distance (5.7 Å) was relatively large. This difference of hydrogen-bonding ability between the D/L-forms may be one reason for the higher affinity of the D-form for plasmin than that of the L-form. The differences of the affinity between the D/L-forms were also shown in P1 Arg substrate (i.e. Ile-Phe-Arg sequence) and chloromethyl ketone inhibitors with sequences of D-Ile-Phe-Lys and D-Ile-Phe-ArgCitation21.
Prediction of YO-2 and PKSI-527 binding modes
YO-2 is a potent plasmin inhibitor with an IC50 of 0.53 μM3; however, the binding mode of YO-2 in plasmin has not yet been solved experimentally. The docking experiments in this study showed that the Tra moiety of YO-2 was bound to the plasmin S1 site and interacted with the side-chain of Asp735 as expected (. The O-picolyl-Tyr moiety adopted an extended conformation along the wall of the S2 site. The terminus of the O-picolyl-Tyr moiety was located in the pocket formed by Glu724, Ser760, Trp761, Leu775, and the S2 wall, hereinafter referred as “extra S2 pocket”. This additional extra S2 pocket was composed mainly of hydrophobic residues and was located at the back side of the S2 site, and appears to correspond with the S4 site of factor XaCitation22.
Figure 5. Predicted complex of plasmin with YO-2. The structure of plasmin is displayed as a surface model (grey) and the inhibitor YO-2 is shown as a stick model (green). The side-chains of residues in the binding and catalytic sites are shown as stick models (white). (a) Identical view as that of . (b) Side-view.
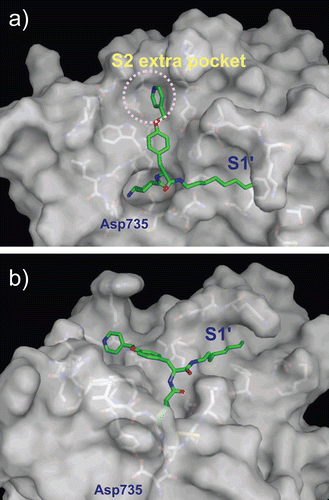
The octylamide moiety of YO-2 adopted an extended conformation and protruded from the area formed by Thr581, Met585, Phe587, Cys588, Cys604, Lys607, Tyr614, and Gln738 (. Based on the binding modes determined for the D/L-Ile-Phe-Lys substrates, this area most likely represents an S1′ site and has a tunnel-like shape. In the original design of YO-2 series inhibitors, the O-picolyl-Tyr and octylamide moieties were assumed to be P1′ and P2′ parts of the molecule, respectivelyCitation23. However, the complex predicted in this study indicates that these moieties are P2 and P1′ parts, respectively. The binding mode of the octylamide moiety, characterized by the long aliphatic chain passing through the hydrophobic tunnel of the S1′ site, may be a dominant force for the tight binding of YO-2 to plasmin. The predicted binding mode is supported by the structure–activity relationship (SAR) data indicating that YO-2 series inhibitors have shorter or branched chains, or hydrophilic functionalities exhibit lower inhibitory activity than YO-2 (Citation3,Citation4,Citation24). For instance, IC50 values of inhibitors with para-ethyl-phenylamide, isopentylamide, and carboxyl-heptylamide groups were 1.7, 1.9, and 5.5 μM, respectively.
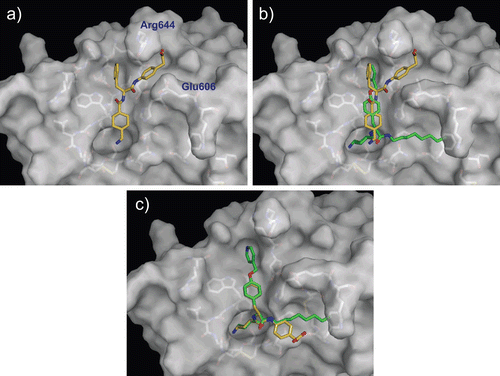
To further understand the SAR of YO-2 series inhibitors, a weak plasmin inhibitor with an IC50 of 630 μM, PKSI-527, was used for the binding mode prediction. In the model structure, surprisingly, the Tra moiety of PKSI-527 was not bound to the S1 site (; rather, PKSI-527 was located at the region around the S2 site. The Phe moiety was located in the extra S2 pocket, and well overlapped with the position of the terminus of the O-picolyl-Tyr moiety of YO-2 (. The 4-carboxymethylanilide moiety of PKSI-527 was bound between the side-chains of Glu606 and Arg644, and the carboxyl group of this moiety formed a hydrogen bond with the side-chain of Arg644. Compared with the modelled binding mode of YO-2, PKSI-527 appears to only bind plasmin weakly, since no S1/P1 interaction was observed in this model. These differences in the modelled binding modes may explain the 1000-fold difference in the inhibitory activities of the two inhibitors. To further analyse and compare the binding modes of PKSI-527 and YO-2, the conformation of PKSI-527 was forced to overlap with that of YO-2 (. The 4-carboxymethylanilide moiety of PKSI-527 may not disturb the binding to plasmin if the Tra moiety forms the S1/P1 interaction since no obvious steric clash was observed in this forced model. This finding indicates that the hydrophobicity of P1′ may be a crucial determinant of potency for YO-2 series inhibitors.
Table 1. SAR of Tra-Tyr(O-Pic)-CONH-X.
Figure 6. Predicted complex of plasmin with PKSI-527. The structure of plasmin is displayed as a surface model (grey) and the inhibitor PKSI-527 is shown as a stick model (yellow). The side-chains of residues in the binding and catalytic sites are shown as stick models (white). (a) Identical view as that of . (b) Comparison of the binding modes of YO-2 (green) and PKSI-527. (c) The structure of PKSI-527 overlapped with YO-2.
Comparison with urokinase complexes
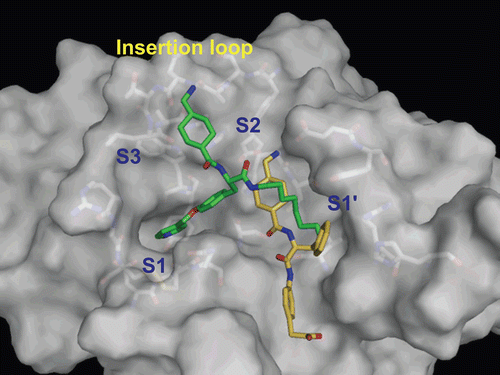
YO-2 exhibits rather low selectivity towards urokinase (IC50 values for plasmin and urokinase are 0.53 and 5.3 μM, respectively), although PKSI-527 shows comparable activity for both enzymes (IC50 values for plasmin and urokinase are 630 and 350 μM, respectivelyCitation3,Citation10). For the development of selective plasmin inhibitors, selectivity to urokinase should be considered. Thus, the binding modes of the two inhibitors to urokinase were predicted and compared (). In contrast with the binding of YO-2 to plasmin, the Tra moiety was located in the S2 site and the O-picolyl-Tyr moiety was bound to the S1 site. This prediction could be due to structural differences in the binding pockets of urokinase: an insertion loop exists between the S2 and S3 sites and the S2 extra pocket is not foundCitation25. The octylamide moiety of YO-2 was located at the S1′ site in urokinase, although the hydrophobic tunnel was not observed. The loss of ionic interaction with Asp189 at the S1 site may weaken the inhibitory activity of YO-2 against urokinase. No S1/P1 interaction was observed in the model structure of the urokinase–PKSI-527 complex either. The superficial interaction of PKSI-527 to urokinase, as seen in plasmin, may explain the similarly weak activity of this inhibitor against urokinase. Based on the structural information obtained for YO-2 and PKSI-527, the introduction of a sterically bulkier group into the terminus of the P2 region of YO-2 series inhibitors to prevent S1-binding may be an appropriate tactic to increase selectivity towards urokinase.
Figure 7. Predicted complex of urokinase with YO-2 and PKSI-527. The structure of urokinase is displayed as a surface model (grey) and the inhibitors YO-2 and PKSI-527 are shown as stick models (green for YO-2 and yellow for PKSI-527). The side-chains of residues in the binding and catalytic sites are shown as stick models (white).
Conclusion
In conclusion, our computational docking experiments have revealed that an extra hydrophobic pocket exists at the back side of the S2 site of plasmin, which YO-2 and PKSI-527 utilise for binding. In addition, the docking experiment and SAR data strongly suggest that hydrophobic interaction of the P1′ moiety with the “tunnel-like” S1′ site is required for tight binding to plasmin. The structural information newly suggested from the modelled structures in this study may aid in the design of potent and selective inhibitors of plasmin.
Declaration of interest
The authors report no declarations of interest.
References
- Petersen TE, Martzen MR, Ichinose A, Davie EW. Characterization of the gene for human plasminogen, a key proenzyme in the fibrinolytic system. J Biol Chem 1990;265:6104–6111.
- Robbins KC, Summaria L, Hsieh B, Shah RJ. The peptide chains of human plasmin. Mechanism of activation of human plasminogen to plasmin. J Biol Chem 1967;242:2333–2342.
- Okada Y, Tsuda Y, Tada M, Wanaka K, Okamoto U, Hijikata-Okunomiya A et al. Development of potent and selective plasmin and plasma kallikrein inhibitors and studies on the structure-activity relationship. Chem Pharm Bull 2000;48:1964–1972.
- Tsuda Y, Tada M, Wanaka K, Okamoto U, Hijikata-Okunomiya A, Okamoto S et al. Structure-inhibitory activity relationship of plasmin and plasma kallikrein inhibitors. Chem Pharm Bull 2001;49:1457–1463.
- Xue F, Seto CT. Structure-activity studies of cyclic ketone inhibitors of the serine protease plasmin: design, synthesis, and biological activity. Bioorg Med Chem 2006;14:8467–8487.
- Okamoto S, Hijikata-Okunomiya A, Wanaka K, Okada Y, Okamoto U. Enzyme-controlling medicines: introduction. Semin Thromb Hemost 1997;23:493–501.
- Judex MO, Mueller BM. Plasminogen activation/plasmin in rheumatoid arthritis: matrix degradation and more. Am J Pathol 2005;166:645–647.
- Kido H, Okumura Y, Yamada H, Le TQ, Yano M. Proteases essential for human influenza virus entry into cells and their inhibitors as potential therapeutic agents. Curr Pharm Des 2007;13:405–414.
- Parry MA, Fernandez-Catalan C, Bergner A, Huber R, Hopfner KP, Schlott B et al. The ternary microplasmin-staphylokinase-microplasmin complex is a proteinase-cofactor-substrate complex in action. Nat Struct Biol 1998;5:917–923.
- Wanaka K, Okada Y, Tsuda Y, Okamoto U, Hijikata-Okunomiya A, Okamoto S. Synthesis of trans-4-aminomethylcyclohexanecarbonyl-L- and -D-phenylalanine-4-carboxymethylanilide and examination of their inhibitory activity against plasma kallikrein. Chem Pharm Bull 1992;40:1814–1817.
- McGrath ME, Sprengeler PA, Hirschbein B, Somoza JR, Lehoux I, Janc JW et al. Structure-guided design of peptide-based tryptase inhibitors. Biochemistry 2006;45:5964–5973.
- Wang X, Lin X, Loy JA, Tang J, Zhang XC. Crystal structure of the catalytic domain of human plasmin complexed with streptokinase. Science 1998;281:1662–1665.
- Hajduk PJ, Boyd S, Nettesheim D, Nienaber V, Severin J, Smith R et al. Identification of novel inhibitors of urokinase via NMR-based screening. J Med Chem 2000;43:3862–3866.
- Case DA, Darden TA, Cheatham TE, III Simmerling, CL, Wang J, Duke RE, Luo R, Crowley M, Walker RC, Zhang W, Merz KM, Wang B, Hayik S, Roitberg A, Seabra G, Kolossváry I, Wong KF, Paesani F, Vanicek J, Wu X, Brozell SR, Steinbrecher T, Gohlke H, Yang L, Tan C, Mongan J, Hornak V, Cui G, Mathews DH, Seetin MG, Sagui C, Babin V, Kollman PA. AMBER 10, University of California, San Francisco, 2008.
- Duan Y, Wu C, Chowdhury S, Lee MC, Xiong G, Zhang W et al. A point-charge force field for molecular mechanics simulations of proteins based on condensed-phase quantum mechanical calculations. J Comput Chem 2003;24:1999–2012.
- Boström J, Greenwood JR, Gottfries J. Assessing the performance of OMEGA with respect to retrieving bioactive conformations. J Mol Graph Model 2003;21:449–462.
- McGann MR, Almond HR, Nicholls A, Grant JA, Brown FK. Gaussian docking functions. Biopolymers 2003;68:76–90.
- Grant JA, Pickup BT, Nicholls A. A smooth permittivity function for Poisson-Boltzmann solvation methods. J Comp Chem 2001;22:608–640.
- Tomoo K, Satoh K, Tsuda Y, Wanaka K, Okamoto S, Hijikata-Okunomiya A et al. Binding diversity of a noncovalent-type low-molecular-weight serine protease inhibitor and function of a catalytic water molecule: X-ray crystal structure of PKSI-527-inhibited trypsin. J Biochem 2001;129:455–460.
- Okada Y, Tsuda Y, Teno N, Wanaka K, Sasaki K, Hijikata A et al. Synthesis of plasmin substrates and relationship between their structure and plasmin activity. Int J Pept Protein Res 1986;27:79–85.
- Tsuda Y, Teno N, Okada Y, Wanaka K, Bohgaki M, Hijikata-Okunomiya A et al. Synthesis of tripeptide chloromethyl ketones and examination of their inhibitory effects on plasmin and plasma kallikrein. Chem Pharm Bull 1989;37:3108–3111.
- Straub A, Roehrig S, Hillisch A. Entering the era of non-basic p1 site groups: discovery of Xarelto (Rivaroxaban). Curr Top Med Chem 2010;10:257–269.
- Teno N, Wanaka K, Okada Y, Taguchi H, Okamoto U, Hijikata-Okunomiya A et al. Development of active center-directed plasmin and plasma kallikrein inhibitors and studies on the structure-inhibitory activity relationship. Chem Pharm Bull 1993;41:1079–1090.
- Tada M. Development of plasmin and plasma kallikrein inhibitors, and development of purification method for plasma kallikrein. Thesis of Kobe Gakuin University, 2002.
- Nienaber VL, Davidson D, Edalji R, Giranda VL, Klinghofer V, Henkin J et al. Structure-directed discovery of potent non-peptidic inhibitors of human urokinase that access a novel binding subsite. Structure 2000;8:553–563.