Abstract
The growth of Mycobacterium tuberculosis is strongly inhibited by weak acids although the mechanism by which these compounds act is not completely understood. A series of substituted benzoic acids, nipecotic acid, ortho- and para-coumaric acid, caffeic acid and ferulic acid were investigated as inhibitors of three β-class carbonic anhydrases (CAs, EC 4.2.1.1) from this pathogen, mtCA 1 (Rv1284), mtCA 2 (Rv3588c) and mtCA 3 (Rv3273). All three enzymes were inhibited with efficacies between the submicromolar to the micromolar one, depending on the scaffold present in the carboxylic acid. mtCA 3 was the isoform mostly inhibited by these compounds (KIs in the range of 0.11–0.97 µM); followed by mtCA 2 (KIs in the range of 0.59–8.10 µM), whereas against mtCA 1, these carboxylic acids showed inhibition constants in the range of 2.25–7.13 µM. This class of relatively underexplored β-CA inhibitors warrant further in vivo studies, as they may have the potential for developing antimycobacterial agents with a diverse mechanism of action compared to the clinically used drugs for which many strains exhibit multi-drug or extensive multi-drug resistance.
Introduction
Mycobacterium tuberculosis has been found to be highly sensitive to weak organic acids, (most of which incorporate aromatic scaffolds) administered as amide-type prodrugsCitation1. Indeed, two first line medications for treating tuberculosis are constituted by hydrazides/amides of two carboxylic acid, i.e. isoniazide (isonicotinoyl hydrazide A) and pyrazinamide BCitation1. Carboxylic acids on the other hand also act as inhibitors of the zinc enzyme carbonic anhydrase (CA, EC 4.2.1.1)Citation2–5. Indeed, simple carboxylates such as formate and acetate were investigated as mammalian CA inhibitors (CAIs) by Scozzafava’s group who showed, by using paramagnetic NMR and UV-VIS spectroscopy of Co(II)-substituted CAs complexed with these anions, the metal ion is pentacoordinated in these adducts by three His residues from the enzyme, a water molecule and a carboxylate anion from the inhibitorCitation6,Citation7. This has been thereafter confirmed by X-ray crystallographic studies which showed these anions to bind to the metal ion in a monocoordinated manner with one oxygen of the carboxylate moiety bound to the Zn(II) ion, which is indeed in a trigonal bipyramidal geometry, since a water molecule is also coordinated to it (in addition to the carboxylate ligand)Citation8. In contrast to the α-class CAs present in mammals, including humansCitation2–5, many β-CAs are found in bacterial/fungal pathogens and their inhibition started to be investigated only recently as a possibility to obtain novel types of antibiotics/antifungalsCitation9,Citation10. For example, bacterial enzymes from Escherichia coli, Helicobacter pylori, M. tuberculosis, Brucella spp., Streptomyces pneumoniae, Salmonella enterica and Haemophilus influenzae were cloned and characterized in the last yearsCitation9. The catalytic/inhibition mechanisms of these CAs are well understood, as X-ray crystal structures are available for some of them, but no adducts of these enzymes with inhibitors were characterized so far by this technique, except the acetate adduct of an enzyme (Can2) from the fungal pathogen Cryptococcus neoformansCitation10. As for the α-class CAs mentioned aboveCitation6–8, acetate is coordinated to the Zn(II) ion from the Can2 active site, substituting the water molecule/hydroxide ion responsible for the catalytic activity. Thus, Zn(II) is in a tetrahedral geometry in this complexCitation10.
The genome of the human pathogen M. tuberculosis encodes at least three β-CAs (EC 4.2.1.1), called mtCA 1, 2 and 3, which are encoded by the genes Rv1284, Rv3588c Rv3273Citation11–16. These enzymes have been cloned and their catalytic activity investigatedCitation11–16. As most CAs described to date, these enzymes are inhibited by sulfonamides and related compoundsCitation17–23. However, although interesting, low nanomolar or subnanomolar sulfonamide inhibitors were detectedCitation3–6, it is not yet clear whether their in vivo inhibition has an antimycobacterial effectCitation17.
Resistance to antibiotics belonging to several different classes is escalating and represents a worldwide problemCitation24–28. Many strains of Gram-negative/-positive bacteria (such as among others Staphylococcus aureus, M. tuberculosis, H. pylori, B. suis, S. pneumoniae, etc.) no longer respond to various classes of antibioticsCitation24–28. The situation is particularly dramatic for M. tuberculosis with the emergence of multi-drug resistant and extensive multi-drug resistant strain all over the worldCitation24–28. It is thus stringent to investigate alternative mycobacterial targets as well as different chemotypes (than the usual ones for these targets) in order to identify novel lead compounds which may help the development of efficient antibiotics.
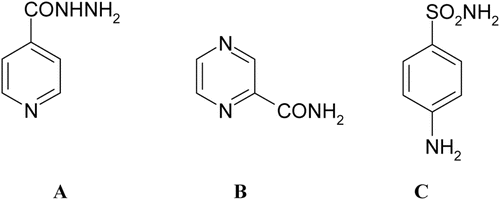
The classical CA inhibitors (CAIs) are the primary sulfonamides, RSO2NH2, which are used clinically for more than 50 years as diuretics or systemically acting antiglaucoma drugsCitation2–5. More than 30 clinically used drugs (or compounds in clinical development) belonging to the sulfonamide or sulfamate class, show significant CA inhibitory activity and are used for the management of a variety of disordersCitation2–5. However, all these drugs target all mammalian CAs, of which 16 different isoforms are known so far, and as thus, they may show undesired side effectsCitation2–5. Many of them also inhibit efficiently the bacterial/fungal β-CAsCitation9. As thus, as mentioned above, a large number of such derivatives started to be investigated as possible antiinfectives with the aim of specifically targeting bacterial/fungal CAs. This field is however still a new one with relevant resistance from the scientific community (not only from bacteria and fungi) in accepting these enzymes as anti-infective targetsCitation9. It should be also mentioned that sulfonamides, of which the prototypical drug is the bacteriostatic drug sulfanilamide C, do not show antimycobacterial effectsCitation17 ().
Figure 1. Structures of compounds isoniazide, pyrazinamide and sulphanilamide.
Thus, exploring various chemotypes, other than the sulfonamide one, may lead to the discovery of β-CA inhibitors with good affinity for enzymes from pathogenic species. Based on the fact that carboxylic acids are reported to show relevant antimycibacterial effectsCitation1 and that many such compounds act as CAIsCitation5–9, here we investigated a series of carboxylic acids incorporating various aromatic/heterocyclic scaffold as inhibitors of the three β-CAs from M. tuberculosis, i.e. mtCA 1 – mtCA 3, evidencing interesting biological activities for many of them.
Materials and methods
Chemistry
Carboxylic acids 1–10 investigated here were commercially available from Sigma-Aldrich, except o-coumaric acid (sodium salt) which was prepared by the procedure of Maresca et al.Citation29 from coumarin by alkaline hydrolysis.
Enzymology
mtCA1–mtCA 3 were recombinant enzymes obtained as described earlierCitation11–17.
CA catalytic activity and inhibition assay
An Applied Photophysics stopped-flow instrument has been used for assaying the CA-catalyzed CO2 hydration activityCitation30. Phenol red (at a concentration of 0.2 mM) has been used as indicator, working at the absorbance maximum of 557 nm with 10–20 mM Hepes (pH 7.5, for α-CAs) or TRIS (pH 8.3 for β-CAs) as buffers, and 20 mM Na2SO4 (for α-CAs) or 20 mM NaCl for β-CAs (for maintaining constant the ionic strength), following the initial rates of the CA-catalyzed CO2 hydration reaction for a period of 10–100 s. The CO2 concentrations ranged from 1.7 to 17 mM for the determination of the kinetic parameters and inhibition constants. For each inhibitor, at least six traces of the initial 5–10% of the reaction have been used for determining the initial velocity. The uncatalyzed rates were determined in the same manner and subtracted from the total observed rates. Stock solutions of inhibitor (10 mM) were prepared in distilled-deionized water and dilutions up to 0.01 nM were done thereafter with distilled-deionized water. Inhibitor and enzyme solutions were preincubated together for 15 min at room temperature prior to assay in order to allow for the formation of the E-I complex. The inhibition constants were obtained by non-linear least-squares methods using PRISM 3, whereas the kinetic parameters for the uninhibited enzymes from Lineweaver–Burk plots, as reported earlierCitation11–17,Citation31, and represent the mean from at least three different determinations.
Results and discussions
We report here the investigation of a series of carboxylic acids of type 1–10 for their interaction with three β-CAs from the bacterial pathogen M. tuberculosis, i.e. mtCA 1 (Rv1284), mtCA 2 (Rv3588c) and mtCA 3 (Rv3273). The rationale for investigating these compounds as inhibitors of these enzymes is based on the fact that many carboxylic acids strongly inhibit the growth of this pathogen by a poorly understood mechanism of actionCitation1. Indeed, the low permeability of the mycobacterial cell wall which is due to the presence of mycolic acid-arabinogalactan complexes is regarded as a significant contributor to the intrinsic drug resistance of mycobacteriaCitation1,Citation17. This particular architecture/composition of the mycobacterial cell wall is probably also responsible for the fact that sulfonamides, poorly membrane-permeable compoundsCitation32, do not show antimycobacterial activity in vitro and in vivoCitation17, although some of them strongly inhibit in vitro β-CAs present in this pathogen, some of which seem to be essential for the life cycle of the bacteriumCitation33,Citation34.
The following structure-activity relationship (SAR) can be observed from data of :
Table 1. Inhibition data of mycobacterial β-CA isoforms mtCA 1–3 with carboxylic acids 1–10 and sulfanilamide SA by a stopped-flow, CO2 hydrase assayCitation30.
| (1) | Against mtCA 1, the carboxylates 1–10 behaved as efficient micromolar inhibitors with inhibition constants in the range of 2.25–7.13 µM, comparable (but slightly better in many cases) to that of the lead compound sulfanilamide, SA which has a KI of 9.84 µM (). SAR is rather straightforward with the simple substituted benzoic acids 1–5 as well as nipecotic acid 6, showing KIs of 3.91–6.20 µM. The benzoic acids possessing electron withdrawing or electron donating moieties in para to the COOH moiety showed a similar behavior, which proves that the pKa of the carboxylic acid is not the main factor governing activity, an observation already seen for the inhibition of fungal β-CAs with aliphatic-, branched- or aromatic carboxylic acidsCitation35,Citation36. The ortho- and para-coumaric acids 7 and 8 showed the best inhibition against this isoform with KIs of 2.25–3.03 µM. The additional OH moiety from caffeic acid 9 or the methoxy one in ferulic acid 10 lead to a roughly 2-fold decrease of the activity compared to p-coumaric acid 8, demonstrating that small structural changes considerably affect the enzyme inhibitory activity. | ||||
| (2) | mtCA 2 was also inhibited efficiently by carboxylates 1–10 with inhibition constants in the range of 0.59–8.10 µM (SA showed a KI of 29.6 µM, being a much less effective CAI compared to the carboxylates investigated here). The aromatic carboxylates 1–4 were efficient mtCA 2 inhibitors with inhibition constants in the range of 2.12–3.67 µM, but the nitrosubstituted compound 5 (the most acidic one in the subseries) was a much weaker inhibitor with a KI of 8.10 µM. Nipecotic acid 5 and o-coumaric acid 6 were also medium potency inhibitors (KIs of 4.17–5.27 µM), whereas p-coumaric and caffeic acid were the best mtCA 2 inhibitors with KIs of 0.59–1.56 µM. An extra methyl group present in 9, as in ferulic acid 10, led to a loss of activity, this compound showing an inhibition constant of 3.50 µM (5.9 times worse than that of p-coumaric acid 9). | ||||
| (3) | The third bacterial CA investigated here, mtCA 3, was the most sensitive to inhibition with carboxylates 1–10, these derivatives showing KIs in the range of 0.11–0.97 µM, an order of magnitude better compared to that of sulfanilmide SA (KI of 7.11 µM). The benzoic acids 1–5 showed a compact behavior of efficient mtCA 3 inhibitors with best compounds being those incorporating iodine and cyano moieties (2 and 3), whereas the least effective one was the bromobenzoic acid 1. Nipectotic acid 6 was the best mtCA 3 inhibitor detected in this study with a KI of 0.11 µM () together with o-coumaric acid 7 (KI of 0.12 µM). The remaining natural product carboxylates 8–10 were slightly less inhibitory compared to 7 (KIs of 0.36–0.60 µM), although they show small structural differences with each other, proving that the inhibition of the enzyme is drastically influenced by small changes in the scaffold of the carboxylic acid. | ||||
| (4) | mtCA 3 was the most prone to be inhibited by carboxylates 1–10 followed by mtCA 2, whereas mtCA 1 was the least inhibited isoform. This is quite different from the inhibition profile of the sulfonamide SA. Indeed, for this compound, the best inhibition was seen against mtCA 3 followed by mtCA 1 and the least inhibited isoform was mtCA 2. | ||||
One of the main problems with mtCA inhibitors investigated so far is that no inhibition of the bacterial growth in vivo has been observed to date (unpublished results from this laboratory and ref. Nishimori et al.Citation17), probably due to the very hydrophilic, membrane impermeant character of sulfonamides and their difficulty to cross the mycobacteria cell wallCitation1. The rationale of this study was just to detect inhibitors belonging to a different chemotype, which may be derivatized easily (as amides/hydrazides) which may allow a better penetrability profile to such compounds, as for the two clinically used (pro)drugs A and B discussed above. Further studies are thus warranted to obtain highly effective and lipophilic mtCA inhibitors and prove their in vivo efficacy.
Conclusion
We evaluated a series of aromatic/heterocyclic carboxylates as inhibitors of three β-CAs from the bacterial pathogen M. tuberculosis, mtCA 1 (Rv1284), mtCA 2(Rv3588c) and mtCA 3 (Rv3273). All three enzymes were inhibited with efficacies between the submicromolar to the micromolar one, depending on the scaffold present in the carboxylic acid. mtCA 3 was the isoform mostly inhibited by these compounds (KIs in the range of 0.11–0.97 µM) followed by mtCA 2 (KIs in the range of 0.59–8.10 µM), whereas against mtCA 1, these carboxylic acids showed inhibition constants in the range of 2.25–7.13 µM. This class of relatively underexplored β-CA inhibitors warrant further in vivo studies, as they may have the potential for developing antimycobacterial agents with a diverse mechanism of action compared to the clinically used drugs for which many strains exhibit multi-drug or extensive multi-drug resistance.
Declaration of interest
This work was supported by an EU FP7 research grant (Metoxia project).
References
- Gu P, Constantino L, Zhang Y. Enhancement of the antituberculosis activity of weak acids by inhibitors of energy metabolism but not by anaerobiosis suggests that weak acids act differently from the front-line tuberculosis drug pyrazinamide. J Med Microbiol 2008;57:1129–1134.
- Supuran CT. Carbonic anhydrases: novel therapeutic applications for inhibitors and activators. Nat Rev Drug Discov 2008;7:168–181.
- Neri D, Supuran CT. Interfering with pH regulation in tumours as a therapeutic strategy. Nat Rev Drug Discov 2011;10:767–777.
- Pastorekova S, Parkkila S, Pastorek J, Supuran CT. Carbonic anhydrases: current state of the art, therapeutic applications and future prospects. J Enzyme Inhib Med Chem 2004;19:199–229.
- Supuran CT. Carbonic anhydrase inhibitors and activators for novel therapeutic applications. Future Med Chem 2011;3:1165–1180.
- Bertini I, Luchinat C, Scozzafava A. Interaction of cobalt-bovine carbonic anhydrase with the acetate ion. Biochim Biophys Acta 1976;452:239–244.
- Bertini I, Luchinat C, Scozzafava A. Binding affinity of bicarboxylate ions for cobalt (II) bovine carbonic anhydrase. Bioinorg Chem 1978;9:93–100.
- Håkansson K, Carlsson M, Svensson LA, Liljas A. Structure of native and apo carbonic anhydrase II and structure of some of its anion-ligand complexes. J Mol Biol 1992;227:1192–1204.
- Supuran CT. Bacterial carbonic anhydrases as drug targets: toward novel antibiotics? Front Pharmacol 2011;2:34.
- Schlicker C, Hall RA, Vullo D, Middelhaufe S, Gertz M, Supuran CT et al. Structure and inhibition of the CO2-sensing carbonic anhydrase Can2 from the pathogenic fungus Cryptococcus neoformans. J Mol Biol 2009;385:1207–1220.
- Suarez Covarrubias A, Larsson AM, Högbom M, Lindberg J, Bergfors T, Björkelid C et al. Structure and function of carbonic anhydrases from Mycobacterium tuberculosis. J Biol Chem 2005;280:18782–18789.
- Covarrubias AS, Bergfors T, Jones TA, Högbom M. Structural mechanics of the pH-dependent activity of β-carbonic anhydrase from Mycobacterium tuberculosis. J Biol Chem 2006;281:4993–4999.
- Minakuchi T, Nishimori I, Vullo D, Scozzafava A, Supuran CT. Molecular cloning, characterization, and inhibition studies of the Rv1284 β-carbonic anhydrase from Mycobacterium tuberculosis with sulfonamides and a sulfamate. J Med Chem 2009;52:2226–2232.
- Nishimori I, Minakuchi T, Vullo D, Scozzafava A, Innocenti A, Supuran CT. Carbonic anhydrase inhibitors. Cloning, characterization, and inhibition studies of a new β-carbonic anhydrase from Mycobacterium tuberculosis. J Med Chem 2009;52:3116–3120.
- Güzel O, Maresca A, Scozzafava A, Salman A, Balaban AT, Supuran CT. Discovery of low nanomolar and subnanomolar inhibitors of the mycobacterial β-carbonic anhydrases Rv1284 and Rv3273. J Med Chem 2009;52:4063–4067.
- Carta F, Maresca A, Covarrubias AS, Mowbray SL, Jones TA, Supuran CT. Carbonic anhydrase inhibitors. Characterization and inhibition studies of the most active β-carbonic anhydrase from Mycobacterium tuberculosis, Rv3588c. Bioorg Med Chem Lett 2009; 19:6649–6654.
- Nishimori I, Minakuchi T, Maresca A, Carta F, Scozzafava A, Supuran CT. The ß-carbonic anhydrases from Mycobacterium tuberculosis as drug targets. Curr Pharm Des 2010;16:3300–3309.
- Supuran CT, Scozzafava A, Casini A. Carbonic anhydrase inhibitors. Med Res Rev 2003;23:146–189.
- Winum JY, Köhler S, Supuran CT. Brucella carbonic anhydrases: new targets for designing anti-infective agents. Curr Pharm Des 2010;16:3310–3316.
- Vullo D, Nishimori I, Minakuchi T, Scozzafava A, Supuran CT. Inhibition studies with anions and small molecules of two novel β-carbonic anhydrases from the bacterial pathogen Salmonella enterica serovar Typhimurium. Bioorg Med Chem Lett 2011;21:3591–3595.
- Nishimori I, Onishi S, Takeuchi H, Supuran CT. The α and β classes carbonic anhydrases from Helicobacter pylori as novel drug targets. Curr Pharm Des 2008;14:622–630.
- Supuran CT. Carbonic anhydrase inhibition with natural products: novel chemotypes and inhibition mechanisms. Mol Divers 2011;15:305–316.
- Supuran CT. Carbonic anhydrase inhibitors. Bioorg Med Chem Lett 2010;20:3467–3474.
- Dye C. Doomsday postponed? Preventing and reversing epidemics of drug-resistant tuberculosis. Nat Rev Microbiol 2009;7:81–87.
- Furtado GH, Nicolau DP. Overview perspective of bacterial resistance. Expert Opin Ther Pat 2010;20:1273–1276.
- Ginsberg AM. Emerging drugs for active tuberculosis. Semin Respir Crit Care Med 2008;29:552–559.
- Showalter HD, Denny WA. A roadmap for drug discovery and its translation to small molecule agents in clinical development for tuberculosis treatment. Tuberculosis (Edinb) 2008;88 Suppl 1:S3–17.
- Tomioka H, Tatano Y, Yasumoto K, Shimizu T. Recent advances in antituberculous drug development and novel drug targets. Expert Rev Respir Med 2008;2:455–471.
- Maresca A, Temperini C, Vu H, Pham NB, Poulsen SA, Scozzafava A et al. Non-zinc mediated inhibition of carbonic anhydrases: coumarins are a new class of suicide inhibitors. J Am Chem Soc 2009;131:3057–3062.
- Khalifah RG. The carbon dioxide hydration activity of carbonic anhydrase. I. Stop-flow kinetic studies on the native human isoenzymes B and C. J Biol Chem 1971;246:2561–2573.
- Kolayli S, Karahalil F, Sahin H, Dincer B, Supuran CT. Characterization and inhibition studies of an α-carbonic anhydrase from the endangered sturgeon species Acipenser gueldenstaedti. J Enzyme Inhib Med Chem 2011;26:895–900.
- Baranauskiene L, Hilvo M, Matuliene J, Golovenko D, Manakova E, Dudutiene V et al. Inhibition and binding studies of carbonic anhydrase isozymes I, II and IX with benzimidazo[1,2-c][1,2,3]thiadiazole-7-sulphonamides. J Enzyme Inhib Med Chem 2010;25:863–870.
- Sassetti CM, Boyd DH, Rubin EJ. Genes required for mycobacterial growth defined by high density mutagenesis. Mol Microbiol 2003;48:77–84.
- Betts JC, Lukey PT, Robb LC, McAdam RA, Duncan K. Evaluation of a nutrient starvation model of Mycobacterium tuberculosis persistence by gene and protein expression profiling. Mol Microbiol 2002;43:717–731.
- Carta F, Innocenti A, Hall RA, Mühlschlegel FA, Scozzafava A, Supuran CT. Carbonic anhydrase inhibitors. Inhibition of the β-class enzymes from the fungal pathogens Candida albicans and Cryptococcus neoformans with branched aliphatic/aromatic carboxylates and their derivatives. Bioorg Med Chem Lett 2011;21:2521–2526.
- Innocenti A, Hall RA, Schlicker C, Mühlschlegel FA, Supuran CT. Carbonic anhydrase inhibitors. Inhibition of the β-class enzymes from the fungal pathogens Candida albicans and Cryptococcus neoformans with aliphatic and aromatic carboxylates. Bioorg Med Chem 2009;17:2654–2657.