Abstract
Non-denaturing electrophoresis can be used to screen enzymes that self-regulate their activities by using a combination of enzymes and their inhibitors. Furthermore, this technique can be applied to develop enzyme reactors that self-regulate their activities. After separation of proteins from mouse liver cytosol by non-denaturing isoelectric focusing, lactate dehydrogense (LDH) and esterase activities were qualitatively and quantitatively examined using a combination of two-dimensional electrophoresis (2-DE) and non-denaturing stacking gel electrophoresis. Activities of mouse liver-derived LDH and carboxylesterase were reversibly inhibited by oxamate and 6,9-diamino-2-ethoxyacridine (acrinol), respectively, in the stacking gels and recovered when the enzymes migrated towards the separation gels. After separation and immobilization of the enzymes, their activities were inhibited by inhibitors and recovered after inhibitor removal. These results indicate that non-denaturing electrophoresis can be applied to select enzymes that self-regulate their activities and subsequently aid in the development of enzyme reactors that can control the enzyme activities.
| Abbreviations: | ||
| LDH, | = | lactate dehydrogense |
| NBT, | = | nitro blue tetrazolium |
| PMS, | = | phenazine methosulphate |
| acrinol, | = | 6,9-diamino-2-ethoxyacridine |
| PVDF, | = | polyvinylidene fluoride |
Introduction
Activities of enzymes are analyzed after separation by non-denaturing two-dimensional electrophoresis (2-DE), and the inhibition of the activities is examined by some inhibitors after enzymes are separated by non-denaturing 2-DECitation1,Citation2. The enzyme activities and their inhibitory actions can be reversed on interaction with other proteins and inhibitors under native conditions. Non-denaturing 2-DE can, therefore, be applied to investigate the changes in enzyme activities and their inhibitions. For example, activities of lactate dehydrogense (LDH) and carboxylesterase are reversibly inhibited by oxamate and 6,9-diamino-2-ethoxyacridine (acrinol), respectivelyCitation3,Citation4. The reversible inhibition of enzyme activity is thought to be related to enzyme-inhibitor interactions. In fact, an inhibitor of LDH, oxamate is reported to be reversibly bound to LDHCitation3. Furthermore, an esterase inhibitor, acrinol binds to esterases, and is reversibly inhibitedCitation4. LDH and carboxylesterase are identified after the enzymes obtained from mouse liver are separated by non-denaturing 2-DECitation1,Citation5. Thus, when inhibitors, such as oxamate and acrinol, are allowed to interact with the enzymes in the non-denaturing gels, reversible inhibition of enzyme activity can be examined by non-denaturing 2-DE. In addition, because the reduced form of nicotinamide adenine dinucleotide (NADH) and the changes of azo dye can be quantitatively assayed, activities of LDH and esterase can be quantitatively analyzed after separation by non-denaturing electrophoresisCitation6–8. Furthermore, using non-denaturing electrophoresis, enzymes can be immobilized to the membrane after separation by non-denaturing 2-DECitation9,Citation10. Thus, after enzymes that are reversibly inhibited by inhibitors are selected using non-denaturing 2-DE, enzyme reactors capable of regulating enzyme activities can be developed. For example, the activities of enzymes such as LDH and esterase are thought to be controlled by addition and release of the inhibitor such as oxamate and acrinol, respectively, after production of the enzyme rectors. Thus, the enzyme reactors can be applied to regulate amounts of lactic acid and lipids.
In this study, reversible inhibition of LDH and esterase activity was qualitatively and quantitatively examined using a combination of 2-DE and non-denaturing stacking gel electrophoresis. Furthermore, enzyme reactors able to regulate enzyme activities by inhibitors were produced after separation and immobilization of enzymes. These results indicate that the technique of non-denaturing electrophoresis can be applied to select enzymes that self-regulate their activities and subsequently aid in development of enzyme reactors with ability to control the enzyme activities.
Materials and methods
Reagents
Acrylamide and carrier ampholyte (Pharmalyte, pH 3–10 and Ampholine, pH 6–8) were purchased from Kishida Chemicals (Osaka, Japan) and GE healthcare (Uppsala, Sweden), respectively. Fast red TR salt, fast violet TR salt, α-cyano-4-hydroxycinnamic acid, phenazine methosulphate (PMS), oxamate and acrinol were purchased from Sigma-Aldrich (St. Louis, MO). Water-soluble fromazan dye (WST) and 1-methoxy-5-methylphenazinium methosulfate (1-methoxy PMS) were purchased from Dojindo Lab. (Kumamoto, Japan). α-Naphtyl acetate was purchased from MP Biomedicals Co. (Solon, OH). NAD was purchased from Oriental yeast Co. (Tokyo, Japan). Polyvinylidene fluoride (PVDF) membrane was purchased from Millipore (Billerica, MA). All other reagents such as ammonium persulfate, N,N,N′, N′-tetramethylenediamine (TEMED), 2-amino-2-hydroxymethyl-1,3-propanediol (Tris), glycine, trifluoroacetic acid (TFA), Coomassie Brilliant Blue (CBB), nitro blue tetrazolium (NBT), ammonium hydrogen carbonate and acetonitrile were purchased from Wako Pure Chemicals (Osaka, Japan) or Nacalai Tesque (Osaka, Japan).
Sample preparation, non-denaturing electrophoresis and electrotransfer to membranes
Mouse livers (Swiss Webster) were purchased from Rockland Inc. (Rockland, MA). 1.4 g of liver obtained from one mouse was homogenized in 5.0 mL of 100 mM Tris–HCl buffer (pH 7.2). The homogenate was centrifuged for 5 min at 10,000g to obtain the cytosolic fraction. Sucrose was added to the liver cytosolic fraction at a concentration of 40% (w/v). Proteins in the cytosolic fraction (70–100 µg) were subjected to microscale non-denaturing 2-DE. First dimensional isoelectric focusing (IEF) was performed on rod gels (35 × 1.3 mm ID) containing 4% acrylamide, 0.2% bisacrylamide, 0.05% ammonium persulphate and 0.029% TEMED and mixture of 1.25% pharmalyte (pH 3–10) and 5% ampholine (pH 6–8), or 6.25% pharmalyte (pH 3–10). The electrode solutions in the first dimensional electrophoresis comprised 0.04 M NaOH (cathode) and 0.01 M H3PO4 (anode). After first dimensional IEF, the gel was placed on top of the second dimension slab gel, which was then run on a 7.5% acrylamide (0.375% bisacrylamide). The electrode buffer in the second dimensional electrophoresis comprised 0.05 M Tris and 0.38 M glycine (pH 8.3). For determination of pI in the first-dimensional IEF separation, the IEF gel was cut into 2 mm length pieces after IEF separation. The gel pieces were put into a 0.2 mL vial. An aliquot (50 µL) of boiled water was then added to each vial, which was sealed and left for 2 h. The pH of the solution was measured by using a microelectrode (Beckman Instruments, Brea, CA). In order to analyze reversible inhibition of LDH isozymes, a 4% acrylamide stacking gel containing 0.2% oxamate (7–10 mm thick) was placed onto two-dimensional slab gel. After IEF, the IEF gel was placed on top of the slab gel and the activities of LDH isozymes were examined. For analyzing reversible inhibition of carboxylesterases, two types of 4% acrylamide stacking gel (5 mm thick stacking gel containing 0.1 M Tris–HCl (pH 6.8) and 8–10 mm thick stacking gel containing 0.1 mM acrinol and 0.1 M acetate buffer (pH 4.8)) were placed on two-dimensional slab gel. After IEF, the gel was placed on top of the slab gel and esterase activity was examined. In order to immobilize enzymes on PVDF membranes, enzymes were transferred to a PVDF membrane by a semi-dry type transblotting apparatus using electrode buffers of 0.05 M Tris and 0.38 M glycine (pH 8.3) with a onstant current of 23 mA per gel for 5 h. For detection of proteins on the PVDF membrane, the membrane was soaked in 0.5% Ponceau S in 10 mL of 0.1 M acetate buffer (pH 5.1).
Analysis of LDH activity
To detect LDH activity in the non-denaturing gel or the membrane, the proteins in the gel or the membrane were incubated in 10 mL of 0.1 M Tris–HCl buffer at pH 7.2 containing 5 mg NAD, 3 mg nitro blue tetrazolium (NBT), 0.3 mg PMS and 0.005% lactic acid. To determine the optimal concentration of oxamate to maximize LDH activity inhibition, the 2-DE gel was incubated in a solution (pH 7.2) containing 0–0.4% oxamate after the separation of the cytosol proteins by non-denaturing 2-DE.
Quantitative analysis of LDH activity was performed as reportedCitation6,Citation7. The proteins in the IEF gel, the stacking gel or the separation gel were incubated in 0.1 M Tris–HCl buffer at pH 7.2 containing 0.05% NAD, 0.5 mM WST-1, 0.02 mM 1-methoxy PMS and 0.005% lactic acid for 10 min at 25°C. The absorbance was measured at 450 nm using an UV-Vis absorbance spectrophotometer (Biospec-1600, Shimazu, Kyoto). The data was statistically analyzed with Student’s t-test.
Analysis of esterase activity
To analyze esterase activity in the non-denaturing gel, the proteins in the gel in 10 mL of 0.1 M Tris–HCl buffer (pH 7.4) containing 0.2 mL of 1% (w/v) α-naphtyl acetate and 4 mg Fast red TR salt in the absence or presence of acrinol.
Hydrolytic analysis of phosphatidylcholine by carboxylesterase was performed as previously describedCitation9. In brief, the carboxylesterase on the membrane was incubated in 10 mM Tris–HCl buffer (pH 7.2) containing 25 µg/mL phosphatidylcholine for 30 min at 25°C. For inhibition of the esterase activity, the esterase on membrane was incubated in a solution of 100 µM acrinol for 30 min. To remove acrinol from esterase, the esterase on the membrane was washed with 400 µL of 10 mM asparatic acid for 3 min. After incubation, 1 µL of liquid was collected and applied to the target plate of MALDI-TOF MS. After drying, 1 µL of a saturated solution of α-cyano 4-hydroxycinnamic acid, 0.1% trifluoroacetic acid and 60% acetonitrile was applied onto the same spot of the target plate, and dried. Mass analysis was performed using MALDI-TOF MS. The instrument was operated in a positive ion linear mode at 25 kV accelerating voltage, with the nitrogen laser at 337 nm and 3 Hz. External calibration was performed using the average mass of angiotensin II (m/z = 1046.19).
Quantitative analysis of esterase activity was performed as reportedCitation8. The proteins in the IEF gel, the stacking gel containing acrinol or the separation gel were incubated in 0.1 M Tris–HCl buffer at pH 7.2 containing 0.0075% α-naphtyl acetate and 0.08% Fast violet TR salt for 3 min at 25°C. The absorbance was measured at 490 nm using an UV-Vis absorbance spectrophotometer. The data was statistically analyzed with Student’s t-test.
Results
Reversible inhibition of LDH activity in non-denaturing electrophoresis
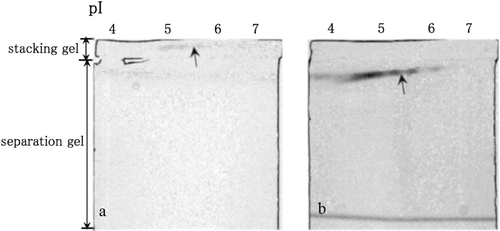
shows LDH activities in the non-denaturing stacking gel containing 0.2% oxamate (a) and in the non-denaturing separation gel (b) after the separation of the cytosolic proteins in the mouse liver by non-denaturing IEF. LDH activity was clearly inhibited by oxamate in the stacking gel (), and the activities were recovered when LDH isozymes migrated to the separation gel (). (LDH activity) shows quantitative analysis of LDH activity in the non-denaturing IEF gel, the stacking gel containing 0.2% oxamate and the separation gel. LDH activity in the presence of oxamate was 7% of that observed in the IEF gel, whereas activity in the non-denaturing separation gel was 20% of that in the IEF gel (). Because little LDH activity remained in the non-denaturing IEF gel after LDH isozymes were separated both in the stacking gel and the separation gel (data not shown), almost all LDH isozymes are thought to have migrated towards the non-denaturing separation gel. The quantitative results indicate that LDH activity was clearly inhibited by oxamate in the stacking gel, and the activity was recovered when the LDH isozymes migrated to the non-denaturing separation gel. Because LDH activity recovered to 20% in the present study, substantial amount of LDH activity can be lost during LDH migration within the non-denaturing stacking and separation gel. Thus, some complexes of LDH and oxamate are thought to remain within the non-denaturing separation gel. This possibility requires further elucidation and confirmation.
Table 1. Quantitative data of LDH and esterase activities within non-denaturing IEF, non-denaturing stacking gel containing inhibitors (oxamate for LDH activity and acrinol for esterase activity) and non-denaturing separation gel after the separation of the cytosolic proteins in the mouse liver by non-denaturing IEF.
Figure 1. LDH activities in the non-denaturing stacking gel containing 0.2% oxamate (a) and in the non-denaturing separation gel (b) after the separation of the cytosolic proteins in the mouse liver by non-denaturing IEF. LDH activity spots are indicated by arrows.
Reversible inhibition of esterase activity in non-denaturing electrophoresis
A stacking gel containing acrinol in acetate buffer (pH 4.8) was placed in between a stacking gel and a separation gel as shown in . Esterase activity was observed (), to be inhibited by acrinol when esterase migrated to the stacking gel containing acrinol (), and recovered when esterase migrated to the separation gel (). (esterase activity) shows quantitative analysis of esterase activity in the non-denaturing IEF gel, the stacking gel containing acrinol and the separation gel. Esterase activity in the presence of acrinol was 37% of that observed in the IEF gel, whereas activity in the non-denaturing separation gel was 86% of that in the IEF gel (). Therefore, esterase activity was clearly inhibited by acrinol in the stacking gel and recovered when the esterase migrated to the non-denaturing separation gel as determined by both esterase activity staining and phosphatidylcholine hydrolysis by esterase.
Figure 2. Esterase activities in the non-denaturing stacking gel containing 0.1 M Tris–HCl (pH 6.8) (a), in the non-denaturing stacking gel containing 0.1 mM acrinol and 0.1 M acetate buffer (pH 4.8) (b) and in the non-denaturing separation gel (c) after the separation of cytosolic proteins in the mouse liver by non-denaturing IEF. Esterase activity spots are indicated by arrowheads.

Production of enzymes reactors able to regulate their activities
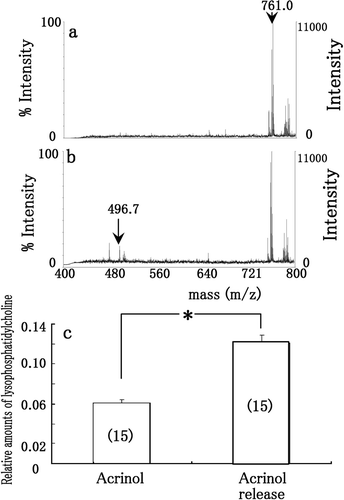
and shows LDH activities staining when the membrane was treated by 0.2% oxamate (a), and washed by Tris–HCl solution (b) after separation by non-denaturing 2-DE and electroblotting onto membrane. After LDH was transferred onto membrane, its activity was inhibited in the presence of 0.2% oxamate (a). After oxamate was released from the enzyme, LDH activity was clearly recovered (b). Thus, the LDH retains the reversible activity even after separation and immobilization. and shows esterase activity staining when the membrane was treated by 0.1 mM acrinol (c), and washed by aspartic acid solution (d) after separation by non-denaturing 2-DE and electroblotting onto membrane. After esterase was transferred onto membrane, its activity was inhibited in the presence of 0.1 mM acrinol (c). After acrinol was released from the enzyme, esterase activity was recovered (d). To confirm the reversible activity of esterase, the reaction from phosphatidylcholine to lysophosphatidylcholine by esterase were examined using MALDI-TOF MS (). When phosphatidylcholine (m/z = 761.0) was incubated with the esterase on membrane (arrowhead in ) treated with 0.1 mM acrinol, the peak of lysophosphatidylcholine (m/z = 496.7) was not clearly obtained (a). When the inhibitor was washed by aspartic solution, the peak of lysophosphatidylcholine (m/z = 496.7) was clearly obtained (b). The relative amounts of lysophosphatidylcholine (m/z = 496.7) are shown in , when pure phosphatidylcholine was applied to esterase on membrane treated with 0.1 mM acrinol (acrinol in c), and washed by aspartic acid solution (Acrinol release in c). The relative amounts of lysophosphatidylcholine (m/z = 496.7) after acrinol release by aspartic acid solution significantly increased those treated with acrinol (). Thus, the esterase retains the reversible activity even after separation and immobilization.
Figure 3. After separation by non-denaturing 2-DE and electroblotting onto membrane, LDH activities staining when the membrane was treated by 0.2% oxamate (a), and washed by Tris–HCl solution (b). After separation by non-denaturing 2-DE and electroblotting onto membrane, esterase activity staining when the membrane was treated by 0.1 mM acrinol (c), and washed by aspartic acid solution (d).
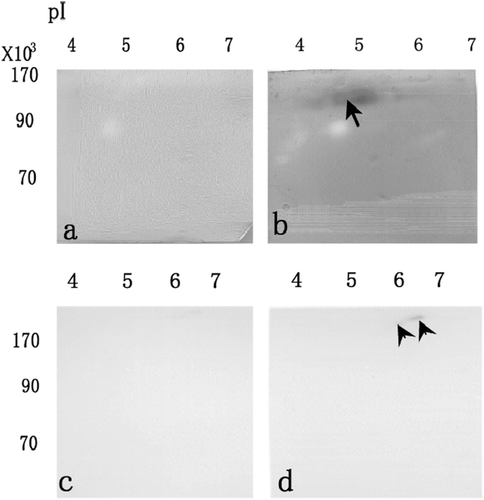
Figure 4. MALDI-TOF MS spectra of phosphatidylcholine and lysophosphatidylcholine when phosphatidylcholine was incubated with the esterase on membrane (arrow in 3d) treated with 0.1 mM acrinol (a), and washed by aspartic acid solution (b) after separation by non-denaturing 2-DE and electroblotting onto membrane. The relative amounts of lysophosphatidylcholine (m/z = 496.7) (c) when pure phosphatidylcholine was applied to esterase on membrane treated with 0.1 mM acrinol (acrinol in c), and washed by aspartic acid solution (acrinol release in c) after separation by non-denaturing 2-DE and electroblotting onto membrane. Each datum indicates mean standard error obtained from 15 individual measurements (number of measurements in c).
Discussion
Present study indicates that activity of LDH isozymes was obtained in the absence () of oxamate, and was inhibited in the presence () of oxamate, after the cytosolic proteins in the mouse liver were separated by non-denaturing 2-DE. It has been reported that isoenzyme activities are selectively inhibited by inhibitorsCitation11–13, and the isoenzymes are separated by IEFCitation14–16. Thus, isoenzyme specific inhibitors can be selected by combining 2-DE and non-denaturing stacking gel electrophoresis, because after isoenzymes are separated by non-denaturing IEF, they interact with inhibitors within the non-denaturing gel. We reported previously that separation of LDH isozymes at pI 4.5–7 is improved with IEF that mainly contained carrier ampholyte (pH 6–8)Citation5. Because the isozymes were separated by the improved IEF separation, it was thought that selective inhibition of LDH isozyme activity could be examined by combining 2-DE and non-denaturing stacking gel electrophoresis. However, selective inhibition of isozyme activity was not obtained in the present study (). In the present study, LDH activity was clearly inhibited by oxamate in the stacking gel, and the activity was recovered when the LDH isozymes migrated to the non-denaturing separation gel ( and ). Since it has been reported that oxamate is bound at the catalytic site of LDHCitation3,Citation14, oxamate is thought to be bound to LDH in the stacking gel, the activity of LDH can be decreased. Further, the reason for the recovery of LDH activity is that oxamate is released from LDH isozymes during electrophoresis in the non-denaturing separation gel. Little activity at pI 4.8–5.6 was detected within non-denaturing IEF containing oxamate (). The explanation for this observation is that some oxamate can be released from LDH isozymes because both proteins have a pI value less than 5.6 and oxamate is negatively charged within the stacking gel at pH 6.8. In addition, present study indicates that LDH activities were inhibited when the membrane was treated by oxamate (), and they were recovered when the membrane was washed by Tris–HCl solution () after separation by non-denaturing 2-DE and electroblotting onto membrane. Thus, a reversible LDH reactor that can regulate LDH activity can be developed by using non-denaturing electrophoresis. It has been reported that lactic acid can be produced using immobilized enzymesCitation17. Because LDH activities are controlled using immobilized LDH isozymes and oxiamate, the amounts of lactic acid could be regulated by the LDH reactor in the present study.
Present study indicates that esterase activity is inhibited by acrinol, and is recovered after removal of acrinol (, , and and ). In the basic research, esterase activity staining was observed in the absence or presence of 0.1 mM acrinol in 0.1 M acetate buffer (pH 4.8) after the cytosolic proteins in the mouse liver were separated by non-denaturing 2-DE (data not shown). However, esterase activity was not inhibited by acrinol in 0.1 M Tris–HCl (pH 7.2) (data not shown). Thus, the inhibition by acrinol appeared to be enhanced in acidic buffer. Changes in esterase activity can be examined in the absence or presence of acrinol in the non-denaturing electrophoresis gels. It has been reported that the active site of human pancreatic lipase is covered by an amphiphilic domain, lid that opens under acidic conditions (pH 4.0)Citation18–20. When esterase migrates to the stacking gel containing acrinol, the esterase active site can open under acidic conditions in the stacking gel. Because acrinol can bind to an enzyme active site, and inhibits its activityCitation4, acrinol in the stacking gel is thought to bind to esterase active site after opening of the active site under acidic conditions. Thus, inhibition of esterase activity can be enhanced in the stacking gel containing acrinol and acidic buffer. However, parts of acidic esterases remain within the non-denaturing stacking gels, because the enzymes possess no or positively charge in acidic conditions ( and ). In addition, after separation by non-denaturing 2-DE and electroblotting onto membrane, esterase activities were inhibited when the membrane was treated by acrinol (), and they were recovered when the membrane was washed by aspartic acid solution (). Thus, a reversible esterase reactor that can self-regulate its activity can be produced using non-denaturing electrophoresis. It has been reported that isopropyl palmitate, an ester, can be synthesized using immobilized lipaseCitation21. Because esterase activity can be controlled by immobilized esterase and acrinol, the amounts of lipids could be regulated by the esterase reactor in the present study.
In summary, present study demonstrated that the technique of non-denaturing electrophoresis can be applied to select enzymes that can self-regulate their activities and subsequently aid in the development of enzyme reactors that can control enzyme activities.
Declaration of interest
The authors report no conflicts of interest.
References
- Shimazaki Y, Sugawara Y, Ohtsuka Y, Manabe T. Analysis of the activity and identification of enzymes after separation of cytosol proteins in mouse liver by microscale nondenaturing two-dimensional electrophoresis. Proteomics 2003;3:2002–2007.
- Shimazaki Y, Sugawara Y, Manabe T. Nondenaturing two-dimensional electrophoresis enzyme profile involving activity and sequence structure of cytosol proteins from mouse liver. Proteomics 2004;4:1406–1411.
- Wong C, Rodríguez-Páez L, Nogueda B, Pérez A, Baeza I. Selective inhibition of the sperm-specific lactate dehydrogenase isozyme-C4 by N-isopropyl oxamate. Biochim Biophys Acta 1997;1343:16–22.
- Bencharit S, Morton CL, Hyatt JL, Kuhn P, Danks MK, Potter PM et al. Crystal structure of human carboxylesterase 1 complexed with the Alzheimer’s drug tacrine: from binding promiscuity to selective inhibition. Chem Biol 2003;10:341–349.
- Shimazaki Y, Shimizu K, Masaoka S. Enzyme separation and isozyme heterogeneity analysis using non-denaturing two-dimensional electrophoresis. Talanta 2010;82:1063–1067.
- Ishiyama M, Shiga M, Sasamoto K, Mizoguchi, M, He PG. A new sulfonated tetrazolium salt that products a highly water-soluble formazan dye. Chem Pharm Bull 1993;41:1118–1122.
- Ishiyama M, Miyazono Y, Sasamoto K, Ohkura Y, Ueno K. A highly water-soluble disulfonated tetrazolium salt as a chromogenic indicator for NADH as well as cell viability. Talanta 1997;44:1299–1305.
- Johnstone KJ, Ashford AE. A simultaneous-coupling azo dye method for the quantitative assay of esterase using α-naphthy acetate as a substrate. Histochem J 1980;12:221–234.
- Shimazaki Y, Kuroda T. Production of enzyme reactors after separation by non-denaturing two-dimensional electrophoresis and immobilization on membrane. Biotechnol Lett 2009;31:1545–1549.
- Shimazaki Y, Sakikawa T. Determination of malic Acid using a malate dehydrogenase reactor after purification and immobilization in non-denaturing conditions and staining with ponceau S. Protein Pept Lett 2010;17:1048–1052.
- Yu Y, Deck JA, Hunsaker LA, Deck LM, Royer RE, Goldberg E et al. Selective active site inhibitors of human lactate dehydrogenases A4, B4, and C4. Biochem Pharmacol 2001;62:81–89.
- Zhu B, Strada SJ. The novel functions of cGMP-specific phosphodiesterase 5 and its inhibitors in carcinoma cells and pulmonary/cardiovascular vessels. Curr Top Med Chem 2007;7:437–454.
- Granchi C, Bertini S, Macchia M, Minutolo F. Inhibitors of lactate dehydrogenase isoforms and their therapeutic potentials. Curr Med Chem 2010;17:672–697.
- Klose J, Spielmann H. Gel isoelectric focusing of mouse lactate dehydrogenase: heterogeneity of the isoenzymes A4 and X4. Biochem Genet 1975;13:707–720.
- Lessard F, Dion R. Analysis for α-amylase isoenzymes by automated isoelectric focusing. Clin Chem 1989;35:2116–2118.
- Wagner L, Wermann M, Rosche F, Rahfeld JU, Hoffmann T, Demuth HU. Isolation of dipeptidyl peptidase IV (DP 4) isoforms from porcine kidney by preparative isoelectric focusing to improve crystallization. Biol Chem 2011;392:665–677.
- Ropchan JR, Barrio JR. Enzymatic synthesis of [1-11C]pyruvic acid, L-[1-11C]lactic acid and L-[1-11C]alanine via DL-[1-11C]alanine. J Nucl Med 1984;25:887–892.
- Winkler FK, D’Arcy A, Hunziker W. Structure of human pancreatic lipase. Nature 1990;343:771–774.
- Ranaldi S, Belle V, Woudstra M, Rodriguez J, Guigliarelli B, Sturgis J et al. Lid opening and unfolding in human pancreatic lipase at low pH revealed by site-directed spin labeling EPR and FTIR spectroscopy. Biochemistry 2009;48:630–638.
- Belle V, Fournel A, Woudstra M, Ranaldi S, Prieri F, Thomé V et al. Probing the opening of the pancreatic lipase lid using site-directed spin labeling and EPR spectroscopy. Biochemistry 2007;46:2205–2214.
- Kee CY, Hassan M, Ramachandran KB. Studies on the kinetics of Isopropyl Palmitate synthesis in packed bed bioreactor using immobilized lipase. Artif Cells Blood Substit Immobil Biotechnol 1999;27:393–398.