Abstract
Virus capsid structure is essential in virion maturation and durability, so disrupting capsid assembly could be an effective way to reduce virion count and cure viral diseases. However, currently there is no known antiviral which affects capsid inhibition, and only a small number of assembly inhibitors were experimentally successful. In this present study, we aimed to find hepatitis B virus (HBV) capsid assembly inhibitor which binds to the HBV core protein and changes protein conformation. Several candidate molecules were found to bind to certain structure in core protein with high specificity. Furthermore, these molecules significantly changed the protein conformation and reduced assembly affinity of core protein, leading to decrease of the number of assembled capsid or virion, both in vitro and in vivo. In addition, prediction also suggests that improvements in inhibition efficiency could be possible by changing functional groups and ring structures.
Introduction
Hepatitis B virus (HBV) is an animal virus which infects human hepatic cells and causes chronic hepatitis. This chronic infection is known to have a strong relationship with the development of cirrhosis and hepatocellular carcinoma. Around 400 million people, especially in Asia and Africa, function as virus carrierCitation1.
HBV contains partial dsDNA genome which is about 3.2 kb long, and this genome codes 5 distinct proteins: surface antigen (HBsAg), two forms of capsid antigen (HBcAg and HBeAg), polymerase and X proteinCitation2. Among these, HBcAg can be assembled to form T = 3 or T = 4 icosahedral nucleocapsid structure and surface antigen is inserted into the host plasma membrane and eventually comprised in an envelope. Polymerase functions as both DNA-dependent DNA polymerase and RNA-dependent DNA polymerase. The function of X protein is hypothesized to be related to carcinogenesisCitation2.
As one of the most prevalent pathogens, HBV has been researched by many biologists and pharmacologists, and several treatments work against HBV proliferation is developed. Nucleoside analogs (e.g. Lamivudine, Adefovir) and α-interferon is used as first-line treatments, and these agents quite efficiently decrease virus titer. Nevertheless, long-term use of nucleoside analogs could develop drug resistanceCitation3. Therefore, it needs to research novel mechanism to inhibit virus proliferation and the establishment of chronic infection.
In this research, target of drug molecule is HBcAg. HBcAg consists of 183–185 AAs and is divided into assembly domain (1–149 AA) and protamine domain (150–183/185 AA)Citation3. Protamine domain interacts with pregenomic RNA (pgRNA) and virus polymerase then regulates reverse transcription of the pgRNA. Assembly domain is a structural domain which interacts with other HBcAgs to construct an icosahedral capsid structureCitation3. Assembly begins with a binding of three HBcAg dimers, and this trimer works as a nucleus, promoting further nucleation without leaving an observable population of intermediatesCitation4. Interaction between HBcAg dimers is weak, but in sum they assemble into a globally stable capsidCitation5. The capsid also exhibits hysteresis to dissociation, so they could persist in the harsh condition which is unfavourable for assemblyCitation6.
We used Cp149, a protein which only contains an assembly domain of HBcAg, instead of full-length HBcAg (Cp183) because of its convenience and structural similarityCitation7. Cp149 is more competent in overexpression in E. coli vector. Also, purification of Cp149 is simpler than process involves HBcAg. Also, it is able to form capsid-like particle spontaneously in vitro and in vivo, so it could be used as an alternative to full-length HBcAgCitation3. Cp exists in dimer form naturally, and dimers interact with each other to form hexameric structure. This hexamer could induce assembly, recruit other dimers to form T = 4 or T = 3 capsidCitation3.
Conformation of HBcAg seems to be important in the assembly process. As supporting evidence, Zn+2, which could alter conformation of HBcAg, also affects both speed of assembly and stability of capsid particleCitation8. Therefore, if we could change the conformation into the unfavourable way, we could also interrupt assembly of HBcAg and ultimately maturation of HBV virion.
Applying in silico methodology, we found several compounds expected to bind to HBcAg and change overall conformation to inhibit capsid assembly. HBcAg is not a receptor protein, but it is able to find a possible binding site by analyzing geometric and physiochemical traits of surfaceCitation9. Also, de novo receptor–ligand binding simulation and chemical evolution simulation could be used to find possible candidate molecules which bind to specific siteCitation10. Screening process as ADMET prediction or conformational change simulation could be applied to predict the traits of molecules such as toxicity, metabolism and inhibition strengthCitation11,Citation12.
The aim of this research is to find a candidate molecule which effectively inhibits assembly of the HBV capsid with low toxicity. We applied in silico methods to find new scaffolds which could work as binding site, and screened molecules probably bind to a given site. We could find some molecules which inhibit capsid assembly. These molecules could lower the number of the capsid in vitro and in vivo. In parallel, we could use these candidates as a part of “extended” molecules, which are predicted to show more powerful inhibition.
Material and methods
Materials
Anti-HBcAb (Polyclonal Rabbit, 1:4000) is from Dako (Glostrup, Denmark). Pepsin (800–2500 units/mg protein), horseradish-conjugated anti-rabbit Ab (1:40,000), mouse monoclonal anti-FLAG Ab (5 Ag/mL) and Dulbecco’s modified Eagle’s medium are from Sigma (St. Louis, MO, USA). Benzenesulfonamide (≥98%) and deuterium oxide (99.98% ± 0.01% D) are from Aldrich. Sulfanilamide (≥99%) is from Sigma-Aldrich. BL21 (DE3) + pLysS E. coli, pET28b vector is from Novagen (Madison, WI, USA). Isopropyl-h-d-1-thiogalactoside (IPTG) is from Duchefa biochemie (Haarkem, Netherlands). Dulbecco’s modified Eagle’s medium (DMEM) is from Welgene (Daegu, Republic of Korea). Fetal bovine serum is from Invitrogen (Carlsbad, CA, USA). Real-time PCR SYBR-Green reaction mixture is from Qiagen (Hilden, Germany).
Modelling monomer and dimer structures of core protein
We built in silico protein model for monomer and dimer structure of cp149/cp183 respectively. Protein structure data were obtained from previous X-ray crystallographic studiesCitation13,Citation14. Water molecules in the crystal lattice were subtracted and lost residues and atoms were added to reconstruct full-length models. Protein structure simulation was done on the basis of AMBER and CHARMM force field sets. Partial charges for each atom were applied by Momany-Rone method. The energy level was minimized with Adopted Basis-set Newton-Raphson algorithm. Obtained structures were equilibrated on 37°C condition for 1,000,000 cycles, and Generalized Born with molecular volume integration (GBMV) method, with dielectric constants of 4 in inside the protein and 80 in the solventCitation15.
Fragment search and conformational change estimation
Probable interaction sites were defined previously to the prediction, and elementary fragment data were obtained from the Standard Ludi Library (1048 entries)Citation16 and ZINC databaseCitation17. De novo receptor program was used to find the small fragments may bind to given binding site, High-scored interaction poses were selected and binding energy was calculated for each protein-fragment complex. Fragments with strong binding energy were selected and structure simulation is done for each protein-fragment complex as described previously. Changes in atom coordinates and protein conformation were estimated by RMSD value. Complexes with high RMSD were selected to identify fragments which cause a significant conformational change.
ADME/toxicity prediction and random binding simulation
High-scored candidate molecules were further screened by ADME/Toxicity screening to examine behaviours and adverse effects. In the ADME prediction step, we estimated aqueous solubility, blood-brain barrier penetration, hepatotoxicity, plasma protein binding and intestinal absorption. Mutagenicity, carcinogenicity, LD50 and chronic LOAEL were examined in toxicity prediction stepsCitation11,Citation12. By estimating properties of each ligand, we screened some molecules which have undesirable traits such as low solubility or high toxicity.
Random binding simulation was done after the screening. Using LibDock program, input molecules were allowed to bind any site on the Cp dimer surface, and binding energy was calculated to screen which binding pose could exist in reality.
Conformational change simulation in oligomer level
For the screened molecules, we used LibDock and CDOCKER programs were used to confirm whether screened molecules could really bind to the binding site, and to construct Cp149-molecule complexes. A Hexamer model for unbound Cp149 and Cp149-molecule complex was constructed by applying the n = 3 rotational symmetry. Structures were equilibrated on 37°C condition for 1,000,000 cycles. RMSD between Cp149 hexamer and Cp149-candidate complex hexamer was calculated and visible conformational change features were analyzed.
Cp149 purification and assembly reaction
Cp149 was cloned directly from pET28b vector (Novagen). The construct was transformed into BL21 (DE3) + pLysS E. coli (Novagen) and purified as describedCitation18. Purified dimers were stored in glycine buffer (pH 9.5) and were set at a 4 µg/µL concentration. For Cp149 assembly, reaction buffer (50 mM HEPES (pH 7.5), 15 mM NaCl, 10 mM CaCl2) was mixed with purified Cp149 (with final concentration of 60 µM) and incubated at 37°C for 1 h. Candidate molecules were added to reaction buffer and incubated with Cp149 for given time. Assembled samples were separated by electrophoresis on a 0.9% native agarose gel, and then the resolved proteins in the gel were transferred overnight onto nitrocellulose membrane. Assembled capsid particles on the membrane were detected by immunoblot analysis with a rabbit polyclonal anti-HBc antibody as described previouslyCitation18.
Sucrose gradient centrifugation
Purified Cp149 was mixed with reaction buffer and incubated at 37°C for 3 hours and stabilized at 4°C overnight. Control samples were prepared by treating 3.5 M urea to dissociate residual oligomers. Sucrose gradient centrifugation and immunoblot analysis were performed as a previouslyCitation18.
Deuterium exchange and MALDI-TOF MS
Hydrogen atoms in peptide bond and amino group were substituted with deuterium in Cp149 dimer and capsid sample. After 1 h/3 h incubation in assembly condition, deuterium oxide is added in pH 7.0, 25°C condition. After 1 minute of exposing, 0.1% TFA was added to adjust the pH of sample into 2.5 ± 0.1 and kept the sample in 0°C. 10 units of pepsin were added to each sample and incubated for 10 minutesCitation19.
Mass spectra were obtained using the Autoflex 2 (Bruker daltonics, Bremen, Germany). The peptide digest was mixed with a 3,5-dimethoxy-4-hydroxycinnamic acid (sinapinic acid) matrix. The mixture was applied to a stainless steel plate. To analyze the peptide mass peak, Flex analysis (Bruker Daltonics) was used.
Surface plasmon resonance
Surface plasmon resonance (SPR) analysis was performed using a Biacore 3000 instrument (Biacore AB). FC1 (flow cell 1) contained a protein-free negative control, while experimental FC2 and FC3 contained Cp149 (WT) and sulfanilamide-bound WT dimeric Cp149 respectively, which were immobilized according to the manufacturer’s instructions. The affinity was measured as described previouslyCitation20. Results were analyzed using BIA evaluation software version 3.1 (Biacore AB).
Electron microscopy
For negative staining, 10 µL of a solution containing the assembled core particles was applied to a carbon-coated grid and incubated for 1 min. The grid was washed with water and stained with 2% uranyl acetate for 1 min. Transmission electron micrographs were taken on a LIBRA 120 (Carl Zeiss, Oberkochen, Germany) operating at 80 kV at the NICEM (National Instrumentation Center for Environmental Management).
In silico improvement of inhibitor molecules
Using the ZINC database, we searched for molecules with benzenesulfonamide substructure to build a 500-molecule library. Molecular docking simulation with LibDock and CDOCKER programs were executed to find molecules bind to Cp structure among the library. 10 molecules with highest binding scores were selected, and similarity search was done for each molecule to build another chemical library. Same docking simulation was done for each library built and molecules with highest binding scores were selected.
Results
Simulation
We first constructed Cp149-molecule model to carry out simulations. On the basis of crystallographic dataCitation13,Citation14, structure models for cp149 dimer and hexamer were constructed and stable conformations were found. We could find a groove-like structure near the hydrophobic core of Cp, which may function as a novel binding pocket (). In this pocket, several residues such as Tyr38 and His52 seemed to be important in binding, so we kept these residues in mind when we simulated receptor–ligand interaction.
Figure 1. Constructed protein model and receptor–ligand interaction simulation. (A) Constructed Cp149 dimer model. Lateral view. (B) Constructed Cp149 dimer model. Ventral view. (C) Binding site candidate. Residues Ser35~His52 are depicted in stick model, and binding site is depicted as a coloured dot and pink sphere. (D) Binding simulation of sulfanilamide. Atoms in sulfanilamide are depicted as the ball, and nearby residues are depicted as lines. (E) Surfaces are added to residues described in (D). Sulfanilamide ligand is almost completely interlocked in the protein structure.
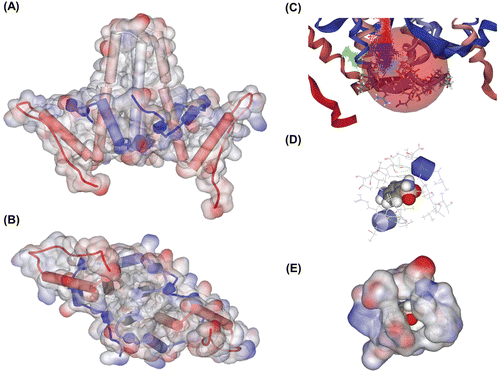
By the de novo receptor–ligand interaction simulation, we could find 145 fragments that could probably bind to the groove. After that, we applied binding energy calculation and ADMET prediction, and screened six molecules which exhibited preferable traits. We then examined the specificity of binding of a molecule by random binding simulation. In every case, around 80% of poses with positive binding strength were located near the binding site ( and ), and the most preferred poses were always at the binding site. After all steps of screening, we chose two of them, benzenesulfonamide and sulfanilamide, which have relatively high binding strength and low toxicity.
Figure 2. Random binding simulation and similarity search. (A)–(B) In this simulation, input molecules were allowed to bind at any site in the protein, thereby simulating random binding of the chemicals. Then we calculated binding energy to predict which poses could really be allowed and sustained. In sum, among the all possible binding poses, only a small number of poses were possible and about 80% of binding poses were located in the binding site we expected as in this figure. (A) Lateral view. (B) Ventral view. (C)–(D) Results of similarity search with molecular docking simulation. We used sulfanilamide as a lead compound, and several molecules expected to have better inhibition strength were detected. (C) Docked state of molecules. These molecules contain sulfanilamide structure as its substructure, and have higher binding strength. Total 125 molecule poses are depicted. (D) 10 molecules with highest binding strength. These molecules share similar conformation as seen above. They contain two aromatic ring structures and these rings are put on a roughly same plane, which means this two-plane structure could work as a scaffold for effective assembly inhibitor.
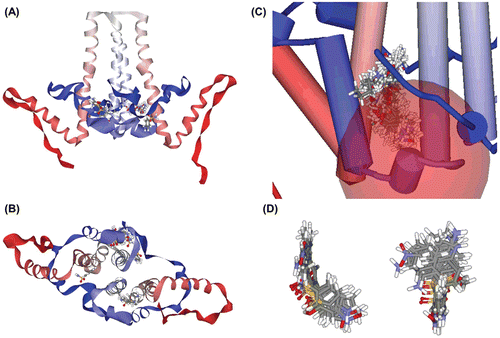
Similarity search and molecular docking
We used the ZINC database to find molecules with benzenesulfonamide substructure, which may enable these molecules to act as assembly inhibitor. As a result, we could find several molecules with maximum 2.89-fold binding score and 2.54-fold binding energy, which may reflect the inhibition strength ( and ). They preferably dock to Cp149 structure, especially in the binding pocket we predicted, and form stable Cp149-molecule complexes.
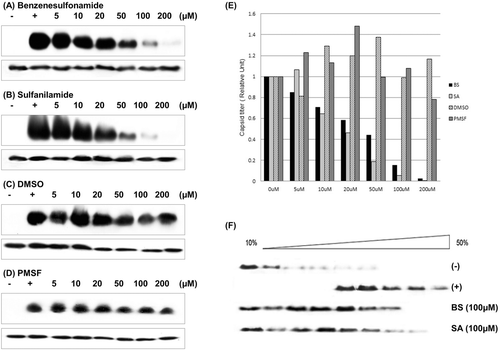
Benzenesulfonamide and sulfanilamide inhibit formation of capsid particles
To find the inhibition effects of benzenesulfonamide and sulfanilamide, we performed in vitro assembly assay. The Cp149 dimers were interacted with these two molecules and made complex with it, resulting in a capsid level decrease ( and ). In the absence of benzenesulfonamide and sulfanilamide, particles made capsid well during 1h incubation time. Structurally similar molecules, DMSO and PMSF, was used to test the specificity of benzenesulfonamide and sulfanilamide, and it was obvious that DMSO and PMSF don’t affect capsid assembly process ( and ). The assembly inhibition caused by these two molecules could be more clearly seen in the graph ().
Figure 3. In vitro assay of Cp149 assembly. (A)–(D) In vitro Cp149 assembly was used to determine the inhibition efficiency of predicted candidates. We used DMSO and PMSF as a control, which could simulate benzenesulfonamide and sulfanilamide structure. Cp149 dimers were incubated for 1 h in 37°C condition with a given concentration of molecules (described above each gel image), and the products were resolved in 0.9% agarose gel (upper panel). Immunoblot analysis was carried out with Anti-HBcAg. Also, as a control, the products were resolved in SDS/15% polyacrylamide gel (lower panel), and immunoblot analysis was carried out with Anti-HBcAg. (A) Benzenesulfonamide, (B) Sulfanilamide, (C) DMSO, (D) PMSF. (E) Analysis of band intensity. The intensity of the band in agarose gel was divided to the intensity of the corresponding band in the SDS-Polyacrylamide gel, and these ratios were normalized to the band intensity ratios of nontreated Cp149 (Positive control) lanes. In each case, positive control lane was standard. (F) Sucrose gradient (10–50 % sucrose in 50 mM Hepes buffer, pH 7.5) centrifugation was carried out using samples assembled in vitro with or without given molecule. (−): Negative control (dimer form), (+): Positive Control (Capsid form).
Assembly intermediates were detected with sucrose gradient centrifugation
Sucrose gradient centrifugation was used to detect both dimer and capsid separately, which enables to confirm the effects of benzenesulfonamide and sulfanilamide (). In the control groups, fully assembled capsids were found mainly found in fractions with sucrose concentration around 30% and dimers were found in upper fractions, consistent with former researchCitation1.
In the benzenesulfonamide-/sulfanilamide-treated samples, Cps were mainly found on the upper fractions, which mean assembly process is hindered by molecule treatment. We could also find some intermediate-mass particles, which were found in the fractions between dimer fraction and capsid fraction. In the control groups, these intermediate fractions were negligible, but benzenesulfonamide-/sulfanilamide-treatment made a tendency of making intermediate-mass assembled particle, which is found in fractions which sucrose concentration is around 20%.
Core–core interaction rate is decreased by treating molecules
To confirm the mechanism how these candidate molecules inhibit the assembly process, we applied SPR technique to measure protein–protein interaction affinity. Sensorgrams of the interaction between dimeric Cp149 are shown ( – 0 µM, – 5 µM, – 10 µM and – 20 µM), and the values for kinetic parameters of the association between the dimers are listed (). The sensorgram shows that the effects of inhibitor are more drastic when the protein concentration in the flow is higher.
Figure 4. Affinity analysis with surface plasmon resonance. FC1: Negative control (blank), FC2: Cp149 (WT), FC3: Cp149 (BS-treated), FC4: Cp149 (SA-treated). Four concentrations of Cp149 (WT) and Cp149-molecule complexes were passed through the flow cell with the flow rate of 60 µL/min. Sensorgrams were analyzed using the BIA evaluation software and the results are shown in .
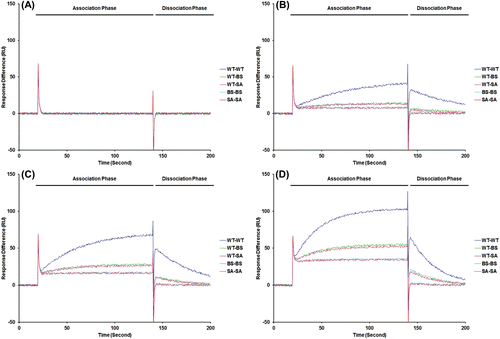
Table 1. Sensorgrams of the interaction between free and molecule-bound dimeric Cp149.
It was shown that the molecule effects both association step and dissociation step. With the candidate molecules, maximum 19.02-fold decrease in ka value was found, which means this molecules block dimer from binding to other dimer. Also, the dissociation constant kd value was increased from twofold to fivefold. Thus the equilibrium dissociation constant, which is defined as kd/ka, was decreased. Maximum 102.9-fold decrease in Kd was detected. Since Kd directly represents affinity between protein dimers, this result could be interpreted that the candidate molecules inhibit assembly processes. Though we could observe not further assembly process but only dimer–dimer interaction, we expect these candidate molecules might impede overall assembly steps. In addition, this result suggests that drug candidate which works by interrupt protein assembly could be designed efficiently by simulation based on structure model.
DXMS reveals specific molecule binding site
We used DXMS (Deuterium Exchange – Mass Spectrometry) to find which site in protein interacts with inhibitor molecules. This technique uses mass difference caused by differential deuterium substitution, which is affected by solvent accessibility on amino group hydrogen and peptide conformation.
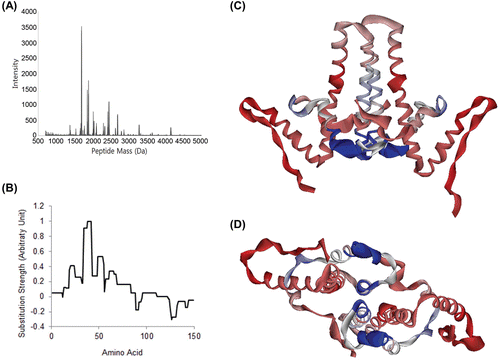
Several peptide peaks that correspond to benzenesulfonamide/sulfanilamide binding site is detected and these peaks have shown evidence of effect of candidate molecules in deuterium substitution (). For example, in the case of the heptapeptide SPHHTAL (AA 49–55, M.W. = 762.843 Da), corresponding mass peak found in candidate-treated samples are about 2–3 Da lighter. Also, several peaks correspond to AA 38–55/60/65/68/71 (C-terminal cleavage site varies) exhibited similar tendency in deuterium substitution. For these fragments, the peaks were 2–4 Da lighter in the samples treated with benzenesulfonamide or sulfanilamide. With the deuterium substitution difference values each peptide fragment has, we calculated the rate of substitution difference caused by treating molecule for each amino acids (). Then, we mapped these signals in our Cp model ( and ). We found that the intensity of the signal was significantly intense at the binding site.
Figure 5. Determination of binding site with deuterium exchange-mass spectrometry. (A) Mass peak of partially digested Cp149. Mass profiles of samples were analyzed to identify mass shift caused by deuterium substitution and its difference. When the molecule was treated, the deuterium substitution rate is decreased and mass of fragments is lowered. (B) Mass shift difference values were normalized to the strongest value, and substitution rate for each AA was calculated for each. (C) and (D) Cp149 dimer model, coloured with DXMS mass shift difference values. Strongest signals were found in AA 34–42 and AA 49–54, which is nearly identical to the location of the binding site we proposed. This mass shift difference was expected to be caused by shadowing effect of sulfanilamide.
EM analysis
We visualized effect of inhibitor molecules with transmission electron microscopy. By counting the number of capsid particle per same area of micrograph (), we examined the effects of molecule on capsid assembly. Normal HBV capsids could be visualized as a white circle in the micrograph, so we could numerate the number of capsids to check the effects and strength of inhibitor molecules. As seen in the table (), in the control group, 1277 assembled particles were found in given 2 µm2 area. Among them, 1204 capsids were normal particles, and 59 capsids were partially broken. 14 capsids left were ‘abnormal’ particles, such as small particle or nonspherical aggregates. Most of assembled particles seemed to be typical T = 4 capsid, but some of the spherical particles were slightly small, about 15%, so it is expected to be T = 3 capsids. Only a small portion of T = 3 capsids was detected in normal condition.
Figure 6. Electron micrographs of assembled core particles. Electromicrographs of nontreated Cp149 and molecule-treated Cp149 capsids (Magnification: ×80,000). Assembled core particles were negatively stained with 2 % uranyl acetate. (A) Nontreated cp149 was assembled in reaction buffer (50 mM Hepes, 15 mM NaCl and 10 mM CaCl2 in pH 7.5). (B) and (C) Benzenesulfonamide-/sulfanilamide-treated cp149 core was assembled in reaction buffer. (B) Benzenesulfonamide, (C) Sulfanilamide. (D) Types of capsids are described. 1: Normal capsids. Black outer masses are considered to be uranyl acetate remnants. 2: Broken capsid. The arrow indicates a broken site. 3: Small capsid (expected to be T = 3 capsid). Red arrow indicates small capsid, which is relatively smaller than the normal capsid indicated by black arrows. 4: Aggregate. The arrow indicates Cp aggregate.
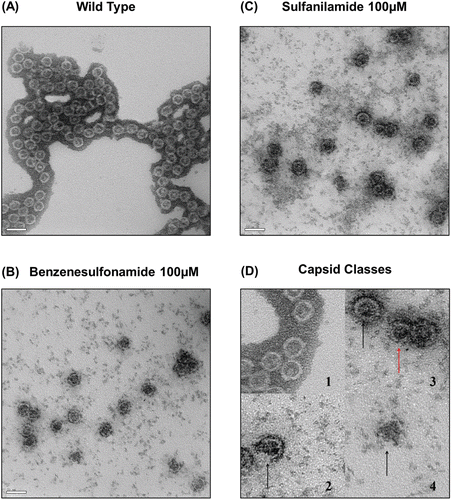
Table 2. Capsid particles in each case.
In the electron micrographs of samples treated with candidate molecules, the number of normal capsid particle is decreased more than tenfold. Other assembled forms, such as incomplete capsids or broken capsids, were more prevalent in these micrographs. It was also able to detect smaller capsids, which is predicted to be T = 3 isoform of capsid. If these assembled forms were included in the count, the number of assembled particles was about 25%, compared to control. Other 75% of protein molecules were predicted to exist in dimer form or oligomer form, which could not be detected properly by microscopy. Rather, it seems to cause the background noises in micrograph, which were significantly higher in treated samples.
Discussion
HBV seems to be related to many hepatic diseases, including chronic hepatitis, cirrhosis and hepatocellular carcinomaCitation2. Capsid assembly is crucial in virion maturation: it is important not only for stabilization of virion in the external conditions, but also for completion of genome duplication, as reverse transcription of HBV pgRNA is done inside of the nascent capsidCitation3,Citation21. So inhibition of assembly could also hinder genome duplication, ultimately helps to deal with HBV infection more efficientlyCitation22.
Capsid assembly inhibition has been a subject of some previous researchesCitation3,Citation23,Citation24. In these researches, several inhibitor candidates with nanomole scale IC50 values were found. However, none of them were approved to be used for clinical purposes yet. Therefore, we aimed to find another group of molecules which might have plausible inhibitory performance and relatively low toxicity. In this research, sulfanilamide structure was chosen as a molecular backbone, as sulfanilamide itself exhibited inhibitory effects on capsid assembly, and it has quite low toxicity.
In this research, we could design several compounds which are predicted to bind to Cp, based on sulfanilamide structure (). By using in vitro assembly assay and DXMS, we clearly demonstrated that binding of the candidate molecules into specific site of Cp could inhibit the capsid assembly. As benzenesulfonamide or sulfanilamide binds to Cp, the binding site is covered by ligand and becomes harder to interact with solvents. Since binding decreases deuterium substitution rate, it could be detected with a mass shift differenceCitation19,Citation25,Citation26. An interaction site could be revealed with this phenomenon.
To investigate the mechanism of inhibition, we used the SPR technique to measure association and dissociation constantsCitation1,Citation20. We revealed that the inhibition of capsid assembly is caused by decreased rate of association of Cp and increased rate of dissociation of Cp, as a result of binding of candidate molecule. However, we could not make sure how these molecules inhibit the assembly process. We assumed that the inhibitory effects are caused by conformational difference, which is already known to affect capsid assemblyCitation8, and to confirm this hypothesis, we analyzed DXMS data, which might contain the signals from conformational changesCitation25. Nevertheless, we could not find any evidences of conformational changes but only the direct effects of molecular binding. This could mean that conformational changes were not big enough to affect the substitution signal, or the molecules do not act by changing overall conformation but in some other way.
In the EM micrograph, the increase of the number of broken capsids could be interpreted as an effect of conformational changes caused by treating molecules, as we assumed in previous experiments. This is also consistent with simulation which predicted decrease in binding affinity between dimers, and experiments such as sucrose gradient centrifugation and SPR, which also detected assembly intermediates in samples treated with candidate molecules.
Since we tested only a limited range of molecules, we could not find molecules with low IC50, preferably in the nanomole level. As some of the predecessor molecules have more powerful effects on assemblyCitation3,Citation23,24, improvements are needed to make these molecules to be compared to other inhibitor candidates. We could design some molecules which are expected to show greater inhibition ( and ), however, because of poor availability, we could not test any molecules we designed. Nevertheless, we could find the possibility of becoming efficient inhibitors, and by applying algorithms such as biological evolution modelCitation10 may help these test molecules to be changed into more powerful assembly inhibitor.
Conclusion
We could find and test some candidate molecules with a possibility of becoming efficient anti-HBV by further optimization. If the molecule with plausible inhibitory performance and low toxicity is obtained, this molecule would be a novel option for anti-HBV treatment. Also, with the combination of anti-HBV agents, such as nucleoside analogs, it will show synergistic effects as its mechanism does not overlap with other anti-HBV agents now we useCitation26. It will ultimately help people to prevent chronic infection of HBV, thereby reducing the prevalence rate of mortal hepatic diseases.
Declaration of interest
This work was supported by the National Research Foundation of Korea (NRF) grant funded by the Korea government (MEST) (No. 2008–2004202).
References
- Kang H, Yu J, Jung G. Phosphorylation of hepatitis B virus core C-terminally truncated protein (Cp149) by PKC increases capsid assembly and stability. Biochem J 2008;416:47–54.
- Yokosuka O, Arai M. Molecular biology of hepatitis B virus: Effect of nucleotide substitutions on the clinical features of chronic hepatitis B. Med Mol Morphol 2006;39:113–120.
- Stray SJ, Zlotnick A. BAY 41-4109 has multiple effects on Hepatitis B virus capsid assembly. J Mol Recognit 2006;19:542–548.
- Zlotnick A, Johnson JM, Wingfield PW, Stahl SJ, Endres D. A theoretical model successfully identifies features of hepatitis B virus capsid assembly. Biochemistry 1999;38:14644–14652.
- Ceres P, Zlotnick A. Weak protein-protein interactions are sufficient to drive assembly of hepatitis B virus capsids. Biochemistry 2002;41:11525–11531.
- Singh S, Zlotnick A. Observed hysteresis of virus capsid disassembly is implicit in kinetic models of assembly. J Biol Chem 2003;278:18249–18255.
- Wingfield PT, Stahl SJ, Williams RW, Steven AC. Hepatitis core antigen produced in Escherichia coli: Subunit composition, conformational analysis, and in vitro capsid assembly. Biochemistry 1995;34:4919–4932.
- Stray SJ, Ceres P, Zlotnick A. Zinc ions trigger conformational change and oligomerization of hepatitis B virus capsid protein. Biochemistry 2004;43:9989–9998.
- Leis S, Schneider S, Zacharias M. In silico prediction of binding sites on proteins. Curr Med Chem 2010;17:1550–1562.
- Zoete V, Grosdidier A, Michielin O. Docking, virtual high throughput screening and in silico fragment-based drug design. J Cell Mol Med 2009;13:238–248.
- Segall M, Champness E, Obrezanova O, Leeding C. Beyond profiling: Using ADMET models to guide decisions. Chem Biodivers 2009;6:2144–2151.
- Valerio LG, Yang C, Arvidson KB, Kruhlak NL. A structural feature-based computational approach for toxicology predictions. Expert Opin Drug Metab Toxicol 2010;6:505–518.
- Wynne SA, Crowther RA, Leslie AG. The crystal structure of the human hepatitis B virus capsid. Mol Cell 1999;3:771–780.
- Packianathan C, Katen SP, Dann CE 3rd, Zlotnick A. Conformational changes in the hepatitis B virus core protein are consistent with a role for allostery in virus assembly. J Virol 2010;84:1607–1615.
- Smith JR, Evans KJ, Wright A, Willows RD, Jamie JF, Griffith R. Novel indoleamine 2,3-dioxygenase-1 inhibitors from a multistep in silico screen. Bioorg Med Chem 2012;20:1354–1363.
- Böhm HJ. LUDI: Rule-based automatic design of new substituents for enzyme inhibitor leads. J Comput Aided Mol Des 1992;6:593–606.
- Irwin JJ, Shoichet BK. ZINC–a free database of commercially available compounds for virtual screening. J Chem Inf Model 2005;45:177–182.
- Choi Y, Gyoo Park S, Yoo JH, Jung G. Calcium ions affect the hepatitis B virus core assembly. Virology 2005;332:454–463.
- Englander JJ, Del Mar C, Li W, Englander SW, Kim JS, Stranz DD et al. Protein structure change studied by hydrogen-deuterium exchange, functional labeling, and mass spectrometry. Proc Natl Acad Sci USA 2003;100:7057–7062.
- Kang HY, Lee S, Park SG, Yu J, Kim Y, Jung G. Phosphorylation of hepatitis B virus Cp at Ser87 facilitates core assembly. Biochem J 2006;398:311–317.
- Ganem D. Assembly of hepadnaviral virions and subviral particles. Curr Top Microbiol Immunol 1991;168:61–83.
- Deres K, Schröder CH, Paessens A, Goldmann S, Hacker HJ, Weber O et al. Inhibition of hepatitis B virus replication by drug-induced depletion of nucleocapsids. Science 2003;299:893–896.
- Zlotnick A, Ceres P, Singh S, Johnson JM. A small molecule inhibits and misdirects assembly of hepatitis B virus capsids. J Virol 2002;76:4848–4854.
- Stray SJ, Bourne CR, Punna S, Lewis WG, Finn MG, Zlotnick A. A heteroaryldihydropyrimidine activates and can misdirect hepatitis B virus capsid assembly. Proc Natl Acad Sci USA 2005;102:8138–8143.
- Woods VL Jr, Hamuro Y. High resolution, high-throughput amide deuterium exchange-mass spectrometry (DXMS) determination of protein binding site structure and dynamics: Utility in pharmaceutical design. J of Cell Biochem 2011;84:89–98.
- Hoofnagle JH, di Bisceglie AM. The treatment of chronic viral hepatitis. N Engl J Med 1997;336:347–356.