Abstract
The synthesis, in vitro antimicrobial activities of some novel hydroxy pyridines supported with various pharmacophores is described. Twenty-six out of the tested 58 compounds exhibited variable inhibitory effects on the growth of the tested Gram positive and Gram negative bacteria. The tested compounds revealed better activity against the Gram positive rather than the Gram negative strains. The synthesized hydroxypyridones have shown very significant inhibitory effect against Staphylococcus aureus and Bacillus subtilis. Twelve compounds namely; 5d, 5f, 6a, 6b, 8b, 18b, 18c, 19c, 21d, 22b, 22d and 23d were able to produce appreciable growth inhibitory activity against Candida albicans when compared to Clotrimazole. Among these, 22d proved to be the most potent antifungal agent.
Introduction
For the last 10 years, we have established a research program aiming at the synthesis and characterization of different heterocyclic ring systems endowed with potential chemotherapeutic activitiesCitation1–11. A wide range of biological activities have been ascribed to pyridine derivatives including antimicrobialCitation12,Citation13, antitubercularCitation14,Citation15, antiamoebicCitation16, antiparasiticCitation17,Citation18, antimalarialCitation19 and antiviralCitation20,Citation21 activities. Particular interest has been given to pyrones and pyridine ring systemsCitation3,Citation6,Citation8,Citation10 which have been subjected to various chemotherapeutic investigations either as antimicrobial, antiviral or anticancer agents. The results revealed that some of the newly synthesized derivatives exhibited promising broad spectrum antitumor and antimicrobial activities. The substitution pattern of the target compounds include various functionalities such as the amino, derived imine, ureido, thioureido and sulfonamido groups that are reported to contribute to various chemotherapeutic activities. It was considered also of interest to cyclize some of the thioureido derivatives to the corresponding thiazolidinones and thiazolines which are known for their potential chemotherapeutic activities. Such structure hybridization was planned with the hope of adding some synergism to the anticipated bioactivities of the target molecules. The variation in the nature and size of substituents at the prementioned functionalities was thought to be of interest as it would offer variable electronic, lipophilic and steric environment that would influence the targeted biological activities. In view of the above-mentioned facts, and in continuation of our interest in studying the synthesis and biological properties of some heterocyclic ring systems as novel structure leads that might be of use in designing new potent and selective chemotherapeutic agents, it was thought worthwhile to synthesize and investigate anticancer and antimicrobial activities of some novel hydroxypyridine derivatives. The target compounds have been subjected to the NCI in vitro disease-oriented human cells screening panel assayCitation22–24 and in vitro antibacterial and antifungal screeningCitation25 tests.
Experimental
Chemistry
Melting points were determined on a Gallenkamp melting point apparatus and are uncorrected. The infrared (IR) spectra were recorded on Shimadzu FT-IR 8400S infrared spectrophotometer using the KBr pellet technique. 1H and 13C NMR spectra were recorded on a Bruker DPX-400 FT NMR spectrometer using tetramethylsilane as the internal standard (Chemical shifts in δ, ppm). CDCl3 was used as the solvent however, in case of insoluble compounds CDCl3 and few drops of DMSO-d6 was used. Splitting patterns were designated as follows: s: singlet; m: multiplet. Mass spectra were measured on a GCM-Q 1000 Ex spectrometer. The IR and mass spectra of some compounds are recorded in Supplementary File S8. Elemental analyses were performed on a 2400 Perkin Elmer Series 2 analyzer and the found values were within ±0.4% of the theoretical values. Follow-up of the reactions and checking the homogeneity of the compounds were made by TLC on silica gel-protected aluminium sheets (Type 60 F254, Merck) and the spots were detected by exposure to UV-lamp at λ 254. The starting compounds 4-hydroxy-6-methylpyran-2-one (1) and 3-hydroxy-2-methylpyran-4-one (16) have been purchased from ACROS Organics Geel-Belgium.
1-amino-4-hydroxy-6-methyl-1H-pyridin-2-one (2)
A solution of 4-hydroxy-6-methyl-2-pyrone 1 (1.26 g, 10 mmol) in ethanol was refluxed with hydrazine hydrate (0.75 mL, 15 mmol) for 1h. The reaction mixture was then cooled, poured into ice cold water; the solid which separated was recrystallized from ethanol as needles. Physicochemical and analytical data are listed in Supplementary File S1. 1H &13C-NMR spectral data are recorded in Supplementary Files S4 and S7, respectively.
N-(4-hydroxy-6-methyl-2-oxo-2H-pyridin-1-yl) substituted sulfonamides (3)
A mixture of the 1-Amino-4-hydroxy-6-methyl-1H-pyridin-2-one 2 (1.4 g, 10 mmol) and appropriate sulfonyl chloride (10 mmol) in pyridine (10 mL) was heated under reflux for 4 h. After cooling to room temperature, the reaction mixture was poured on crushed ice and the separated solid product was filtered, washed with water, dried and recrystallized from ethanol.
N1-(4-hydroxy-6-methyl-2-oxo-2H-pyridin-1-yl)-N3-substituted urea (4) and thiourea derivatives (5)
A mixture of 2 (10 mmol) and anhydrous K2CO3 (20 mmol) in dry acetone (25 mL) was stirred and treated with the appropriate isocyanate or isothiocyanate (12 mmol). After the mixture was stirred and refluxed for 18 h, acetone was removed under pressure, and the solid mass dissolved in water and acidified with 2 N HCl. The crude product was purified by recrystallization from ethanol as needles.
4-Hydroxy-6-methyl-1-(4-oxo-3-substituted thiazolidin-2-ylideneamino)-1H-pyridin-2-one (6)
A mixture of 5 (10 mmol), ethyl bromoacetate (10 mmol) and sodium acetate (20 mmol) in absolute ethanol (30 mL) was refluxed for 2 h. The reaction mixture was then filtered while hot, concentrated and allowed to cool. The product obtained was recrystallized from ethanol as needles.
1-(3-substituted-4-phenyl-3H-thiazol-2-ylideneamino)-4-hydroxy-6-methyl-1H-pyridin-2-one (7)
A solution of the corresponding thiourea derivative 5 (10 mmol) in absolute ethanol (25 mL) was refluxed with phenacyl bromide (10 mmol) and sodium acetate (20 mmol) for 2 h. The reaction mixture was then cooled and poured into water; the precipitated thiazoline derivative was recrystallized from ethanol as needles.
1-(Arylideneamino)-4-hydroxy-6-methyl-1H-pyridin-2-one (8)
1-Amino-4-hydroxy-6-methyl-1H-pyridin-2-one 2 (1.4 g, 10 mmol) was refluxed with the appropriate aldehyde (10 mmol) for 3 h. The reaction mixture was then concentrated and allowed to cool. The product obtained was recrystallized from ethanol as needles.
4-Hydroxy-6-methyl-1-(4-oxo-2-substituted thiazolidin-3-yl)-1H-pyridin-2-one (9)
A mixture of the appropriate arylidene derivative 8 (10 mmol) and thioacetic acid (15 mmol) in dry dioxan (25 mL) was refluxed for 12 h, cooled then poured onto aqueous K2CO3. The obtained solid was recrystallized from ethanol as needles.
p-(4-Hydroxy-6-methyl-2-oxo-2H-pyridin-1-yl)benzenesulfonamide (10)
A solution of 4-hydroxy-6-methyl-1H-pyridin-2-one 2 (2.5 g, 20 mmol) in ethanol (20 mL) was refluxed with p-hydrazinobenzenesulfonamide hydrochloride (4.9 g, 22 mmol) for 4 h. On concentration, the separated product was filtered, washed with cold ethanol and recrystallized from a mixture of ethanol as needles. Physicochemical and analytical data are listed in Supplementary File S2. 1H & 13C-NMR spectral data are recorded in Supplementary Files S5 and S7, respectively.
p-(4-Hydroxy-6-methyl-2-oxo-2H-pyridin-1-yl)benzenesulfonylureas (11a,b; X = O) and benzenesulfonylthioureas (11c; X = S)
A mixture of the pyridine derivative 10 (10 mmol) and anhydrous K2CO3 (20 mmol) in dry acetone (25 mL) was stirred and treated with the appropriate isocyanate (12 mmol). After the mixture was stirred and refluxed for 18 h, acetone was removed under pressure, and the solid mass dissolved in water and acidified with 2 N HCl. The crude product was purified by recrystallization from ethanol as needles.
4-hydroxy-6-methyl-1H-pyridin-2-one (12)
To an ice-cooled stirred solution of 1-Amino-4-hydroxy-6-methyl-1H-pyridin-2-one 2 (1.4g, 10 mmol) in acetic acid (15 mL), was added dropwise, a solution of sodium nitrite (1.05 g, 15 mmol) in water (5 mL) over a period of 2 h. Stirring was maintained for further 2 h, and the reaction mixture was then poured into water; the precipitated solid product was filtered, washed with water, dried and recrystallized from ethanol as needles.
1-Substituted sulfonyl-4-hydroxy-6-methyl-1H-pyridin-2-one (13)
A mixture of the 4-hydroxy-6-methyl-1H-pyridin-2-one 12 (1.1 g, 10 mmol) and appropriate sulfonyl chloride (10 mmol) in pyridine (10 mL) was heated under reflux for 4 h. After cooling to room temperature, the reaction mixture was poured on crushed ice and the separated solid product was filtered, washed with water, dried and recrystallized from ethanol.
1-Substituted carbamoyl-4-hydroxy-6-methyl-1H-pyridin-2-one (14) and 1-substituted thiocarbamoyl-4-hydroxy-6-methyl-1H-pyridin-2-one (15)
A mixture of 4-hydroxy-6-methyl-1H-pyridin-2-one 12 (1.1g, 10 mmol) and anhydrous K2CO3 (20 mmol) in dry acetone (25 mL) was stirred and treated with the appropriate isocyanate or isothiocyanate (12 mmol). After the mixture was stirred and refluxed for 18 h, acetone was removed under pressure, and the solid mass dissolved in water and acidified with 2 N HCl. The crude product was purified by recrystallization from ethanol as needles.
4-(3-Hydroxy-2-methyl-4-oxo-4H-pyridin-1-yl)benzenesulfonamide (17)
A solution of 3-hydroxy-2-methylpyran-4-one 16 (2.5 g, 20 mmol) in ethanol (20 mL) was refluxed with p-hydrazinobenzenesulfonamide hydrochloride (4.9 g, 22 mmol) for 4 h. On concentration, the separated product was filtered, washed with cold ethanol and recrystallized from a mixture of ethanol as needles. Physicochemical and analytical data are listed in Supplementary File S3. 1H & 13C-NMR spectral data are recorded in Supplementary Files S6 and S7, respectively.
N-Arylidene-4-(3-hydroxy-2-methyl-4-oxo-4H-pyridin-1-yl)benzenesulphonamides (18)
4-(3-Hydroxy-2-methyl-4-oxo-4H-pyridin-1-yl)benzenesulfonamide 17 (2.8 g, 10 mmol) was refluxed with the appropriate aldehyde (10 mmol) for 3 h. The reaction mixture was then concentrated and allowed to cool. The product obtained was recrystallized from ethanol as needles.
3-Hydroxy-2-methyl-1-[4-(4-oxo-2-phenylthiazolidine-3-sulfonyl)phenyl]-1H-pyridin-4-one (19)
A mixture of the appropriate arylidene derivative 18 (10 mmol) and thioacetic acid (15 mmol) in dry dioxan (25 mL) was refluxed for 12 h, cooled then poured into aqueous K2CO3. The obtained solid was recrystallized from ethanol as needles.
p-(3-Hydroxy-2-methyl-4-oxo-4H-pyridin-1-yl)benzenesulfonylureas (20) and thioureas (21)
A mixture of the 4-pyrone derivative 17 (10 mmol) and anhydrous K2CO3 (20 mmol) in dry acetone (25 mL) was stirred and treated with the appropriate isocyanate (12 mmol). After the mixture was stirred and refluxed for 18 h, acetone was removed under pressure, and the solid mass dissolved in water and acidified with 2 N HCl. The crude product was purified by recrystallization from ethanol as needles.
4-(3-Hydroxy-2-methyl-4-oxo-4H-pyridin-1-yl)-N-(4-oxo-3-substituted thiazolidin-2-ylidene)benzenesulphonamide (22)
A mixture of the thiourea derivative 21 (10 mmol), ethyl bromoacetate (10 mmol) and sodium acetate (20 mmol) in absolute ethanol (30 mL) was refluxed for 2 h. The reaction mixture was then filtered while hot, concentrated and allowed to cool. The product obtained was recrystallized from ethanol as needles.
N-(3-substituted-4-phenyl-3H-thiazol-2-ylidene)-4-(3-Hydroxy-2-methyl-4-oxo-4H-pyridin-1-yl)benzenesulphonamide (23)
A solution of the corresponding thiourea derivative 21 (10 mmol) in absolute ethanol (25 mL) was refluxed with α-bromoacetophenone (10 mmol) and sodium acetate (20 mmol) for 2 h. The reaction mixture was then cooled and poured into water; the precipitated thiazoline derivative was recrystallized from ethanol as needles.
Biological activity
In vitro antibacterial and antifungal activities
Standard sterilized filter paper discs (5 mm diameter) impregnated with a solution of the test compound in DMSO (1 mg/mL) was placed on an agar plate seeded with the appropriate test organism in triplicates. The utilized test organisms were: Staphylococcus aureus (ATCC 6538), Bacillus subtilis (NRRL B-14819) and Micrococcus luteus (ATCC 21881) as examples of Gram positive bacteria and Escherichia coli (ATCC 25922), Pseudomonas aeruginosa (ATCC 27853) and Klebsiella pneumoniae (clinical isolate) as examples of Gram negative bacteria. They were also evaluated for their in vitro antifungal potential against Candida albicans (ATCC 10231) and Aspergillus niger (recultured) fungal strains were utilized as representatives for fungi. Ampicillin trihydrate and Clotrimazole were used as standard antibacterial and antifungal agents, respectively.
DMSO alone was used as control at the same above-mentioned concentration. The plates were incubated at 37°C for 24 h for bacteria and for 7 days for fungi. Compounds that showed significant growth inhibition zones (≥14 mm) using the two-fold serial dilution technique, were further evaluated for their minimal inhibitory concentrations (MICs).
Minimal inhibitory concentration measurement
The micro-dilution susceptibility test in Müller–Hinton Broth (Oxoid) and Sabouraud Liquid Medium (Oxoid) was used for the determination of antibacterial and antifungal activity, respectivelyCitation25. Stock solutions of the tested compounds, Ampicillin trihydrate and Clotrimazole were prepared in DMSO at concentration of 800 µg/mL followed by two-fold dilution at concentrations of (400, 200,.....6.25 µg/mL). The microorganism suspensions at 106 CFU/mL (Colony Forming Unit/mL) concentrations were inoculated to the corresponding wells. Plates were incubated at 36°C for 24–48 h and the MICs were determined. Control experiments were also done.
Preliminary in vitro anticancer screening
Out of the newly synthesized compounds, 20 derivatives namely; 5c,d,f, 6a,b, 7c, 8b, 9b, 11c, 15a, 18b, 19b, 20b, 21a,b,d, 22b,d and 23b,d; were selected by the National Cancer Institute (NCI) in vitro disease-oriented human cells screening panel assay to be evaluated for their in vitro antitumor activity. Primary in vitro one-dose anticancer assay was performed using the full NCI 60 cell panel in accordance with the current protocol of the Drug Evaluation Branch, NCI, Bethesda. These cell lines were incubated with one concentration (10 μM) for each tested compound. A 48 h continuous drug exposure protocol was used, and a Sulphorhodamine B (SRB) protein assay was employed to estimate cell viability or growth.
Results and discussion
Chemistry
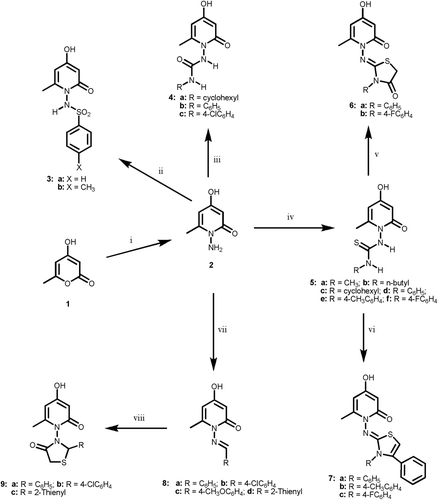
The synthetic strategies adopted for the preparation of the intermediate and target compounds are described in . In , the key intermediates 4-hydroxy-6-methylpyran-2-one 1 was reacted with hydrazine hydrate to afford the desired 1-amino-4-hydroxy-6-methylpyridin-2-one 2 which was utilized as the key intermediate in this part. The IR spectrum revealed strong carbonyl absorption at 1651 cm−1 as well as two bands at 2873–2936 cm−1 and 3121–3279 cm−1 for the OH and NH2 groups, respectively. The 1H-NMR spectrum displayed two singlets at δ 5.88 and δ 5.48 for H-3 and H-5, respectively (Supplementary File S4). The structure was further confirmed from its 13C-NMR spectra (Supplementary File S7). Reaction of the amine 2 with benzenesulfonyl chloride and p-toluenesulfonyl chloride in pyridine gave the corresponding N-substituted benzenesulfonylamino derivatives 3. Moreover, condensation of 2 with substituted isocyanates or isothiocyanates in pyridine as alkaline medium, afforded the corresponding substituted ureido and thioureido analogs 4 and 5. The IR spectra of these compounds showed beside the 2H-pyran carbonyl absorption at 1652–1663 cm−1, two bands at 1310–1366 cm−1 and 1163–1178 cm−1 due to SO2N function as well as distinguishable urea carbonyl absorption at 1665–1672 cm−1 for 4 and thiourea carbonyl absorption at 1125–1155 cm−1 in case of compound 5. The structures were further confirmed from their 1H-NMR and 13C-NMR spectra (Supplementary Files S4 and S7). Cyclization of the thiouriedo derivatives 5 with ethyl bromoacetate in the presence of anhydrous sodium acetate afforded the corresponding thiazolidin-4-ones 6. The IR of these compounds was characterized by a strong band at 1720–1735 cm−1 for thiazolidine carbonyl, whereas, their 1H-NMR spectra revealed a new singlet at δ 4.02–4.11 ppm attributed to the oxothiazolidine H-5 (Supplementary File S4). The 13C-NMR spectrum of 6a exhibited the C-5 of the thiazolidine at δ 32.7 (Supplementary File S7). Analogously, reacting the same derivatives 5 with phenacyl bromide under similar reaction conditions resulted in the formation of the desired 1,3-thiazolines 7. Their 1H-NMR spectra showed a broad singlet at δ 6.29–6.55 ppm due to thiazoline H-5 proton. Finally, condensation of the key intermediate 2 with different aldehyde gave corresponding 1-(arylideneamino)-4-hydroxy-6-methylpyridin-2-ones 8. The IR spectra of these arylidenes 8 revealed strong 2-pyridone carbonyl absorption at 1655–1660 cm−1, their 1H-NMR spectra revealed a new singlet at δ 7.92–8.16 ppm attributed to the CH=N proton (Table 4). Reaction of arylidene derivatives 8 with mercaptoacetic acid yielded the corresponding thiazolidinone derivatives 9. Their IR spectra revealed two strong absorptions at 1655–1664 cm−1 and 1720–1735 cm−1 for the 2H-pyran and thiazolidine carbonyls, respectively. The 1H-NMR and 13C-NMR spectra of the above prepared compounds were recorded in Supplementary Files S4 and S7. Reaction of the 4-hydroxy-6-methylpyran-2-one 1, with 4-aminobenzenesulfonamide gave the target 1-(4-aminosulfonylphenyl)-4-hydroxy-6-methylpyridin-2-one 10. The IR spectrum revealed two bands at 1151 cm−1 and 1324 cm−1 for the SO2N function and a strong carbonyl absorption at 1669 m−1 as well as two bands at 2768–2845 cm−1 and 3287–3365 cm−1 for the OH and NH2 groups, respectively. Reaction of 10 with different isocyanates and isothiocyanates in the presence of pyridine afforded the corresponding substituted ureido and thioureido derivatives (11; X = O or S), respectively. The IR spectra of these compounds showed beside the 2H-pyran carbonyl absorption at 1669–1673 cm−1 two bands at 1324–1352 cm−1 and 1167–1179 cm−1 due to SO2N function as well as distinguishable urea carbonyl absorption at 1665–1672 cm−1 for (11; X = O) and thiourea carbonyl absorption at 1126–1115 cm−1 in case of compound (11; X = S). The structures were further confirmed from their 1H-NMR and 13C-NMR spectra (Supplementary Files S5 and S7). However, reaction of 1-amino-4-hydroxy-6-methylpyridin-2-one 2 with nitrous acid yielded the corresponding 4-hydroxy-6-methylpyridin-2-one 12, which was employed as the key intermediate in this part. The IR spectrum of 12 exhibited strong carbonyl absorption at 1751 cm−1 as well as two bands at 2860 cm−1 and 3292 cm−1 for the OH and NH2 groups, respectively. Reaction of 12 with benzenesulfonyl chloride or p-toluenesulfonyl chloride in the presence of pyridine, leads to the formation of the N-substituted benzenesulfonyl derivatives 13 (X = H or CH3). Furthermore, condensation of the 2H-pyran-2-one 12 with isocyanate and isothiocyanate derivatives in pyridine afforded the corresponding 1-substituted carbamoyl and thiocarbamoyl derivatives 14 and 15, respectively. The IR spectra of these compounds showed beside the 2H-pyran carbonyl absorption at 1662–1669 cm−1 a characteristic urea carbonyl absorption at 1654–1658 cm−1 for (14; X = O) and a thiourea carbonyl absorption at 1122–1136 cm−1 in case of compound (15; X = S). The 1H-NMR and 13C-NMR spectra of the above prepared compounds were recorded in Supplementary Files S5 and S7.
Scheme 1. Reagents and reaction conditions: (i) ethanol, NH2NH2·H2O, ref lux, 1 h; (ii) arylsulfonyl chloride, pyridine, ref lux, 4 h; (iii) anhyd. K2CO3, dry acetone, RNCO, ref lux, 18 h; (iv) anhyd. K2CO3, dry acetone, RNCS, ref lux, 10 h; (v) ethyl bromoacetate, Na acetate, ethanol, ref lux 2 h; (vi) ethanol, phenacyl bromide, Na acetate, ref lux 2 h; (vii) approp. aldehyde, ref lux, 3 h; (viii) thioacetic acid, dry dioxan, ref lux 12 h.
Scheme 2. Reagents and reaction conditions: (i) ethanol, sulfanilamide, ref lux, 4 h; (ii) anhyd. K2CO3, dry acetone, approp. RNCO or RNCS, ref lux, 10–18 h; (iii) NaNO2/CH3COOH, stirring, 2 + 2 h; (iv) arylsulfonyl chloride, pyridine, ref lux, 4 h; (v) anhyd. K2CO3, dry acetone, RNCO, ref lux, 18 h; (vi) anhyd. K2CO3, dry acetone, RNCS, ref lux, 10 h.
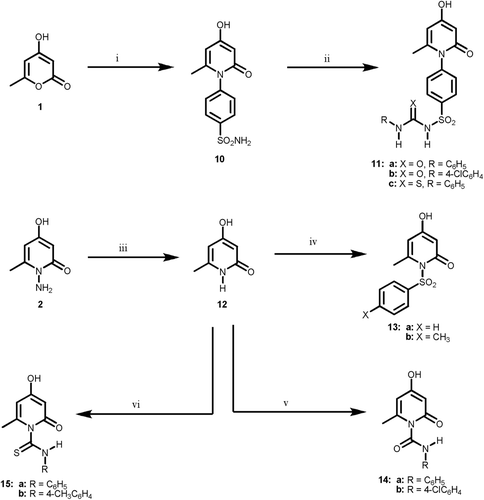
Scheme 3. Reagents and reaction conditions: (i) sulfanilamide, ethanol, ref lux, 4 h; (ii) RCHO, ethanol, ref lux, 3 h; (iii) HSCH2COOH, dry dioxan, ref lux, 12 h; (iv) RNCO, anhyd. K2CO3, dry acetone, ref lux 18 h; (v) RNCS, anhyd. K2CO3, dry acetone, ref lux 10 h; (vi) ethyl bromoacetate, anhyd. NaOAc, abs. ethanol, ref lux, 2 h; (vii) phenacyl bromide, anhyd. NaOAc, abs. ethanol, ref lux, 2 h.
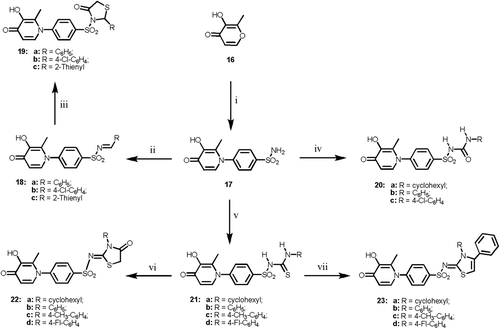
Likewise, amination of the target compound 3-hydroxy-2-methylpyran-4-one 16 with sulfanilamide afforded the corresponding 1-(4-aminosulfonylphenyl)-3-hydroxy-2-methylpyridin-4-one 17, which was utilized as a key intermediate in . The IR spectrum of 17 revealed two bands at 1318 cm−1 and 1155 cm−1 for the SO2N group and a strong carbonyl absorption at 1653 m−1 as well as two bands at 2868–2924 cm−1 and 3325–3372 cm−1 for the OH and NH2 groups, respectively. Condensation of the 4-aminosulfonyl derivative 17 with different aldehydes gave the corresponding azomethines 18. The IR spectra of these arylidenes showed strong carbonyl absorption at 1652–1656 cm−1, while their 1H-NMR spectra revealed a new singlet at δ 7.97–8.26 ppm attributed to the CH=N proton. Reaction of the arylidene derivatives 18 with mercaptoacetic acid afforded the corresponding thiazolidinone derivatives 19. Their IR spectra revealed two strong carbonyl absorption bands at 1658–1662 cm−1 and 1720–1735 cm−1 for the 4H-pyran and thiazolidinone moieties, respectively. Moreover, reaction of 17 with isocyanate and isothiocyanate derivatives in pyridine, gave the corresponding substituted sulfonylureido and sulfonylthioureido analogs 20 and 21, respectively. The IR spectra of these compounds showed beside the 4H-pyran carbonyl absorption at 1651–1656 cm−1, two bands at 1315–1377 cm−1 and 1168–1182 cm−1 due to SO2N function as well as distinguishable urea carbonyl absorption at 1660–1667 cm−1 for 20 and thiourea carbonyl absorption at 1122–1165 cm−1 for compound 21. Cyclization of the sulfonylthioureido derivatives 21 with ethyl bromoacetate in the presence of anhydrous sodium acetate afforded the corresponding thiazolidin-4-ones 22. The IR of these compounds displayed a carbonyl band at 1718–1722 cm−1, whereas, their 1H-NMR spectra revealed a broad singlet at δ 4.11–4.18 ppm attributed to the oxothiazolidine H-5. The 13C-NMR spectra of 22 exhibited the C-5 of the thiazolidine moity at δ 32.5–32.9 (Supplementary File S7). Analogously, reacting the same derivatives 21 with phenacyl bromide under similar reaction conditions resulted in the formation of the desired 1,3 -thiazolines 23. Their 1H-NMR spectra showed a new singlet at δ 6.25–6.34 ppm due to the thiazoline H-5 proton. The 1H-NMR and 13C-NMR spectra of 1–23 are recorded in Supplementary Files S6 and S7.
Biological evaluation
In vitro antibacterial and antifungal activities
The antibacterial activity of the synthesized compounds revealed that 26 out of the tested 58 compounds displayed variable inhibitory effects on the growth of the tested Gram positive and Gram negative bacterial strains. In general, most of the compounds showed better activity against the Gram positive rather than the Gram negative bacteria. Among the Gram positive bacteria, S. aureus and B. subtilis showed relatively high sensitivity towards the tested compounds. In this view, compounds 6b and 22d were equipotent to Ampicillin (MIC 6.25 μg/mL) against S. aureus, whereas the analogs 5f, 6a 15a, 18b, 22b and 23d (MIC 12.5 μg/mL) were 50% less active than Ampicillin. Moreover, compounds 5c, 5d, 7a, 8b, 9b, 11c, 18c and 21d (MIC 25 μg/mL) showed 25% of the activity of Ampicillin against the same organism. With regard to the activity against B. subtilis, the best activity was displayed by compound 22d, which was equipotent to Ampicillin (MIC 12.5 μg/mL), whereas the analogs 6b, 7a, 11c, 14b, 15a, 18b, 21d and 22b (MIC 25 μg/mL) which represented half the potency of Ampicillin. The analogs 5c, 5d, 5f, 6a, 8a, 9b, 18c, 19c, 21b and 23d (MIC 50 μg/mL) exhibited 25% of the potency of Ampicillin against the same species. Micrococcus luteus proved to be the least sensitive Gram positive microorganism towards most of the tested compounds. Only two compounds namely; 6b and 22d (MIC 25 μg/mL) exhibited moderate growth inhibitory effect towards M. luteus which was 50% of the activity of Ampicillin (MIC 12.5 μg/mL).
On the other hand, investigation of the antibacterial potency against the three tested Gram negative strains revealed that most of the active compounds showed better growth inhibitory activity against E. coli when compared with the other two strains. Compound was equipotent to Ampicillin (MIC 6.25 μg/mL), whereas the analogs 6b, 15a and 21d produced noticeable growth inhibitory activity (MIC 12.5 μg/mL) which was 50% of the activity of Ampicillin. Moreover, compounds 5f, 6a, 14b, 18b, 19c and 23d (MIC 25 μg/mL), exhibited moderate activity against the same organism. Meanwhile, the tested P. aeruginosa and K. pneumoniae strains proved to be weakly sensitive to most of the tested compounds. Among these, compound 22d showed moderate growth inhibitory profile against P. aeruginosa (MIC 25 μg/mL), which was about 50% of the activity of Ampicillin (MIC 12.5 μg/mL).
Concerning the antifungal activity of the tested compounds, the results revealed that eleven compounds namely; 5d, 5f, 6a, 6b, 8b, 18b, 18c, 19c, 21d, 22b, 22d and 23d were able to produce appreciable growth inhibitory activity against C. albicans (MIC values 12.5–100 μg/mL, respectively) when compared to Clotrimazole (MIC 6.25 μg/mL). Among these, compound 22d proved to be the most potent antifungal agent with MIC value 12.5 μg/mL and is 50% of Clotrimazole. The analogs 6b, 18b, 19c and 21d exhibited recognizable antifungal activity (MIC 25 μg/mL) that represented 25% of the standard’s activity. It has been found that all of the tested compounds lacked antifungal activity against A. niger ().
Table 1. Minimal inhibitory concentrations (MIC, μg/mL) of the active newly synthesized compounds.
A further investigation into the structures of the active compounds revealed that, the tested compounds belong to two main series namely; 4-hydroxy-6-methyl-2-oxo-1-substituted-2H-pyridines 2–15 ( and ) and -hydroxy-2-methyl-4-oxo-1-substituted-4H-pyridines 17–23 ().
Within the first series (), the obtained data in showed that the parent pyridine-2-one 1 was totally inactive against all the tested microorganisms. However, the antimicrobial potential of the derived compounds is obviously dependant on the nature of the substituent of the amino function at position-1. In this respect, sulfonylation of this amino group as seen in compound 3b (X = CH3) which was weakly active against the three tested Gram positive strains. Derivatization of the N1-amino substituent into an ureido functionality yielded a mildly active compound 4c (R = 4-Cl-C6H4). However, the preparation of substituted thioureido derivatives led to three active compounds 5c, 5d and 5f with a better antimicrobial profile, among which the analog 5f (R = 4-F-C6H4) revealed broad spectrum of antimicrobial activity against all the tested organisms, with a remarkable activity against S. aureus (MIC 12.5 μg/mL) and E. coli (MIC 25 μg/mL), together with an appreciable antifungal activity against C. albicans (MIC 25 μg/mL). Cyclization of the thioureido functionality into thiazolidine derivatives (6a,b) resulted in a remarkable improvement in the overall antimicrobial profile. Compound 6a (Y = H) showed two-fold increase in the activity against S. aureus, E. coli and C. albicans when compared with the parent 5d, whereas the analog 6b (Y = F) revealed a distinctive antimicrobial activity against all the tested organisms. It showed equipotent activity to Ampicillin against S. aureus (MIC 6.25 µg/mL) and 50% of its activity against B. subtilis, M. luteus, E. coli and P. aeruginosa (MIC 12.5 µg/mL), together with an appreciable antifungal activity against C. albicans (MIC 25 µg/mL). Meanwhile, cyclization of the thioureido function into thiazolines as in 7a (Y = H) led to a limited change in the overall antimicrobial activity when compared with the parent 5d.
On the other hand, condensation of the N1-amino group with different aryl and hetaryl aldehydes afforded two active azomethines 8a,b among which the analog 8b (R = 4-Cl-C6H4) showed better activity than 8a (R = C6H5), especially against S. aureus, M. Luteus, E. coli and C. albicans. However, cyclization into a thiazolidine ring as in 9b (R = 4-Cl-C6H4) did not offer a noticeable improvement in the overall antimicrobial potential over the parent azomethine.
The compounds presented in , the N1-benzenesulfonamido analog 10 showed weak activity only against the Gram positive S. aureus and B. subtilis. Derivatization of the sulfonamido group into sulfonylureido functionality as in 11b (X = O, R = Cl) resulted in a limited enhancement in the anti-Gram positive activity and an observable improvement in the activity against E. coli. On the contrary, the sulfonylthioureido 11c (X = S, R = H) exhibited better spectrum of activity against the Gram positive and Gram negative strains except K. pneumoniae when compared with the parent sulfonamide 10. On the other hand, in spite of the inactivity of the key intermediate pyridone 12, introducing a benzenesulfonyl moiety at position-1 as in 13a,b did not offer any advantage. However, incorporating a substituted carbamoyl counterpart as in 14b (R = Cl) resulted in an increase in antimicrobial activity against B. subtilis and E. coli. Whereas, introducing a thiocarbamoyl moiety at position-1 led to a potentially active compound 15a (R = H) which exhibited 50% of the activity of Ampicillin against S. aureus, B. subtilis and E. coli, however with no antifungal efficacy.
Shifting to the second series of pyridines 17–23 illustrated in , the key intermediate sulphonamide 17 showed mild activities only against the Gram positive S. aureus and B. subtilis (MIC 50 and 100 µg/mL, respectively). Condensation of the N1-sulfonamido group with different aryl and hetaryl aldehydes resulted into two active azomethines 18b,c, among which the analog 18b (R = 4-Cl-C6H4) showed better activity than 18c (R = 2-thienyl), against S. aureus and B. subtilis which was 50% of that of Ampicillin, and E. coli. It has also exhibited a remarkable antifungal activity against C. albicans which was 25% of that of Clotrimazole. However, cyclization into a thiazolidine ring as in 19c (R = 2-thienyl) led to a decrease in the Gram positive efficacy and a two-fold increase in the activity against Gram negative organisms when compared to the parent 18c. Furthermore, derivatization of the parent sulphonamide 17 into sulfonylureido function gave rise to a moderately active compound 20c (R = 4-Cl-C6H4) with MIC range of 50–100 µg/mL. However, bioisosteric replacement of the sulfonylureido function with a sulfonylthioureido as in 21a–d resulted in a remarkable improvement in the antimicrobial profile. Among these, the analog 21d (R = 4-F-C6H4) exhibited an improvement in the antibacterial potentials against both Gram positive and Gram negative organisms (B. subtilis and E. coli) together with an appreciable antifungal activity against C. albicans (25% of the activity of Clotrimazole). Cyclization of the former sulfonylthioureido compounds into thiazolidine derivatives (22a–d) resulted in a remarkable enhancement in the overall antimicrobial potential. Among these, compound 22d (R = 4-F-C6H4) proved to be equipotent to Ampicillin against S. aureus, E. coli and B. subtilis (MIC values 6.25, 6.25 and 12.5 µg/mL), and 50% of its activity against M. luteus and P. aeruginosa (MIC 25 µg/mL), together with a remarkable antifungal activity against C. albicans (MIC 25 µg/mL) which represents 50% of that of Clotrimazole. Finally, cyclization of the sulfonylthioureido function into thiazolines 23d (R = 4-F-C6H4) offered a limited enhancement in the activity against the Gram positive strains when compared with the parent 21d.
Preliminary in vitro anticancer screening
Out of the newly synthesized compounds, twenty derivatives 5c,d,f, 6a,b, 7c, 8b, 9b, 11c, 15a, 18b, 19b, 20b, 21a,b,d, 22b,d and 23b,d, were selected by the National Cancer Institute (NCI) in vitro disease-oriented human cells screening panel assay to be evaluated for their in vitro antitumor activity. Effective one-dose assay has been added to the NCI 60 Cell screen in order to increase compound throughput and reduce data turnaround time to suppliers while maintaining efficient identification of active compounds. All compounds submitted to the NCI 60 Cell screen, are now tested initially at a single high dose (10 μM) in the full NCI 60 cell panel including leukaemia, non-small cell lung, colon, CNS, melanoma, ovarian, renal, prostate and breast cancer cell lines. However, the data obtained from the one-dose assay revealed that none of the tested compounds showed significant antitumor activity and consequently, none of them was promoted to the 5-dose assay.
Conclusion
The results revealed that twenty six out of the tested fifty eight compounds displayed variable inhibitory effects on the growth of the tested Gram positive and Gram negative bacterial strains. In general, most of the tested compounds revealed better activity against the Gram positive than the Gram negative bacteria. Among the Gram positive bacteria tested, two strains namely; S. aureus and B. subtilis showed high sensitivity towards the tested compounds. However, most of the active compounds displayed better antibacterial activity against E. coli when compared with P. aeruginosa and K. pneumoniae. Moreover, eleven compounds were able to produce appreciable growth inhibitory activity against C. albicans fungus when compared with Clotrimazole. However, all of the tested compounds lacked antifungal activity against A. niger.
Acknowledgements
The authors are very grateful to the authorities of the Institute of Research and Consultation, King Abdulaziz University, Jeddah, Saudi Arabia, as well as SABIC group of companies, for the financial support of this research. Extendable thanks are due to the staff members of the Department of Health and Human Services, National Cancer Institute (NCI), Bethesda, Maryland, U.S.A. for carrying out the anticancer screening of the newly synthesized compounds.
Declaration of interest
The authors report no conflicts of interest.
References
- Faid-Allah HM, Al-Saadi MS, Rostom ShAF. Synthesis of new coumarin-3-carbonylurea, thiourea analogs and some derived ring systems of possible therapeutic interest. J Saudi Chem Soc (JSCS) 2003;7:367–372.
- Al-Saadi MSM, Rostom ShAF, Faid-Allah HM. In vitro antitumor screening of some polysubstituted pyrazole analogs. Saudi Pharm J (SPJ) 2005;13:89–96.
- Al-Saadi MSM, Rostom ShAF, Faid-Allah HM. Synthesis and biological evaluation of some 3-cyano-4-(1-methyl-1H-pyrrol-2-yl)-6-substituted-2(1H)-pyridinones and their 2-imino isosters. Alex J Pharm Sci 2005;19:15–21.
- Faid-Allah HM, Al-Saadi MS, Rostom ShAF, Fahmy HT. Synthesis of some sulfonamides, disubstituted sulfonylureas or thioureas and some structurally related variants. A class of promising antitumor agents. Med Chem Res 2007;16:300–318.
- Al-Saadi MS, Rostom SA, Faidallah HM. 3-methyl-2-(4-substituted phenyl)-4,5-dihydronaphtho[1,2-c]-pyrazoles: synthesis and in vitro biological evaluation as antitumour agents. Arch Pharm (Weinheim) 2008;341:181–190.
- Faid-Allah HM, Al-Saadi MS, Rostom ShAF. Synthesis and biological evaluation of certain 2H-pyran-2-ones and some derived 1H-pyridin-2-one analogs as antimicrobial agents. Saudi Pharm J (SPJ) 2008;16:33–42.
- Al-Saadi MSM, Faid-Allah HM, Rostom ShAF. Synthesis and biological evaluation of some 2,4,5-trisubstituted thiazole derivatives as potential antimicrobial and anticancer agents. Arch Pharm Chem Life Sci 2008;341:424–434.
- Rostom ShAF, Faidallah HM, Al Saadi MSM. A Facile synthesis of some 3- cyano-1,4,6-trisubstituted-2-(1H)-pyridines and their biological evaluation as anticancer agents. Med Chem Res 2011;20:1260–1272.
- Faidallah HM, Khan KA, Asiri AM. Synthesis and biological evaluation of new 3-trifluoromethyl-pyrazolesulfonylurea and thiourea derivatives as antidiabetic and antimicrobial agents. J Fluorine Chem 2011;132:131–137.
- Faidallah HM, Khan KA, Asiri AM. Synthesis and characterization of a novel series of benzenesulfonylurea and thiourea derivatives of 2H-pyran and 2H-pyridine-2-ones as antibacterial, antimycobacterial and antifungal agents. Eur J Chem 2011;2:243–250.
- Faidallah HM, Khan KA, Asiri AM. Synthesis and biological evaluation of new 3,5-di(trifluoromethyl)-1,2,4-triazoles-ulfonylurea and thiourea derivatives as antidiabetic and antimicrobial agents. J Fluorine Chem 2011;132:870–877.
- Aanandhi MV, George S, Vaidhyalingam V. Synthesis and antimicrobial activities of 1-(5-substituted-2-oxoindolin-3-ylidene)-4-(substituted pyridin-2-yl)thiosemicarbazide. ARKIVOC 2008;(xi):187–194.
- Narayana BL, Rao ARR, Rao PS. Synthesis of new 2-substituted pyrido[2,3-d]pyrimidin-4(1H)-ones and their antibacterial activity. Eur J Med Chem 2009;44: 1369–1376.
- Ranft D, Seyfarth T, Schaper K-J, Lehwark-Yvetot G, Bruhn C, Buege A. New N1-hetarylmethylene-substituted amidrazones with potential antimycobacterial activity. Arch der Pharm-Chem Life Sci 1999;332:427–430.
- Khoshneviszadeh M, Edraki N, Javidnia K, Alborzi A, Pourabbas B, Mardaneh J et al. Synthesis and biological evaluation of some new 1,4-dihydropyridines containing different ester substitute and diethyl carbamoyl group as anti-tubercular agents. Bioorg Med Chem 2009;17:1579–1586.
- Abid M, Husain K, Azam A. Synthesis and antiamoebic activity of new oxime ether derivatives containing 2-acetylpyridine/2-acetylfuran. Bioorg Med Chem Lett 2005;15:4375–4379.
- Saudi MNS, El-Sayed MH, Abou El-Hoda M. 3-Cyanopyridine derivatives: novel fasciolicidal candidates. Alex J Pharm Sci 2002;16:75–82.
- Goebel T, Ulmer D, Projahn H, Kloeckner J, Heller E, Glaser M et al. In search of novel agents for therapy of tropical diseases and human immunodeficiency virus. J Med Chem 2008;51:238–250.
- Rodrigues T, Guedes RC, dos Santos DJ, Carrasco M, Gut J, Rosenthal PJ et al. Design, synthesis and structure-activity relationships of (1H-pyridin-4-ylidene)amines as potential antimalarials. Bioorg Med Chem Lett 2009;19:3476–3480.
- Gudmundsson KS, Johns BA, Wang Z, Turner EM, Allen SH, Freeman GA et al. Synthesis of novel substituted 2-phenylpyrazolopyridines with potent activity against herpesviruses. Bioorg Med Chem 2005;13:5346–5361.
- Allen SH, Johns BA, Gudmundsson KS, Freeman GA, Boyd FL Jr, Sexton CH et al. Synthesis of C-6 substituted pyrazolo[1,5-a]pyridines with potent activity against herpesviruses. Bioorg Med Chem 2006;14:944–954.
- Monks A, Scudiero D, Skehan P, Shoemaker R, Paull K, Vistica D et al. Feasibility of a high-flux anticancer drug screen using a diverse panel of cultured human tumor cell lines. J Natl Cancer Inst 1991;83:757–766.
- Grever MR, Schepartz SA, Chabner BA. The National Cancer Institute: cancer drug discovery and development program. Semin Oncol 1992;19:622–638.
- Boyd MR, Paull KD. Practical considerations and applications of the national cancer institute in vitro anticancer drug discovery screen. Drug Rev Res 1995;34:91–109.
- Conte JE, Barriere SL. (1988). Manual of Antibiotics and Infectious Disease. 1st Edition. USA: Lea and Febiger, 135–138.