Abstract
The standardization of HIV-1 strains for vaccine tests include the use of viral reference panels. We determined a series of pharmacological descriptors (molecular surfaces, volumes, electrostatic energies, solvation energies, number of atoms, number of hydrogen donors or acceptors and number of rigid bonds) for the gp120 CD4-binding sites structures in the unliganded state from a reference panel of 60 diverse strains of HIV-1. We identified the descriptors that varied significantly between the strains, the outliner strains for each descriptor set and the possible correlations between the descriptors. Our results improve the knowledge about gp120, its molecular and possible neutralization properties.
Introduction
Entry of human immunodeficiency virus type 1 (HIV-1) into the T-helper lymphocytes is mediated by its envelope glycoproteins: gp120 and gp41, named according to their molecular mass and coded by the same viral gene (Env). The structure of HIV-1 gp120 glycoprotein is comprised of five variable regions (V1–V5) interposed among more conserved regions (C1-C5) that form the protein core region. The variable regions form exposed loops anchored at their bases by disulphide bondsCitation1. Gp120 determines the HIV tropism through a very strong and specific binding to the CD4 receptor present mainly on T-helper lymphocytes surface. Gp120 binding to CD4 is the first step for virus internalization. Binding to CD4 induces ample conformational changes in gp120 which allows it to bind one of the chemokine receptors with co-receptor role (CCR5 or CXCR4)Citation2.
The development of a vaccine is considered the only viable long-term solution to the AIDS pandemic. Given the huge variability of the virus, it is widely believed that the induction of a broadly neutralizing antibody response will be crucial for the success of a vaccine against the HIV. The development of an immunogen that elicits it still remains elusiveCitation3. Any rational attempt to design anti-HIV inhibitors or vaccines must therefore involve the characterization of the two significantly different conformational states of gp120: prior to CD4 binding and CD4-bound.
The variable regions of HIV-1 elicit a high abundance of strain-specific antibodies, whereas antibodies that target conserved regions are rare and only develop in a subset of individualsCitation4. Therefore, only a few antibody species have been identified, so far, as effective in neutralizing primary HIV-1 isolates. The CD4-binding site is included in the C2-C5 conserved regions of gp120 and elicits a few known broadly neutralizing antibodies: B12Citation4,Citation5, HJ16 and VRC01Citation3,Citation6.
In the prefusion state, as revealed by a fully glycosylated simian immunodeficiency virus (SIV) gp120 core structure, gp120 presents a distinct antigenic surface than that from the bound structuresCitation2. Comparison of the new structure of unliganded gp120 with that of the CD4-bound protein shows that part of the molecule (the “inner domain,” which includes most of the CD4 binding site) undergoes extensive conformational rearrangement upon receptor binding. Compared to the CD4-bound stateCitation7, its inner domain does not shift as a rigid body relative to the almost unchanged “outer domain,” but rather a few of its distinct regions move independently relative to each otherCitation2.
The 60 strain panel whose pharmacological descriptors we modelled in the present study contains a combination of sensitive and resistant isolates and of CCR5 or CXCR4. The early transmitted CCR5 viruses are arguably the key for HIV-1 vaccine development and in the assessment of vaccine immunogenicity. Most of the isolates obtained for the panel were non-syncytium inducing (NSI) and utilize CCR5 as a coreceptor for viral entry. However, syncytium-inducing, CXCR4-utilizing viruses were also included in the panelCitation8.
Each group of 10 sequences is included in its respective clade or circulating recombinant form (CRF) with a bootstrap value of 100%. The strains within each clade or CRF were typical of within-clade diversity and were without significant associations. The diversity of strains within each clade was similar to that observed with the complete genome sequences. The genetic analysis of the virus panel established that 40 strains from clades A through D were nonrecombinant and could be assigned to their respective clades, representing diversity within that clade with high confidence. The CRF01_AE and CRF02_AG groups strains likewise faithfully represented their respective CRF and mirrored the diversity recognized for these CRF, currently low for CRF01_AE and higher for CRF02_AG. The 60 gp120 sequences provide an envelope sequence proximal to the virus stock for neutralization analysisCitation8.
A previous study proved 3D-QSAR molecular shape analysis (MSA) technique to be effective for the study of the inhibitory activity of styrylquinoline derivatives on HIV-1 integrase. The best model obtained from stepwise regression and GFA techniques shows 51.6% predicted variance (leave-one-out) and 57.3% explained variance. These models show the importance of several sterical descriptors, like total polar surface area, relative polar surface area, relative hydrophobic surface area and relative positive charge, for the design and characterization of potent HIV-1 integrase inhibitorsCitation9.
The volume of a selected group in a protein describes the space occupied by the group and therefore is a sterical descriptor of that groupCitation10. Protein accessible surface area is a sterical descriptor of proteins that has possible relationships to solvent excluded volume and thus to hydrophobic interaction free energy and transfer free energy of solute molecules. The surface is affected by the molecule’s conformation, but is independent of atomic chargesCitation11.
The aim our study is to model through homology modelling the structures for gp120 CD4-binding sites in the unliganded form corresponding to the 60 representative strains of HIV-1, to calculate for them a broad array of pharmacological descriptors (molecular volume and solvent accessible area, hydrogen donor/acceptor bond character, number of hydrophobic centres, hydrophobic/polar surface area, number of flexible/rigid bonds, internal dipole moment) and then to identify the descriptors that present significant variation between the strains.
Materials and methods
We modelled the structures of gp120 and calculated the pharmacological descriptors of their CD4bs for the collection of 60 HIV-1 isolates that includes strains with very different infectivity and neutralization properties (see )Citation8. The sequences for the gp120 corresponding to the 60 strains were extracted from UniprotCitation12 or GenBankCitation13. If both the full-length and the envelope (Env) sequences of a strain were known, we modelled the Env sequence. The 60 all-atom models of free HIV-1 strains gp120 were constructed based on the free structure of the highly related SIV gp120 structure (pdb code: 2BF1)Citation2. Regions missing from SIV structure were modelled using as template corresponding regions from available CD4-bound gp120 structuresCitation14,Citation15. Thus, the V3 loop and the 205–216 region were built using as template a V3-loop containing CD4-bound HIV gp120 structure (pdb code: 2B4C)Citation14, while the C5 segment of gp120 was modelled using as template the NMR solution structure of HIV gp120 C5 in 40% trifluoroethanol (pdb code: 1MEQ)Citation15. The V1/V2 loop and the C1 region are distant to the CD4 binding site and were not included in the models.
Table 1. The strain codes, clades, sequences, TCID50 values (median tissue culture infective dose) and percentages of neutralization in soluble CD4 (sCD4) (10 µg/mL) of the CD4-binding sites for the 60 gp120 strain panel considered.
The alignment and sequence analysis (including the CD4bs consensus sequence of the 60 strains, see ) was done with Jalview 2.7 softwareCitation16. Homologous Cys involved in disulfide bonds formation were preferentially aligned. The sequence alignment between one of the 60 HIV-1 gp120 strains considered (AY713410), the main SIV gp120 template (PDB ID: 2BF1) and the other templates used is presented in . The model was built using a homology modelling method and optimized by short molecular dynamics simulations using MODELLER 9v3 softwareCitation17.
Figure 1. Homology modelling of free gp120 corresponding to strain code AY71341 (sequence number 11 out of the 60 strains considered). The 41 amino-acids of the CD4-binding site are bolded and underlined. The alignment follows Chen et al.Citation2.
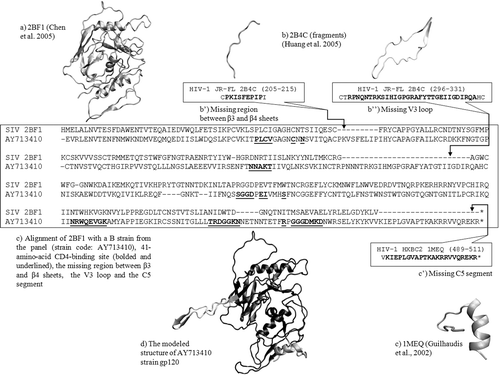
The CD4 binding site (CD4bs) was defined in a previous studyCitation18 as the residues being in contact with CD4 in the bound gp120 structure along with residue S375 (the disposition of these residues through gp120 sequence for the AY713410 strain of the panel is presented in ). Even if S375 is more than 5 Å apart from CD4, it was included in the binding site, as the S375W mutation is known to partially stabilize the CD4-bound stateCitation19. CD4bs includes a total of 41 amino-acids that can be grouped in six segments from the C2, C3, C4, C5 conserved regions of the protein. The corresponding positions in the HIV-1 HXB2 reference strain are: 124–127, 196, 198, 279–283, 365–368, 370, 371, 375, 425–432, 455–461, 469 and 471–477. The regions don’t present insertions or deletions among the 60 strain-panel considered. For our analysis of protein surfaces, we chose all the 41 amino acids of CD4bs.
We calculated the descriptors for all the 41 amino-acids of CD4bs. The analysis of structures was performed using CHARMM c34b1Citation20 and VMD 1.8.7Citation21 software. The electrostatic surface potential energy (UE) of solvated gp120 CD4 binding site regions were calculated using the PBEQ moduleCitation22 implemented in CHARMMCitation20 using a grid centred in the centre of the molecule, with a minimum distance between the solute and the grid boundary of 10 Å and a coarse grid spacing of 1.5 Å before focusing and 1 Å after focusing. For the dielectric boundary, we considered the molecular contact and reentrant surface created using the water radius (1.4 Å) and the atomic radii of protein atoms from the Charmm27 force fieldCitation23. We performed calculations using a protein dielectric constant of 1, a bulk-solvent dielectric constant of 80 and a salt concentration of 0.15 M/L, at 310 K. The solvation free energy of the binding site regions were calculated as the difference between the electrostatic surface potential energy of the structure in water and the electrostatic surface potential energy in a reference environment, with a dielectric constant of 1.
The number of hydrophobic and polar atoms, and of flexible and rigid bonds CD4-binding site region were calculated with CHARMMCitation20. We considered as rigid bonds the bonds within aromatic (Phe, Tyr, Trp) and non-aromatic (Pro) amino-acid cycles. We considered as hydrophobic atoms the carbon and hydrogen atoms and as polar atoms the oxygen, nitrogen, and sulfur atoms.
The solvent accessible surface (SAS) of gp120 CD4-binding site is important in defining the hydrophobic term of the interaction with CD4. We used CHARMMCitation20 to calculate SAS for the 41-amino acid CD4-binding site regions by rolling a probe radius of 1.4 Å along the target surface of the binding site. We thus measured for all the 60 strains the SAS of all atoms, of the positively and the negatively-charged atoms, as well as of the hydrophobic and the polar atoms (as defined above).
Results
The studied 60 strain panel data is presented in (the codes, clades CD4-binding site sequences). For reference, includes two measured biological propertiesCitation8: the viral 50% toxicity citotoxic concentration (TCID50) and the percentage of virus neutralized by a 10 µg/mL soluble CD4 (sCD4). The rank of the 60 models of gp120 strain variants is the same as in the original panelCitation8. The CD4 binding site considered in our study (41 amino-acids) has a global identity conservation of ~84%, with 17 identical amino-acid positions in all the strains and 24 positions with at least one amino-acid variation. We calculated three types of pharmacological descriptors for the 60-strain panel: sterical, energetical and structural descriptors.
Sterical descriptors
We calculated solvent accessible surface (SAS) for all atoms, for positive and negative atoms and for hydrophobic and polar atoms, and the total volume. We also calculated the total dipole moment and its components on the X, Y and Z axes (see ). We analyzed the variation of the calculated molecular descriptors that characterize the scale of their variations compared to the median values.
Table 2. Significant structural, energetical and sterical descriptors of the CD4-binding sites for the 60 gp120 strains and their statistics.
Solvent accessible surface of positive atoms (see ) has as the five lowest outliners the strains AY713425 (631.48 Å2) (lowest), AY713422, AY713421, AY736812 and AY736848 and as the five highest outliers the strains AY736823, AY736830, AY713410, AY736839 and AY736828 (857.41 Å2) (highest). AY713425 also presents the lowest SAS of negative atoms (see below), the lowest total SAS (data not shown) and the second lowest SAS of hydrophobic atoms. The presence in AY713425 strains’ CD4bs of small amino-acids in several variable positions (one treonine and two alanine residues instead of a valine and two arginine residues present in most strains) is in good correlation with its small surfaces.
Figure 2. Variation of significant structural descriptors of the CD4-binding sites for the 60 gp120 strains. (a) SAS of positive atoms; (b) SAS of and negative atoms; (c) SAS of hydrophobic atoms; (d) SAS of polar atoms. The mean values and the standard deviations are noted in the higher right corner. The Y-axes are adjusted for a clearer representation of the descriptor variation.
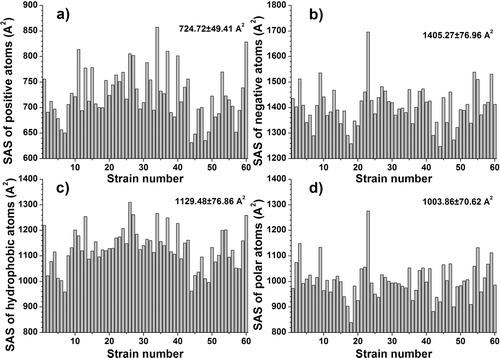
Solvent accessible surface of negative atoms (see ) has as the five lowest outliners the strains AY713425 (1247.97 Å2) (lowest), AY713408, AY713422, AY736812 and AY736837, and as the five highest outliers the strains AY736809, AY736841, AY736813, AY736845 and AY713413 (1695.97 Å2) (highest).
Solvent accessible surface of hydrophobic atoms (see ) has as the five lowest outliners the strains AY736812 (958.69 Å2) (lowest), AY713425, AY713420, AY736814 and AY713422, and as the five highest outliers the strains AY713409, AY736839, AY713417, AY736828, AY736823 (1310.25 Å2) (highest). As can be seen from the above values, the recorded changes are significant.
Solvent accessible surface of polar atoms (see ) has as the five lowest outliners the strains AY713408 (839.13 Å2) (lowest), AY736837, AY713422, AY736819 and AY736844, and as the five highest outliers the strains AY736841, AY736846, AY736813, AY736809, and AY713413 (1275.96 Å2) (highest). The recorded changes are therefore significant.
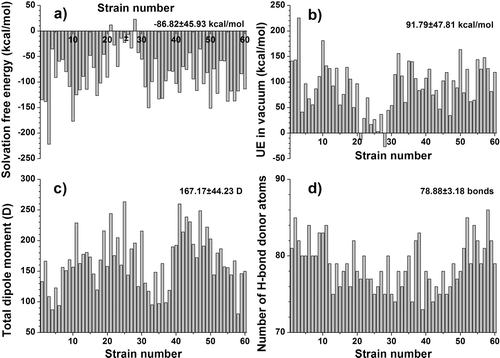
The total dipole moment (see ) has as the five lowest outliners the strains AY736842 (80.33 debyes) (lowest), AY736810, AY736814, AY736834 and AY736827, and as the five highest outliers the strains AY736838, AY713414, AF070707, AY713424 and AY833060 (263.52 debyes) (highest), with very significant changes between strains. The highest dipole moment presented by strain AY833060 is associated to the additional presence of two acidic residues compared to other strains (one aspartic acid and one glutamic acid residues instead of glycine and asparagine in the consensus sequence) (see ).
Figure 3. Variation of significant energetical and sterical descriptors of the CD4-binding sites for the 60 gp120 strains. (a) free solvation energy; (b) electrostatic surface potential energy (UE) in vacuum; (c) total dipole moment; (d) the number of hydrogen-bond donors. The mean values and the standard deviations are noted in the higher right corner. The Y-axes are adjusted for a clearer representation of the descriptor variation.
Energetical descriptors
We measured the solvation free energy, electrostatic surface potential energy in water, electrostatic surface potential energy in vacuum (see and ).
Solvation free energy (see ) has as the five lowest outliners the strains AY736809 (−221.74 kcal/mol) (lowest) AY736816, AY713419, AY713418 and AY713406, and as the five highest outliers the strains AY833060, AY833059, AY736823, AY713414 and AY736826 (23.31 kcal/mol) (highest). As we can see, the largest value is positive, indicating a slightly energetically unfavourable solvation for the AY736826 strain’s gp120 CD4-binding site. Unsurprisingly, the AY736826 strain also has the lowest (most favourable) electrostatic potential energy in vacuum (see below). Its CD4bs sequence contains non-polar amino-acids (two additional valine residues and a glycine residue compared to the consensus sequence).
Electrostatic surface potential energy in vacuum (UE in vacuum) (see ) has as the five lowest outliners the strains AY736826 (−26.73 kcal/mol) (lowest) AY713414, AY736823, AY833059 and AY833060, and as the five highest outliers the strains AY736848, AY713418, AY713419, AY736816 and AY736809 (225.6 kcal/mol) (highest).
Structural descriptors
We determined the following structural descriptors: total charge, the number of hydrophobic atoms, number of polar atoms, number of bonds that allow free rotation, number of rigid bonds, number of H-bond donor atoms and H-bond acceptor atoms (see and ).
The number of hydrogen bond donors (see ) has as the four lowest outliners the strains AY736833 (73) (lowest) AY736829, AY736837 and AY736831, and as the six highest outliers the strains AY736845, AY736814, AY713410, AY713406, AY736843 and AY736842 (86) (highest). The presence of small non-polar alanine and threonine stands out in the sequence of AY736833 CD4bs, while AY736842 CD4bs sequence is characterized by the presence of two additional asparagine residues compared to the consensus sequence.
Discussion
Descriptors calculation in the CD4 binding sites of gp120 strains has shown that significant variations were obtained for: solvent accessible surface (SAS) of positive atoms (from 631.48 Å2 (in AY713425 strain) to 857.41 Å2 (in AY736828 strain). The descriptor SAS of negative atoms recorded quite large variations (from 1247.97 Å2, AY713425 to 1695.97 Å2, AY713413). The same strain (AY713413) also presented the largest solvent SAS of polar atoms (which varied from 839.13 Å2 for AY713408, to 1275.96 Å2 for AY713413), and the largest SAS on all atoms (data not shown). Its CD4bs sequence differences from other strains studied are several bulky polar residues (glutamic acid and lysine).
Significant variations were recorded when the descriptor SAS of hydrophobic atoms (from 958.69 Å2, AY736812 to 1310.25 Å2, AY736823) was considered. AY736823 strain CD4bs also presents the third lowest electrostatic energy in vacuum and solvation free energy, and the lowest electrostatic energy in water (data not shown). The presence of three hydrophobic isoleucine residues stands out compared to the CD4bs consensus sequence.
The solvation free energy and electrostatic surface potential energy in vacuum presents very large and inversely correlated variations (see above and and ). The total dipole moment presents large variations among the 60 protein sites (see ). The electrostatic surface potential energy in water varied in a very narrow range (but with a standard deviation of more than half the mean value), owing to the shielding effect of water on an electric fieldCitation24 (data not shown).
Our analysis has shown that among the structural descriptors, the number of hydrogen bond donors presents significant variations (see above and ). The variations present in the other sterical descriptors are relatively small and can be considered non-significant (data not shown). Given the constant number of amino-acids of the CD4-binding site (41), and the relatively high overall identity rate (~84%) this fact is explainable despite the numerous mutations present.
Conclusion
Molecular simulation techniques will continue to give important information about vaccine and inhibitor design). We identified eight pharmacological descriptors for the unliganded gp120 CD4-binding sites (SAS of positive and negative atoms, hydrophobic and polar SAS, free solvation energy, electrostatic surface potential energy in vacuum, total dipole moment and number of hydrogen-bond donors) that vary significantly between the strains of a 60 member HIV-1 strain collection. We also identified the outliner strains for each descriptor set and the correlations between the descriptors and sequence for a better calibration of both simulations and experiments. Our results improve the knowledge about gp120 and its weaknesses and are useful together with the data provided by other molecular simulation techniques in the design of broad-spectrum anti-HIV-1 inhibitors and vaccines.
Declaration of interest
The authors acknowledge the financial support of the Romanian Ministry of Education, Research, Youth and Sport through the “IDEAS” grant 137/2011.
References
- Hartley O, Klasse PJ, Sattentau QJ, Moore JP. V3: HIV’s switch-hitter. AIDS Res Hum Retroviruses 2005;21:171–189.
- Chen B, Vogan EM, Gong H, Skehel JJ, Wiley DC, Harrison SC. Structure of an unliganded simian immunodeficiency virus gp120 core. Nature 2005;433:834–841.
- Walker LM, Burton DR. Rational antibody-based HIV-1 vaccine design: Current approaches and future directions. Curr Opin Immunol 2010;22:358–366.
- Stamatatos L, Morris L, Burton DR, Mascola JR. Neutralizing antibodies generated during natural HIV-1 infection: Good news for an HIV-1 vaccine? Nat Med 2009;15:866–870.
- Lin G, Nara PL. Designing immunogens to elicit broadly neutralizing antibodies to the HIV-1 envelope glycoprotein. Curr HIV Res 2007;5:514–541.
- Corti D, Langedijk JP, Hinz A, Seaman MS, Vanzetta F, Fernandez-Rodriguez BM et al. Analysis of memory B cell responses and isolation of novel monoclonal antibodies with neutralizing breadth from HIV-1-infected individuals. PLoS ONE 2010;5:e8805.
- Kwong PD, Wyatt R, Robinson J, Sweet RW, Sodroski J, Hendrickson WA. Structure of an HIV gp120 envelope glycoprotein in complex with the CD4 receptor and a neutralizing human antibody. Nature 1998;393:648–659.
- Brown BK, Darden JM, Tovanabutra S, Oblander T, Frost J, Sanders-Buell E et al. Biologic and genetic characterization of a panel of 60 human immunodeficiency virus type 1 isolates, representing clades A, B, C, D, CRF01_AE, and CRF02_AG, for the development and assessment of candidate vaccines. J Virol 2005;79:6089–6101.
- Leonard JT, Roy K. Exploring molecular shape analysis of styrylquinoline derivatives as HIV-1 integrase inhibitors. Eur J Med Chem 2008;43:81–92.
- Richards FM. The interpretation of protein structures: Total volume, group volume distributions and packing density. J Mol Biol 1974;82:1–14.
- Richmond TJ. Solvent accessible surface area and excluded volume in proteins. Analytical equations for overlapping spheres and implications for the hydrophobic effect. J Mol Biol 1984;178:63–89.
- Reorganizing the protein space at the Universal Protein Resource (UniProt). Nucleic Acids Res 2012;40:D71–D75.
- Benson DA, Karsch-Mizrachi I, Lipman DJ, Ostell J, Sayers EW. GenBank. Nucleic Acids Res 2011;39:D32–D37.
- Huang CC, Tang M, Zhang MY, Majeed S, Montabana E, Stanfield RL et al. Structure of a V3-containing HIV-1 gp120 core. Science 2005;310:1025–1028.
- Guilhaudis L, Jacobs A, Caffrey M. Solution structure of the HIV gp120 C5 domain. Eur J Biochem 2002;269:4860–4867.
- Waterhouse AM, Procter JB, Martin DM, Clamp M, Barton GJ. Jalview Version 2–a multiple sequence alignment editor and analysis workbench. Bioinformatics 2009;25:1189–1191.
- Sali A, Blundell TL. Comparative protein modelling by satisfaction of spatial restraints. J Mol Biol 1993;234:779–815.
- Zhou T, Xu L, Dey B, Hessell AJ, Van Ryk D, Xiang SH et al. Structural definition of a conserved neutralization epitope on HIV-1 gp120. Nature 2007;445:732–737.
- Xiang SH, Kwong PD, Gupta R, Rizzuto CD, Casper DJ, Wyatt R et al. Mutagenic stabilization and/or disruption of a CD4-bound state reveals distinct conformations of the human immunodeficiency virus type 1 gp120 envelope glycoprotein. J Virol 2002;76:9888–9899.
- Brooks BR, Bruccoleri RE, Olafson BD, States DJ, Swaminathan S, Karplus M. CHARMM: A Program for macromolecular energy, minimization, and dynamics calculations. J Comp Chem 1983;4:187–217.
- Humphrey W, Dalke A, Schulten K. VMD: Visual molecular dynamics. J Mol Graph 1996;14:33–8, 27.
- Im W, Beglov D, Roux B. Continuum solvation model: Computation of electrostatic forces from numerical solutions to the Poisson-Boltzmann equation. Comput Phys Comm 1998;111:59–75.
- Mackerell AD Jr, Feig M, Brooks CL 3rd. Extending the treatment of backbone energetics in protein force fields: Limitations of gas-phase quantum mechanics in reproducing protein conformational distributions in molecular dynamics simulations. J Comput Chem 2004;25:1400–1415.
- Levy Y, Onuchic JN. Water and proteins: A love-hate relationship. Proc Natl Acad Sci USA 2004;101:3325–3326.