Abstract
The development of specific inhibitors of the proteasome represents an important opportunity for new drug therapies. The central role of the multicatalytic complex in the intracellular proteolysis mediated by ubiquitin-proteasome pathway goes to discovery many molecules able to selectively inhibits enzymatic active subsites. Now, we report synthesis and activity of a new partial retro-inverso oligopseudopeptide derivatives bearing a trans,trans-muconic acid ethyl ester pharmacophoric unit at the C-terminal. Some analogues selectively inhibited in µM range the caspase-like (C-L) activity in the β1 subunit of the proteasome.
Introduction
The degradation of intracellular proteins plays a central role in regulating cellular function and maintaining homeostasis. Most of the cellular proteins are degraded through the ubiquitin proteasome pathway (UPP) including those involved in main processes such as apoptosis, cell cycle, transcription, antigen presentation by Major histocompatibility complex (MHC) and othersCitation1. Defects within this system are associated with several diseases, including cancer. Proteins are first tagged by a polyubiquitin chain in a complex enzymatic process and then recognized and degraded by the proteasome 26SCitation2.
This 26S multicatalytic complex consists of a 20S proteolytic core particle, which has a cylindrical shape, with the seven different α and seven different β subunits forming four stacked rings, and two 19S regulatory caps which recognize polyubiquitinated protein substrates and promote their entry into the central catalytic chamberCitation3–5. Three major proteolytic activities of proteasome can be distinguished as trypsin-like (T-L) located in β2, chymotrypsin-like (ChT-L) in β5 and peptidyl-glutamyl peptide hydrolase (PGPH) or caspase-like (C-L) activities in β1 subunitsCitation6. The catalytic residue of the 20S proteasome is a threonine, responsible for the cleavage of substrates through nucleophilic attackCitation7. Inhibition of enzymatic activity with specific proteasome inhibitors may provide an anti-tumoral and other therapeutic effectsCitation8,Citation9.
Several classes of synthetic and natural compounds which inhibit the proteolytic activities of the multicatalytic complex have been developed, and have contributed significantly to the identification of the essential functions of the 26S proteasome in various processes and pathways in eukaryotic cellsCitation10.
Most proteasome inhibitors are short synthetic peptides bearing a C-terminal pharmacophoric groups, such as aldehyde (e.g. MG132), boronic acid (Bortezomib) applied in multiple myeloma therapy, epoxyketone (Carfilzomib) and vinyl sulphone, or natural molecules (epoxomicin, lactacystin, salinosporamide, syringolins A and B) that form covalent adducts with catalytic threonineCitation11–19. Non-covalent inhibitors, such as TMC-95A (a naturally-constrained cyclic tripeptide) have been investigated in less detail, although they are thought to have weaker side effects in therapeutic applicationsCitation20,Citation21.
In previous studies, we reported the design, synthesis and activity of several class of peptide-based proteasome inhibitors containing a variety of pharmacophoric units as a electrophilic group potentially able to interact by the catalytic threonineCitation22–24. Herein we describe the synthesis and proteasome inhibition of novel C-terminal trans,trans-muconic acid ethyl ester partial retro-inverso (RI) pseudopeptides.
In recent years, a large number of peptide-based molecules have been identified and are currently available as a potential drug. Peptide limitations to the application in therapy are determined by unfavourable pharmacokinetic properties as low oral bioavailability, enzymatic degradation and difficulties to permeate biological membranes and cellular barriers. Chemical modifications of the canonical peptide structure are widely developed to improve bioavailability with the scope to obtain compounds that can be used in therapyCitation25. Bioisosteric replacement of peptide bond is one of the most applied manners to increase proteolytic stability. In particular, reversal carboamidic function in which the CO and NH were replaced by NH and CO, respectively, would maintain the chemical and stereochemical nature of the original peptide bond. RI modification, in many cases, allowed to obtain bioactive pseudopeptides with a favourable pharmacokinetic propertiesCitation26.
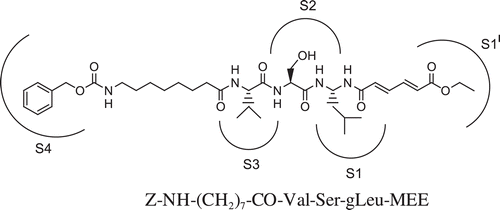
In this report, we have developed a new series of molecules with a reversed amidic bond between tripeptidic sequence and the potential pharmacophoric unit trans,trans-6-oxo-esa-2,4-dienoic acid ethyl ester (MEE). Differently from our previous classes of proteasome inhibitors, the new compounds contain a C-terminal extended conjugation as electrophylic trap for catalytic threonine. The aim of this study was to evaluate whether this conjugated system, potentially able to interact with proteasomal catalytic subunits, can strengthen the interaction between enzyme/inhibitor and consequently increase the inhibitory capacity. The oligopeptidic sequences (2 or 3 residues) of the analogs 1–16 are either functionalized at the N-terminal with 3-hydroxy-2-methylbenzoyl (HMB), Z-protected 6-aminohexanoyl and 8-aminooctanoyl groups or carrying the free α-amino function, according to the biological response obtained for the previous series ().
Figure 1. Structure of the C-terminal retro-inverso pseudotripeptide 16 and hypothetical pockets of the proteasomal substrate binding catalytic site that are occupied by the pharmacophore, residue side chains and N-terminal function.
Materials and methods
Chemistry-general
Amino acids, amino acid derivatives, and chemicals were purchased from Bachem, Novabiochem, and Fluka (Switzerland). Crude products were purified by preparative reversed-phase HPLC using a Water Delta 600 system with a Phenomenex column C18 (30 × 3 cm, 15 μm spherical particle size column). The column was perfused at a flow rate of 30 mL/min, with a mobile phase-containing solvent A (10%, v/v, acetonitrile in 0.1% TFA), and a linear gradient from 0 to 100% of solvent B (60%, v/v, acetonitrile in 0.1% TFA); 30 min was the time adopted for elution of the compounds. HPLC analysis was performed using a Beckman System Gold with a Hypersil BDS C18 column (5 μm; 4.6 × 250 mm). Analytical determination and capacity factor (K′) of the peptides were assayed via HPLC conditions in the above solvent system (solvents A and B), programmed at flow rates of 1 mL/min, using the following linear gradients: (a) from 0 to 90% B for 25 min and (b) from 30 to 100% B for 25 min. No pseudopeptide showed more than 1% impurity when monitored at 220 and 254 nm. The molecular weights of the compounds were determined by electrospray ionisation (ESI) (MICROMASS ZMD 2000), and the values are expressed as [MH]+. TLC was performed on precoated plates of silica gel F254 (Merck, Darmstadt, Germany), exploiting the following solvent systems: (c) AcOEt/n-hexane (1:1, v/v), (d) CH2Cl2/methanol (9.5:0.5, v/v), (e) CH2CL2/methanol (9:1, v/v), and (f) CH2CL2/methanol/toluene (17:2:1, v/v/v). Ninhydrin (1%) or chlorine iodine spray reagents were employed to detect the peptides. Melting points were determined by a Kofler apparatus and are uncorrected. Optical rotations were determined by a Perkin–Elmer 141 polarimeter with a 10 cm water-jacketed cell. 1H NMR spectroscopy was obtained using a Varian 400 spectrometer.
Synthesis
Butadienyl ethyl ester partial RI peptides 1–16 were prepared following the strategy reported in . C-Terminal RI dipeptide carrying pharmachoporic unit was synthetized starting by acylation of leucine amide with Fmoc (9-fluorenylmethoxycarbonyl)-protected residue (Leu or Ser) using WSC (water soluble carbodiimide)/HOBt (N-hydroxybenzotriazole). The oxidative rearrangement employs IBTFA (iodobenzene-bis-trifluoroacetate) as reagent for the conversion of the N-protected dipeptide amide to the corresponding pseudodipeptide containing gem-diamino leucine (gLeu). It has been shown that this rearrangement retains the chiral integrity of the asymmetric centre at the reacting residueCitation27. Pharmacophoric unit was introduced by reaction between RI dipeptide and the trans,trans-muconic acid ethyl ester using again WSC/HOBt. Fmoc was removed by 20% piperidine in DMF to give derivatives 1 and 2 and other pseudodipeptide analogues were obtained by acylations with 3-hydroxy-2-methylbenzoic (3,4), Z-protected 6-aminohexanoic (5,6) or 8-aminooctanoic acids (7,8) always with WSC/HOBt as coupling reagent.
Scheme 1. Synthesis of new trans,trans-muconic acid ethyl ester derivatives 1–16.
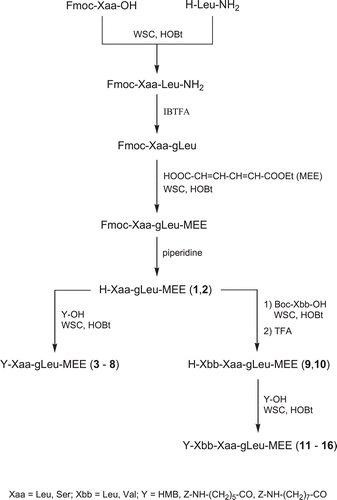
Starting from compounds 1 and 2 by acylation with Boc (tert-butoxycarbonyl)-Leu-OH or Boc-Val-OH and after TFA (trifluoroacetic acid) treatment permitted to obtain the corresponding free N-terminal analogues 9 and 10. Finally, the other pseudotripeptidic derivatives were obtained from 7 and 8 respectively by acylation via WSC/HOBt always with 3-hydroxy-2-methylbenzoic (11,12), Z-protected 6-aminohexanoic (13,14) or 8-aminooctanoic acids (15,16).
All products were purified and isolated by preparative RP-HPLC, and lyophilized molecules was analyzed by HPLC and characterized by electrospray ionisation (ESI) mass spectrometry () and 1H-NMR spectroscopy.
Table 1. Analytical data and physicochemical properties of C-terminal retro-inverso peptides 1–16.
General synthetic procedures
Fomc deprotection
Fmoc deprotection was obtained treating RI dipeptidic intermediate (1 mmol) with piperidine/DMF 20% (5 mL) mixture for 60 min. After evaporation, the residue was utilized for next synthetic steps.
TFA deprotection
Boc was removed by treating intermediates (1 mmol) with 1.5 mL of the aqueous 90% TFA (1:10, w/v) for 30–40 min. After evaporation, the residue was triturated with Et2O, centrifuged, and the resulting solid was collected and dried.
Coupling with WSC/HOBt
The deprotected α-amine intermediate (1 mmol), NMM (2 mmol) WSC (1 mmol) and HOBt (1 mmol) were added to a solution of carboxylic component (1 mmol) in DMF (3 mL) at 0°C. The reaction mixture was stirred for 1 h at 0°C and 18 h at RT; then the solution was diluted with AcOEt (80 ml) and washed consecutively with HCl 0.1 N, NaHCO3 and brine. The organic phase was dried (MgSO4) and evaporated to dryness. The residue was treated with Et2O and the resulting solid separated by centrifugation.
IBTFA rearrangement
To a stirred suspension of Nα-protected amide dipeptide (1 mmol) in a mixture of acetonitrile/water (3:2 v/v, 5 mL) was added iodobenzene bis-trifluoroacetate (IBTFA) (1.2 mmol) dissolved in acetonitrile (2 mL). The mixture was stirred at room temperature under argon. After evaporation the residue was purified by preparative RP-HPLC.
1H NMR of the Fmoc-protected intermediates and selected compounds
Fmoc-Leu-gLeu-MEE: 1H NMR (CDCl3): δ 0.89–0.92 (m, 12H); 1.27–1.29 (m, 4H); 1.49 (m, 1H); 1.66–1.68 (m, 2H); 1.87–1.90 (t, 1H, J = 4 Hz); 2.24 (t, 1H, J = 4 Hz); 2.30 (m, 2H); 4.14–4.19 (m, 3H); 4.87–4.90 (m, 1H); 4.93–4.99 (m, 3H); 5.55 (d, 1H, J = 15 Hz); 5.69 (d, 1H, J = 15 Hz); 7.28-7.46 (m, 6H); 7.58 (dd, 2H, J = 0.5 Hz, J = 5 Hz).
Fmoc-Ser-gLeu-MEE: 1H NMR (CDCl3): δ 0.90–0.93 (m, 6H); 1.26 (t, 3H, J = 7 Hz); 1.36 (m, 1H); 1.44 (m, 1H); 1.69 (m, 1H); 4.05 (m, 1H); 4.15–4.19 (m, 3H); 4.74–4.77 (t, 1H, J = 6.5 Hz); 4.90 (m, 1H); 5.08 (d, 2H, J = 7 Hz); 5.50 (d, 1H, J = 15 Hz); 5.63-5.71 (m, 2H); 7.27–7.82 (m, 10H).
HMB-Val-Ser-gLeu-MEE (12): 1H NMR (CDCl3): δ 0.90–0.97 (m, 12H); 1.28 (m, 4H); 1.55 (m, 1H); 2.00 (m, 1H); 2.15 (s, 3H); 2.63 (m, 1H); 4.12–4.19 (m, 3H); 4.43 (m, 1H); 4.65 (m, 1H); 4.75 (d, 1H, J = 6 Hz); 5.35–5.38 (d, 1H, J = 16 Hz); 5.61–5.64 (d, J = 16.0 Hz, 1H); 5.70 (m, 1H); 6.91–7.80 (m, 5H).
Z-NH-(CH2)5-CO-Leu-Leu-gLeu-MEE (13): 1H NMR (CDCl3): δ 0.90 (m, 18H); 1.28 (m, 3H); 1.49 (m, 3H); 1.60 (m, 1H); 1.65–1.68 (m, 2H); 1.39 (m, 6H); 1.78–1.85 (m, 2H); 2.22 (t, 2H, J = 6 Hz); 2.34 (m, 1H); 3.30 (m, 2H); 4.17 (t, 2H, J = 7 Hz); 4.48 (bs, 1H); 4.93 (bs, 1H); 5.05 (s, 2H); 5.35 (m, 1H); 5.49 (d, J = 16.1 Hz, 1H); 5.67 (d, J = 16.1 Hz, 1H); 7.27 (d, J = 16.1 Hz, 1H); 7.33 (s, 5H); 7.39 (d, J = 16.1 Hz, 1H).
Z-NH-(CH2)5-CO-Val-Ser-gLeu-MEE (14): 1H NMR (CDCl3): δ 0.90 (m, 12H); 1.33 (m, 6H); 1.50–1.55 (m, 6H); 1.71 (m, 1H);2.21 (m, 2H); 2.41 (m, 1H); 3.27 (t, 1H); 3.57 (t, 1H); 3.77 (m, 1H); 4.17 (m, 2H); 4.38 (m, 1H); 4.79 (m, 1H); 5.05 (s, 2H); 5.41 (d, J = 16.0 Hz, 1H); 5.46 (m, 1H); 5.64 (d, J = 16.1 Hz, 1H); 7.33 (s, 5H); 7.38 (d, J = 16.1 Hz, 1H); 7.51 (d, J = 16.1 Hz, 1H).
Z-NH-(CH2)7-CO-Val-Ser-gLeu-MEE (16): 1H NMR (CDCl3): δ 0.90 (m, 12H); 1.28 (m, 9H); 1.54 (m, 5H); 1.62 (m, 1H);1.90 (m, 1H); 2.23 (m, 2H); 2.43 (m, 1H); 3.16 (t, 1H); 3.56 (t, 1H); 3.85 (m, 1H); 4.17 (m, 2H); 4.42 (m, 1H); 4.50 (m, 1H); 4.91 (d, 1H); 5.05 (s, 2H); 5.47 (m, 2H); 5.67 (d, J = 16.1, 1H); 7.33 (s, 6H); 7.42 (d, J = 16.1, 1H).
Biological investigation
Proteasome purification and inhibition assays
Inhibition of the proteolytic activities of proteasomes semi-purified from lymphoblastoid cell lines as previously describedCitation28, was determined using fluorogenic substrates specific for the three main proteolytic activities. Suc-LLVY-AMC, Boc-LRR-AMC and Z-LLE-AMC were used at concentration of 100 µM to measure chymotrypsin-like, trypsin-like and caspase-like proteasome activities, respectively. Substrates were incubated, with 10 µg of proteasome pretreated with incremented concentrations (from 0.001 to 10 µM) of the C-terminal butadienyl ethyl ester partially RI peptides, in activity buffer containing 50 mM Tris–HCl (pH 7,5), 5 mM MgCl2, 500 mM EDTA (pH 8), 1 mM dithiothreitol, 2 mM ATP, at 37°C for 30 min (). Activity was evaluated in fluorescence units, and the inhibitory activity of the compounds is expressed as IC50Citation22. The data were plotted as percentage control (the ratio of percentage conversion in the presence or absence of inhibitor) versus inhibitor concentration, and fitted with the equation Y = 100/1+(X/IC 50)A, where IC50 is the inhibitor concentration at 50% inhibition and A is the slope of the inhibition curve.
Table 2. Inhibition of proteasome subunits and metabolic stability of butadienyl ethyl ester derivatives.
Enzymatic stability assays
The biostability of the selected compounds 12, 14 and 16 was studied in human plasma and the degradation half-life (T1/2) was obtained as described previouslyCitation29.
Results and discussion
C-terminal RI butadienyl ester peptides were synthesized following the strategy reported in and tested as proteasome inhibitors.
Biological evaluation of the new pseudopeptides was carried out to assess their capacity to inhibit the trypsin-like, chymotrypsin-like and post-acidic activities of the proteasome. The results obtained show that these new peptide-based molecules are poor inhibitors of the proteasome. In general, none of the compounds inhibit either the chymotrypsin-like or the trypsin-like activities of the proteasomes isolated from LCL. Likewise, inhibition of the β2 subunit was relatively unpronounced, with IC50 values of the order of 10 µM for the analogs 14 and 16. Howerer compounds 14 and 16 showed an appreciable inhibitory capacity of the postacidic activity with IC50 values around the µM range. The different biological results of the new derivatives as regards postacidic activity seem to be independent from the dipeptidic central sequence, although a more prolonged aliphatic hydrocarbon chain at the N-terminal position appears to be preferred. N-terminal substituent seems to promote the interaction with the β1 subunit, thereby confirming that a bulky substituent at the N-terminal favors β1-specific inhibitionCitation30.
The introduction of the new C-terminal pharmacophore unit with an extended conjugation determines an appreciable decrease of the activity when compared to the previous series of compounds which showed a favourable biological profile.
The susceptibility of selected derivatives 12, 14 and 16 to enzymatic hydrolysis was determined by incubation at 37°C in human plasma. The partial RI pseudopeptides showed strong enzymatic resistance to human plasma proteases as easy foreseeable by the structural modification of the canonic peptidic backbone.
Conclusions
In this work, we have designed, synthesized and defined the biological profile of a new series of C-terminal trans,trans-muconic acid ethyl ester partial RI pseudopeptides. The best derivatives of this series inhihit the β1 catalytic site of the proteasome at µM concentration. General analysis of the activity profile shows that the new compounds have a low capacity to interact with catalytic subunits of the enzymatic complex suggesting that, the C-terminal new pharmacophoric group is not a good electrophilic moiety for the nucleophilic attack by catalytic threonine. In our opinion, the major finding of this work is the insertion of a reversed peptide bond between oligopeptide sequence and C-terminal pharmacophoric unit, a structural modification that will be applied in other series of the peptide-based proteasome inhibitors.
Declaration of interest
Financial support of this work was provided by the University of Ferrara and the Ministero dell’Università e della Ricerca Scientifica e Tecnologica (MURST).
References
- Reinstein E, Ciechanover A. Narrative review: protein degradation and human diseases: the ubiquitin connection. Ann Intern Med 2006;145:676–684.
- McGrath JP, Jentsch S, Varshavsky A. UBA 1: an essential yeast gene encoding ubiquitin-activating enzyme. EMBO J 1991;10:227–236.
- Groll M, Ditzel L, Löwe J, Stock D, Bochtler M, Bartunik HD et al. Structure of 20S proteasome from yeast at 2.4 A resolution. Nature 1997;386:463–471.
- Groll M, Bajorek M, Köhler A, Moroder L, Rubin DM, Huber R et al. A gated channel into the proteasome core particle. Nat Struct Biol 2000;7:1062–1067.
- Navon A, Goldberg AL. Proteins are unfolded on the surface of the ATPase ring before transport into the proteasome. Mol Cell 2001;8:1339–1349.
- Groll M, Heinemeyer W, Jäger S, Ullrich T, Bochtler M, Wolf DH et al. The catalytic sites of 20S proteasomes and their role in subunit maturation: a mutational and crystallographic study. Proc Natl Acad Sci USA 1999;96:10976–10983.
- Borissenko L, Groll M. 20S proteasome and its inhibitors: crystallographic knowledge for drug development. Chem Rev 2007;107:687–717.
- Goldberg AL. Functions of the proteasome: from protein degradation and immune surveillance to cancer therapy. Biochem Soc Trans 2007;35:12–17.
- Britton M, Lucas MM, Downey SL, Screen M, Pletnev AA, Verdoes M et al. Selective inhibitor of proteasome’s caspase-like sites sensitizes cells to specific inhibition of chymotrypsin-like sites. Chem Biol 2009;16:1278–1289.
- Kisselev AF, Goldberg AL. Proteasome inhibitors: from research tools to drug candidates. Chem Biol 2001;8:739–758.
- Hines J, Groll M, Fahnestock M, Crews CM. Proteasome inhibition by fellutamide B induces nerve growth factor synthesis. Chem Biol 2008;15:501–512.
- Adams J. The development of proteasome inhibitors as anticancer drugs. Cancer Cell 2004;5:417–421.
- Demo SD, Kirk CJ, Aujay MA, Buchholz TJ, Dajee M, Ho MN et al. Antitumor activity of PR-171, a novel irreversible inhibitor of the proteasome. Cancer Res 2007;67:6383–6391.
- Nazif T, Bogyo M. Global analysis of proteasomal substrate specificity using positional-scanning libraries of covalent inhibitors. Proc Natl Acad Sci USA 2001;98:2967–2972.
- Fenteany G, Standaert RF, Lane WS, Choi S, Corey EJ, Schreiber SL. Inhibition of proteasome activities and subunit-specific amino-terminal threonine modification by lactacystin. Science 1995;268:726–731.
- Groll M, Huber R, Potts BC. Crystal structures of Salinosporamide A (NPI-0052) and B (NPI-0047) in complex with the 20S proteasome reveal important consequences of beta-lactone ring opening and a mechanism for irreversible binding. J Am Chem Soc 2006;128:5136–5141.
- Groll M, Schellenberg B, Bachmann AS, Archer CR, Huber R, Powell TK et al. A plant pathogen virulence factor inhibits the eukaryotic proteasome by a novel mechanism. Nature 2008;452:755–758.
- Kisselev AF, van der Linden WA, Overkleeft HS. Proteasome inhibitors: an expanding army attacking a unique target. Chem Biol 2012;19:99–115.
- Groll M, Huber R, Moroder L. The persisting challenge of selective and specific proteasome inhibition. J Pept Sci 2009;15:58–66.
- Genin E, Reboud-Ravaux M, Vidal J. Proteasome inhibitors: recent advances and new perspectives in medicinal chemistry. Curr Top Med Chem 2010;10:232–256.
- Kohno J, Koguchi Y, Nishio M, Nakao K, Kuroda M, Shimizu R et al. Structures of TMC-95A-D: novel proteasome inhibitors from Apiospora montagnei sacc. TC 1093. J Org Chem 2000;65:990–995.
- Marastoni M, Baldisserotto A, Canella A, Gavioli R, Risi CD, Pollini GP et al. Arecoline tripeptide inhibitors of proteasome. J Med Chem 2004;47:1587–1590.
- Marastoni M, Baldisserotto A, Trapella C, Gavioli R, Tomatis R. P3 and P4 position analysis of vinyl ester pseudopeptide proteasome inhibitors. Bioorg Med Chem Lett 2006;16:3125–3130.
- Baldisserotto A, Ferretti V, Destro F, Franceschini C, Marastoni M, Gavioli R et al. Alpha,beta-unsaturated N-acylpyrrole peptidyl derivatives: new proteasome inhibitors. J Med Chem 2010;53:6511–6515.
- Vlieghe P, Lisowski V, Martinez J, Khrestchatisky M. Synthetic therapeutic peptides: science and market. Drug Discov Today 2010;15:40–56.
- Chorev M. The partial retro-inverso modification: a road traveled together. Biopolymers 2005;80:67–84.
- Pallai P, Goodman M. Preparation of optically pure monoacyl 2-alkyl gem-diamines from peptide amides. J Chem: Soc Chem Commun 1982;5:280–281.
- Gavioli R, Vertuani S, Masucci MG. Proteasome inhibitors reconstitute the presentation of cytotoxic T-cell epitopes in Epstein-Barr virus-associated tumors. Int J Cancer 2002;101:532–538.
- Marastoni M, Baldisserotto A, Cellini S, Gavioli R, Tomatis R. Peptidyl vinyl ester derivatives: new class of selective inhibitors of proteasome trypsin-like activity. J Med Chem 2005;48:5038–5042.
- Lei B, Abdul Hameed MD, Hamza A, Wehenkel M, Muzyka JL, Yao XJ et al. Molecular basis of the selectivity of the immunoproteasome catalytic subunit LMP2-specific inhibitor revealed by molecular modeling and dynamics simulations. J Phys Chem B 2010;114:12333–12339.