Abstract
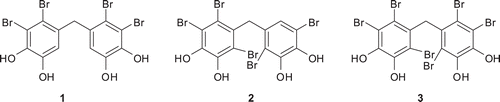
Three novel bromophenols 10–12 were synthesized. Acylation of veratrole (4) with 2,3-dimethoxy benzoic acid (5) gave a kown diarylmethanone 6. Bromination of 6 with different equivalents of molecular bromine afforded new di and tribrominated compounds 7–9 which were converted to their corresponding bromophenols 10–12 via O-demethylation with BBr3. Paraoxonase-1 (PON1) was purified from human serum with approximately 42% and 3584 U × mg−1 specific activity. The synthesized compounds 6–12 showed inhibitory effects on paraoxonase-1 (PON1) which is an organophosphate (OP) hydrolyser and an antioxidant bioscavenger enzyme. IC50 values were determined in the range of 0.123–1.212 mM.
Graphical abstract
Introduction
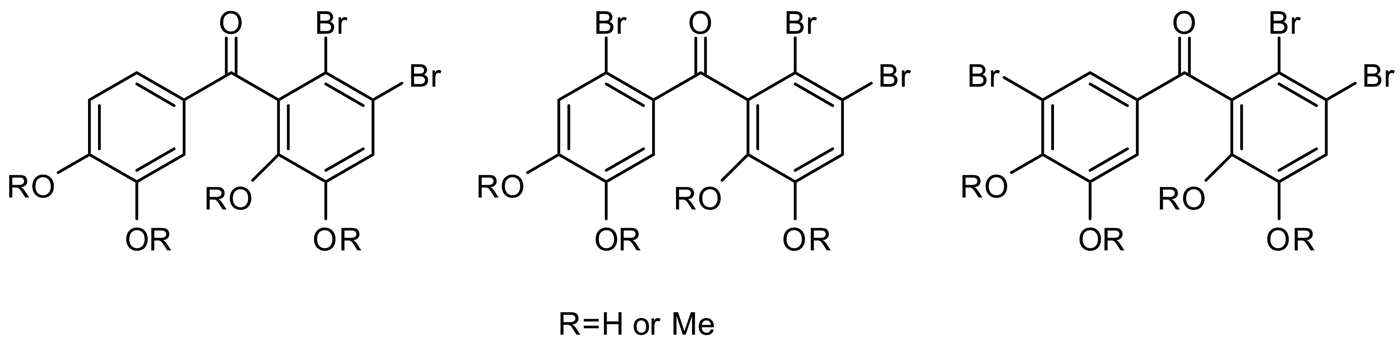
Among natural organohalogen compounds, bromophenols are abundantly found in marine life and frequently isolated from red algae of the family RhodomelaceaeCitation1. Recent studies revealed that the marine bromophenols exhibit a wide spectrum of beneficial biological activitiesCitation2. These compounds have attracted much attention in the field of functional food and pharmaceutical agentsCitation2. It has been reported that naturally occurring bromophenol 1 shows enzyme inhibitionCitation3, cytotoxicityCitation4, feeding deterrentCitation5, anti-microbialCitation6, anti-cancerCitation7 and anti-oxidantCitation8 activities. Aldose reductaseCitation9 and carbonic anhydraseCitation10 inhibitory properties of natural products 2–3 have also been noted ().
Figure 1. Some natural bromophenols.
The high-density lipoprotein associated serum esterase, paraoxonase (arylesterase, EC 3.1.8.1, PON1) is a key enzyme for protection against atherosclerosis in metabolism. The enzyme includes two Ca2+ ions in its 3D-structure. A Ca2+ is in active region of the enzyme and another one is structural. Each Ca2+ ions are important catalytic activity of the enzyme. If one of the Ca2+ ions is removed from enzyme structure, the enzyme loses its catalytic activity, intrestinglyCitation11–13. PON1 with 354 amino acids has three activities including paraoxonase, arylesterase and diazoxon activitiesCitation14. The enzyme is called as paraoxonase since it was first characterized as a hydrolyzer for OPs including paraoxon and its physiological substrate is unknown. PON1 is an esterase enzyme. It hydrolyses O–P ester linkage in paraoxonCitation15. Paraoxon is also a strong acetylcholine esterase inhibitor and is an insecticide. The compound is formed from mitochondrial oxidation of parathion. This process is drived by cytochrome p450 enzyme system in the metabolismCitation16. Moreover, PON1 enzyme has detoxifying, anti-atherogenic and antioxidant effectsCitation17. In addition to paraoxon substrate, the enzyme can hydrolyze many OPs, nerve agents like soman and sarin, insecticides such as chlorpyrifos, oxon and diazoxon and some synthetic esters like phenyl-acetateCitation17. These inhibitions on the enzyme are crucial for especially atherosclerotic lesions. Because, inhibition of PON1 results increasing oxidative stress and oxidized low density lipoproteins (LDL), which is called bad cholesterol. PON1 gain its antioxidant property from cystein 284 in its structureCitation12. Therefore, decreased activity of PON1 has been acknowledged as a risk factor for cardiovascular disease and OP toxicityCitation15.
In the present study we focused on the synthesis and in vitro paroxonase activity of compounds 6–12.
Materials and methods
All chemicals and solvents are commercially available. All solvents (EtOAc, CH2Cl2, hexane) were distilled and dried according to standard procedures. Silica gel (SiO2, 60 mesh; Merck, Darmstadt, Germany) was used for column chromatography (CC). 1 mm of SiO2 60 PF (Merck) on glass plates were used for preparative thick layer chromatography. Mp of all compounds was determined with cap. melting-point apparatus (BUCHI 530; Flawil, Switzerland) and uncorrected. IR Spectra were recorded as solns. in 0.1 mm cells with a Mattson 1000 FT-IR spectrophotometer (Cambridge, England). 1H- and 13C-NMR spectra were recorded on 400 (100)-MHz Varian spectrometer (Danbury, CT) in deuterated solvents (CDCl3 and CD3COCD3) with Tetramethylsilane (TMS) as an internal standard for protons and solvent signals, as internal standard for carbon spectra. Chemical shift values were mentioned δ in ppm. Elemental analyses were recorded on Leco CHNS-932 apparatus (MI). Paroxonase activities of samples were determined on a spectrophotometer (UV-1208, Shimadzu, Japan).
Synthesis of (2,3-dimethoxyphenyl)(3,4-dimethoxyphenyl)methanone (6)
Polyphosphoric acid (PPA), prepared from concentration H3PO4 (85%, 9.40 g, 81.60 mmol) and P2O5 (16.9 g, 120.04 mmol) was heated to 80°C in a beaker (100 mL). Veratrole (4) (2.3 g, 16.47 mmol) and 2,3-dimethoxybenzoic acid (5) (3.00 g, 16.47 mmol) were added to this mixture quickly. The mixture was stirred with a glass stick at 80°C for 45 min. It was poured onto 35 mL of ice/water carefully. The organic phase was extracted with EtOAc (3 × 50 mL). The combined organic layers were dried over Na2SO4 and the solvent was evaporated. Crystallization of the residue with EtOAc-hexane gave 6Citation18 as white crystals (4.2 g, 85% yield). Mp 124–126°C. 1H-NMR (400 MHz, CDCl3), δ 7.56 (d, J = 1.8 Hz, 1H), 7.25 (dd, J = 1,8 Hz, J = 8.4 Hz, 1H), 7.09 (dd, J = 7.7 Hz, J = 8.1 Hz, 1H), 7.01 (dd, J = 1.5 Hz, J = 8.1 Hz, 1H), 6.86 (dd, J = 1.5 Hz, J = 7.7 Hz, 1H), 6.79 (d, J = 8.4 Hz, 1H), 3.91 (s, methoxide, 3H), 3.90 (s, methoxide, 3H), 3.89 (s, methoxide,3H), 3.73 (s, methoxide, 3H). 13C-NMR (100 MHz, CDCl3), δ 195.03 (CO), 153.78 (C), 152.94 (C), 149.2 (C), 146.82 (C), 134.74 (C), 130.67 (C), 126.44 (CH), 124.10 (CH), 120.47 (CH), 114.12 (CH), 111.08 (CH), 110.00 (CH), 61.95 (OCH3), 56.27 (OCH3), 56.20 (OCH3), 56.13 (OCH3). Anal. Calcd for C17H18O5: C 67.54, H 6.00; found: C 67.58, H 6.09.
Synthesis of (2,3-dibromo-5,6-dimethoxyphenyl)(3,4-dimethoxyphenyl)methanone (7)
A solution of bromine (0.64 g, 3.97 mmol) in CH2Cl2 (15 mL) was added to a stirred solution of benzophenone 14 (1.00 g, 3.31 mmol) in CH2Cl2 (30 mL) drop wise at room temperature (RT) over 10 min. After the reaction mixture was stirred at RT for 24 h, the solvent was evaporated. Chromatography of the residue on silica gel (SiO2, 100 g) with ethyl acetate/hexane (1:4) yielded 7 as white crystals (0.72 g, 47%). Mp 138–140°C. 1H-NMR (400 MHz, CDCl3), δ 7.59 (d, J = 2.2 Hz, 1H), 7.24 (s, 1H), 7.17 (dd, A part of AB system, J = 2.2 Hz, J = 8.4 Hz, 1H), 6.92 (d, B part of AB system, J = 8.4 Hz, 1H), 3.94 (s, methoxide, 3H), 3.91 (s, methoxide, 3H), 3.88 (s, methoxide, 3H), 3.70 (s, methoxide, 3H). 13C-NMR (100 MHz, CDCl3), δ 191.87 (CO), 154.45 (C), 152.71 (C), 149.68 (C), 146.20 (C), 137.54 (C), 129.19 (C), 126.10 (CH), 120.14 (C), 117.81 (CH), 111.79 (C), 110.62 (CH), 110.38 (CH), 61.91 (OCH3), 56.52 (OCH3), 56.30 (OCH3), 56.25 (OCH3). Anal. Calcd for C17H16Br2O5: C 44.38, H 3.5; found: C 44.47, H 3.67. IR (CH2Cl2, cm−1): 2938, 1666, 1585, 1513, 1464, 1419, 1300, 1268, 1143, 1003.
Bromination of compound 6 with 4 equiv bromine
A solution of bromine (2.08 g, 13.00 mmol) in CH2Cl2 (20 mL) was added to a solution of benzophenone 14 (1.00 g, 3.31 mmol) in CH2Cl2 (30 mL) drop wise at RT over 20 min. The reaction mixture was stirred at RT for 24 h, and then the solvent was evaporated. Chromatography of the residue on silica gel (SiO2, 100 g) column with ethyl acetate/hexane (1:5) gave 7 (0.61 g, 40% yield), 8 (0.46 g, 26% yield) and 9 (0.36 g, 20% yield), respectively.
(2-Bromo-4,5-dimethoxyphenyl)(2,3-dibromo-5,6-dimethoxyphenyl)methanone (8)
White crystals, Mp 142–144°C. 1H-NMR (400 MHz, CDCl3), δ 7.31 (s, 1H), 7.23 (s, 1H), 7.08 (s, 1H), 3.92 (s, methoxide, 3H), 3.88 (s, methoxide, 3H), 3.84 (s, methoxide, 3H), 3.71 (s, methoxide, 3H). 13C-NMR (100 MHz, CDCl3), δ 190.99 (CO), 153.24 (C), 152.76 (C), 148.47(C), 146.30 (C), 138.22 (C), 128.86 (C), 120.25 (C), 118.05 (CH), 117.50 (CH), 115.23 (C), 114.95 (CH), 112.13 (C) 61.72 (OCH3), 56.61 (OCH3), 56.52 (OCH3), 56.47 (OCH3). Anal. Calcd for C17H15Br3O5: C 37.88, H 2.80; found: C 37.88, H 2.74. IR (CH2Cl2, cm−1): 2938, 2841, 1681, 1588, 1508, 1465, 1419, 1367, 1337, 1298, 1260, 1219, 1199, 1164, 1041, 1005, 925, 841.
(3-Bromo-4,5-dimethoxyphenyl)(2,3-dibromo-5,6-dimethoxyphenyl)methanone (9)
White crystals, Mp 149–151°C. 1H-NMR (400 MHz, CDCl3), δ 7.53 (d, J = 1.8 Hz, 1H), 7.35 (d, J = 1.8 Hz, 1H), 7.27 (s, 1H), 3.93 (s, methoxide, 3H), 3.92 (s, methoxide, 3H), 3.90 (s, methoxide, 3H), 3.71 (s, methoxide, 3H). 13C-NMR (100 MHz, CDCl3), δ 191.23 (CO), 154.08 (C), 152.69 (C), 151.71(C), 146.19 (C), 136.53 (C), 132.52 (C), 128.07 (CH), 120.31 (C), 118.19 (CH), 111.75 (C), 111.62 (CH), 111.42 (C) 61.92 (OCH3), 60.98 (OCH3), 56.54 (OCH3), 56.49 (OCH3). Anal. Calcd for C17H15Br3O5: C 37.88, H 2.80; found: C 37.87, H 2.94. IR (CH2Cl2, cm−1): 2939, 1677, 1562, 1465, 1419, 1403, 1280, 1551, 1082, 1044, 1004.
General procedure for the synthesis of bromophenols 10–12
A solution of BBr3 (0.60 mL) in CH2Cl2 (10.0 mL) was added to a stirred solution of dibromide 7 (0.40 g, 0.87 mmol) in CH2Cl2 (15 mL) drop wise at 0°C under N2(g) over 5–10 min. The reaction mixture was stirred at the same temperature for 2 h and then at RT for 1 day under N2. The reaction was monitored by TLC and after the reaction was completed, methanol (35 mL) was slowly added over 15 min to the mixture at 0°C. The solvent was evaporated, water (45 mL) and EtOAc (40 mL) were added to the residue. The organic layer was separated and the water phase was extracted with EtOAc (2 × 40 mL). The combined organic layers were dried over Na2SO4 and the evaporation of the solvent gave bromophenol 10 (0.34 g, 96%). Compounds 11 (from 8, 98% yield) and 12 (from 9, 98% yield) were also synthesized by the same procedure.
(2,3-Dibromo-5,6-dihydroxyphenyl)(3,4-dihydroxyphenyl)methanone (10)
Pale yellow solid. Mp 228–230°C. 1H-NMR (400 MHz, CD3COCD3), δ 9.36 (bs, OH, 1H), 8.80 (bs, OH, 1H), 8.45 (bs, OH, 1H), 8.26 (bs, OH, 1H), 7.35 (d, J = 1.8 Hz, 1H), 7.29 (s,1H), 7.23 (dd, A part of AB system, J = 1.8 Hz, J = 8.4 Hz, 1H), 6.92 (d, B part of AB system, J = 8.4 Hz, 1H). 13C-NMR (100 MHz, CD3COCD3), δ 191.18 (CO), 151.20 (C), 145.79 (C), 145.32 (C), 143.41 (C), 131.53 (C), 129.05 (C), 123.77 (CH), 119.52 (CH), 116.08 (CH), 115.42 (CH), 113.69 (C), 110.34 (C). Anal. Calcd for C13H8Br2O5: C 38.65, H 2.00; found: C 38.55, H 2.09. IR (Acetone, cm−1): 3198, 2947, 1644, 1589, 1522, 1443, 1402, 1300, 1122, 953, 846.
(2-Bromo-4,5-dihydroxyphenyl)(2,3-dibromo-5,6-dihydroxyphenyl)methanone (11)
Brownish solid. Mp 227–229°C. 1H-NMR (400 MHz, CD3COCD3), δ 9.40 (bs, OH, 1H), 9.15 (bs, OH, 1H), 8.63 (bs, OH, 1H), 8.41 (bs, OH, 1H), 7.29 (s, 1H), 7.20 (s, 1H), 7.16 (s, 1H). 13C-NMR (100 MHz, CD3COCD3), δ 190.13 (CO), 150.44 (C), 145.92 (C), 144.43 (C), 143.67 (C), 131.72 (C), 127.62 (C), 121.97 (CH), 120.17 (CH), 119.81 (CH), 113.89 (C), 112.46 (C), 110.54 (C). Anal. Calcd for C13H7Br3O5: C 32.33, H 1.46 found: C 32.28, H 1.50. IR (Acetone, cm−1): 3228, 2955, 1659, 1585, 1505, 1403, 1364, 1284, 1217, 1053, 972, 853, 810, 707, 672.
(3-Bromo-4,5-dihydroxyphenyl)(2,3-dibromo-5,6-dihydroxyphenyl)methanone (12)
Brownish solid. Mp 237–239°C. 1H-NMR (400 MHz, CD3COCD3), δ 9.43 (bs, OH, 1H), 9.25 (bs, OH, 1H), 9.05 (bs, OH, 1H), 8.38 (bs, OH, 1H), 7.49 (d, J = 1.8 Hz, 1H), 7.33 (d, J = 1.8 Hz, 1H), 7.32 (s,1H). 13C-NMR (100 MHz, CD3COCD3), δ 190.37 (CO), 148.76 (C), 145.88 (C), 145.85 (C), 143.53 (C), 130.63 (C), 129.25 (C), 126.38 (CH), 119.86 (CH), 115.03 (CH), 113.84 (C), 110.13 (C), 109.37(C). Anal. Calcd for C13H7Br3O5: C 32.33, H 1.46; found: C 32.32, H 1.46. IR (Acetone, cm−1): 3230, 2963, 1645, 1582, 1489, 1426, 1403, 1302, 966, 852, 727.
X-ray structure determination
For the crystal structure determination, the single-crystal of the compounds 7, 8 and 9 was used for data collection on a four-circle Rigaku R-AXIS RAPID-S diffractometer (equipped with a 2D-area IP detector). The graphite-monochromatized Mo Kα radiation (λ = 0.71073 Å) and oscillation scans technique with Δω = 5° for one image were used for data collection. The lattice parameters were determined by the least-squares methods on the basis of all reflections with F2 > 2σ(F2). Integration of the intensities, correction for Lorentz and polarization effects and cell refinement was performed using CrystalClear (Rigaku/MSC Inc., 2005) software. The structures were solved by direct methods using SHELXS-97 and refined by a full-matrix least-squares procedure using the program SHELXL-97. H atoms were positioned geometrically and refined using a riding model. Final difference Fourier maps showed no peaks of chemical significance. Crystal data for dibromide 7: C17H16Br2O5, crystal system, space group: monoclinic, P21/n; (no:14); unit cell dimensions: a = 8.4968(2), b = 9.4886(2), c = 22.2020(6) Å, β = 92.02(2)˚; volume: 1788.88(8)Å3; Z = 4; calculated density: 1.708 g/cm3; absorption coefficient: 4.554/mm−1; F(000): 912; θ range for data collection 2.3–30.6°; refinement method: full-matrix least-square on F2; data/parameters: 4195/218; goodness-of-fit on F2: 1.271; final R indices [I > 2σ(I)]: R1 = 0.083, wR2 = 0.189; largest diff. peak and hole: 0.458 and −0.463 e Å−3; CCDC-877660. Crystal data for tribromide 8: C17H15Br3O5, crystal system, space group: monoclinic, P21/c; (no:14); unit cell dimensions: a = 9.4465(7), b = 21.0003(18), c = 9.6253(9) Å, α = 90 β = 106.08(4), γ = 90°; volume: 1834.8(3)Å3; Z = 4; calculated density: 1.951 g/cm3; absorption coefficient: 6.620/mm−1; F(000): 1048; θ range for data collection 2.4–26.4°; refinement method: full-matrix least-square on F2; data/parameters: 2604/228; goodness-of-fit on F2: 1.252; final R indices [I > 2σ(I)]: R1 = 0.0865, wR2 = 0.197; largest diff. peak and hole: 0.773 and −0.111 e Å−3; CCDC-878580. Crystal data for tribromide 9: C17H15Br3O5, crystal system, space group: monoclinic, P21/c; (no:14); unit cell dimensions: a = 13.5992(5), b = 9.4949(2), c = 14.7357(7) Å, α = 90 β = 104.37(2), γ = 90°; volume: 1843.23(12)Å3; Z = 4; calculated density: 1.942 g/cm3; absorption coefficient: 6.590/mm−1; F(000): 1048; θ range for data collection 2.6–30.9°; refinement method: full-matrix least-square on F2; data/parameters: 2668/226; goodness-of-fit on F2: 1.078; final R indices [I > 2σ(I)]: R1 = 0.0957, wR2 = 0.108; largest difference peak and hole: 0.438 and −0.766 e Å−3; CCDC-879013.
Paroxonase activity of compounds 6, 7, 8, 9, 10, 11, 12
Enzyme activity assay
The enzyme assay was based on the estimation of p-nitrophenol at 412 nm. Paraoxonase activity was determined spectrophotometrically using the same procedure in our previous studiesCitation19. One enzyme unit was defined as the amount of enzyme that catalyzes the hydrolysis of 1 μmol of substrate at 25°C.
Purification of PON1
Twenty five milliliters of human serum was precipitated with ammonium sulphate. The precipitation intervals for paraoxonase were 60–80%Citation20. The precipitate was collected by centrifugation at 15,000 rpm for 20 min and redissolved in 100 mM Na-phosphate buffer (pH 7.0). The enzyme solution, which had been dialyzed in the presence of 1 mM Na-phosphate buffer (pH 7.0) at 4°C, was loaded onto the DEAE-Sephadex A50 anion exchange column (3 cm2 × 30 cm), which had been equilibrated with 100 mM Na-phosphate buffer (pH 7.0). The column procedure was applied like previous studyCitation21. Enzyme solution from the DEAE-Sephadex A50 column were mixed with glycerol and loaded onto the Sephadex G-200 chromatography (60 cm × 2 cm2). For each fraction qualitative protein assay was performed at 280 nm and activity determination at 412 nm spectrofotometrically. Active fractions were combined for other kinetic studiesCitation12.
Quantitative protein assay
Quantitative presence of protein in the solution was determined at 595 nm spectrophotometricallyCitation22.
SDS polyacrylamide gel electrophoresis
The purifiy of PON1 was controlled by SDS polyacrylamide gel electrophoresis as in previous studyCitation23.
In vitro studies of synthesized compounds
We examined the inhibitory effects of novel synthesized compounds 6, 7, 8, 9, 10, 11, 12. PON activities were measured in the presence of different compound concentrations. Control activity was assumed to be 100% in the absence of inhibitor. Experiments of all compounds were tested in triplicate at each concentration used and Activity%-[Chemical] graphs were drawn for each compoundsCitation12.
Results and discussion
Alkylation or acylation of aromatic compounds with PPA (plyphosphoric acid) prepared from phosphoric acid and P2O5 have been given in the literature clearlyCitation24. By applying the same procedure, acylation of veratrole (4) with 2,3-dimethoxybenzoic acid (5) in PPA at 80°C furnished 6Citation18 with a yield of 85%. As it is known, bromination of aromatic compounds can be achieved by either N-bromosuccinimide (NBS)Citation25, and LiBr/Ceric ammonium nitrateCitation8, or molecular bromine (Br2)Citation26. Here, Br2 was chosen as brominating agent. Bromination of benzophenon 6 with 1.2 eq Br2 in CH2Cl2 at RT followed by chromatographic separation of the residue gave 7 in 47% yield. The bromination of 6 with excess amount of Br2 (4 eq) in CH2Cl2 at RT was also carried out. Dibrominated product 7, three brominated pruducts 8 and 9 were synthesized in yields of 40, 26 and 20%, respectively by the bromination of 6 with 4 eq Br2 (). NMR analysis of compounds 7, 8 and 9 did not allow us to determine their exact structures. Therefore, the structures of these compounds were determined by X-ray diffraction analysis ().
Scheme 1. Synthesis of novel bromophenols (i) PPA, 80°C, 85%; (ii) 1.2 eq Br2/CH2Cl2, 25°C, 47%; (iii) 4 eq Br2/CH2Cl2, 25°C; (iv) 1. BBr3/CH2Cl2, 0–25°C, 2. MeOH or H2O, 0–25°C.
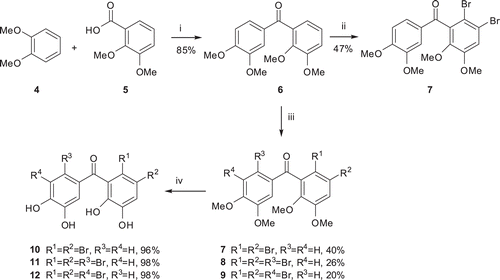
Figure 2. The molecular structures of the compounds 7, 8 and 9, showing the atom numbering scheme.
Interestingly, further analysis of the X-ray crystal structure revealed the presence of oxygen bromine halogen bonding. This intermolecular attraction was observed between the bromine atom and the oxygen atom of the carbonyl (for 9) and OMe groups (for 7 and 8). The geometrical parameters of the contacts included an interatomic distance (Br…O) of 3.36, 3.29 and 3.05 Å and a bond angle (C–Br…O) of 146.7, 152 and 172.1° for the structure 7, 8 and 9, respectively. The Br…O distances were smaller than the sum of the van der Waals radius (3.37 Å) for Br…O, especially for the structure 9 suggesting the presence of halogen bonding. The halogen bonding is typically observed between a polarized halogen atom (a Lewis acid) and negatively-charged oxygen, nitrogen or sulfur atom (a Lewis base). Oxygen-derived Lewis bases include the carbonyl, hydroxyl, charged carboxylate and phosphate groups. Halogen bonding has been reported to exist in many biological molecules, such as protein and nucleic acid structures, and has also been demonstrated to play significant roles in intermolecular recognition and self-assembly processesCitation27.
Cleavage of ethers is an integral functional group transformation, as it is important for the synthesis of natural products, pharmaceuticals and fine chemicals. Cleavage of alkyl aryl ethers with HBrCitation28 or BBr3Citation8,Citation10,Citation24 are well-known in the literature. In the present study, O-demethylation of aryl methyl ethers was carried out according to the procedure described by us previouslyCitation8,Citation10,Citation24. Reaction of 7–9 with BBr3 in CH2Cl2 at 0–25°C under N2 followed by addition of H2O or MeOH provided novel synthetic bromopehols 10, 11 and 12 with yields of 96, 98 and 98%, respectively ().
PON1 were purified with three steps from human serum by ammonium sulfate precipitation, DEAE-Sephadex A50 ion exhange chromatography and Sephadex G-200 gel filtration chromatography (). The purification results for PON1 depicted a 42.1% yield, 3584 U × 1/mg−1 proteins specific activity and ~230 purification fold. However, we did not encounter any study about paraoxonase and novel synthesized compounds interactions. In the present study, our compounds indicate inhibitory effects on PON1. All compounds inhibited the enzyme activity and IC50 values for 6, 7, 8, 9, 10, 11, 12 were calculated as 1.212, 0.174, 0.123, 0.125, 0.832, 1.110, 0.701 mM, respectively (). Inhibition rank can expressed as 8>9>7>12>10>11>6.
Table 1 Purification steps of PON1 enzyme from human serum by Ammonium sulfate precipitation, anion exchange chromatography and gel filtration chromatography.
Table 2. IC50 values of chemicals on PON1.
Conclusions
In summary, starting from veratrole (4) and 2,3-diemthoxybenzoic acid (5) three bromophenols 10–12 were synthesized for the first time, the procursores of these compounds can be important synthons for further biological and synthetic purposes. In addition, PON1 enzyme was purified by simple chromatographic methods, rapidly. Enzyme-chemical compound interaction experiments were performed and IC50 values were calculated from Activity%-[bromophenol] curves. All synthesized compounds 6–12 used in this study affect the activity of PON1 because of the presence of the different functional groups (OMe, OH and Br) present in their aromatic scaffold. Our findings here indicate that these compounds and their derivatives may be used as PON1 inhibitors.
Acknowledgment
Authors are indebted to the Department of Chemistry (Atatürk University) for research conditions.
Declaration of interest
This research was financed by grants from TÜBİTAK (The Scientific and Technological Research Council of Turkey) (Project no: TBAG-107T348).
References
- Gribble GW. The diversity of naturally occurring organobromine compounds. Chem Soc Rev 1999;28:335–346.
- Liu M, Hansen PE, Lin X. Bromophenols in marine algae and their bioactivities. Mar Drugs 2011;9:1273–1292.
- Lee HS, Lee TH, Lee JH, Chae CS, Chung SC, Shin DS et al. Inhibition of the pathogenicity of Magnaporthe grisea by bromophenols, isocitrate lyase inhibitors, from the red alga Odonthalia corymbifera. J Agric Food Chem 2007;55:6923–6928.
- Xu X, Song F, Wang S, Li S, Xiao F, Zhao J et al. Dibenzyl bromophenols with diverse dimerization patterns from the brown alga Leathesia nana. J Nat Prod 2004;67:1661–1666.
- Kurata K, Taniguchii K, Takashima K, Hayashi I, Suzuki M. Feeding-deterrent bromophenols from Odonthalia corymbifera. Phytochemistry 1997;45:485–487.
- Oh KB, Lee JH, Chung SC, Shin J, Shin HJ, Kim HK et al. Antimicrobial activities of the bromophenols from the red alga Odonthalia corymbifera and some synthetic derivatives. Bioorg Med Chem Lett 2008;18:104–108.
- Shi D, Li J, Guo S, Su H, Fan X. The antitumor effect of bromophenol derivatives in vitro and Leathesia nana extract in vivo. Chin J Ocean Lim 2009;27:277–282.
- Balaydin HT, Gülçin Í, Menzek A, Göksu S, Sahin E. Synthesis and antioxidant properties of diphenylmethane derivative bromophenols including a natural product. J Enzyme Inhib Med Chem 2010;25:685–695.
- Wang W, Okada Y, Shi H, Wang Y, Okuyama T. Structures and aldose reductase inhibitory effects of bromophenols from the red alga Symphyocladia latiuscula. J Nat Prod 2005;68:620–622.
- Balaydin HT, Soyut H, Ekinci D, Göksu S, Beydemir S, Menzek A et al. Synthesis and carbonic anhydrase inhibitory properties of novel bromophenols including natural products. J Enzyme Inhib Med Chem 2012;27:43–50.
- Harel M, Aharoni A, Gaidukov L, Brumshtein B, Khersonsky O, Meged R et al. Structure and evolution of the serum paraoxonase family of detoxifying and anti-atherosclerotic enzymes. Nat Struct Mol Biol 2004;11:412–419.
- Alici HA, Ekinci D, Beydemir S. Intravenous anesthetics inhibit human paraoxonase-1 (PON1) activity in vitro and in vivo. Clin Biochem 2008;41:1384–1390.
- Ates O, Azizi S, Alp HH, Kiziltunc A, Beydemir S, Cinici E et al. Decreased serum paraoxonase 1 activity and increased serum homocysteine and malondialdehyde levels in age-related macular degeneration. Tohoku J Exp Med 2009;217:17–22.
- Isgör MM, Beydemir S. Some cardiovascular therapeutics inhibit paraoxonase 1 (PON1) from human serum. Eur J Pharmacol 2010;645:135–142.
- Aviram M, Rosenblat M, Bisgaier CL, Newton RS, Primo-Parmo SL, La Du BN. Paraoxonase inhibits high-density lipoprotein oxidation and preserves its functions. A possible peroxidative role for paraoxonase. J Clin Invest 1998;101:1581–1590.
- La Du BN. (1992). Pharmo genetics of drug metabolism. New York: Pergamon Press, 51–91.
- Mackness MI, Durrington PN. HDL, its enzymes and its potential to influence lipid peroxidation. Atherosclerosis 1995;115:243–253.
- Hosangadi BD, Kasbekar AB, Nabar MJ, Desai RC. Novel reactions with polyphosphoric acid II. Decarboxylative acetylation, trans-carbonylation, and other reactions of substituted aromatic carboxylic acids. Ind J Chem 1973;11:711–713.
- Ekinci D, Beydemir S. Evaluation of the impacts of antibiotic drugs on PON 1; a major bioscavenger against cardiovascular diseases. Eur J Pharmacol 2009;617:84–89.
- Ekinci D, Beydemir S. Effect of some analgesics on paraoxonase-1 purified from human serum. J Enzyme Inhib Med Chem 2009;24:1034–1039.
- Senturk M, Ekinci D, Alıcı HA, Beydemir S. Paraoxonase-1, an organophosphate detoxifier and cardioprotective enzyme, is inhibited by anesthetics: An in vitro and in vivo insight. Pestic Biochem Phys 2011;101:206–211.
- Yilmaz H, Ciftci M, Beydemir S, Bakan E. Purification of glucose 6-phosphate dehydrogenase from chicken erythrocytes. investigation of some kinetic properties. Prep Biochem Biotechnol 2002;32:287–301.
- Soyut H, Beydemir S, Hisar O. Effects of some metals on carbonic anhydrase from brains of rainbow trout. Biol Trace Elem Res 2008;123:179–190.
- Balaydin HT, Akbaba Y, Menzek A, Sahin E, Göksu S. First and short syntheses of biologically active, naturally occurring brominated mono- and dibenzyl phenols. Arkivoc 2009;XIV:75–87.
- Göksu S, Seçen H. Concise syntheses of 2-aminoindans via indan-2-ol. Tetrahedron 2005;61:6801–6807.
- Göksu S, Uğuz MT, Özdemir H, Seçen H. A concise synthesis and the antibacterial activity of 5,6-dimethoxynaphthalene-2-carboxylic acid. Turk J Chem 2005;29:199–205.
- Lu Y, Shi T, Wang Y, Yang H, Yan X, Luo X et al. Halogen bonding–a novel interaction for rational drug design? J Med Chem 2009;52:2854–2862.
- Göksu S, Seçen H, Sütbeyaz Y. An alternative synthesis of the dopaminergic drug 2-amino-1,2,3,4-tetrahydronaphthalene-5,6-diol (5,6-ADTN). Helv Chim Acta 2006;89:270–273.