Abstract
In this study, the synthesis and potential enzyme interactions of new Pyrrolo[2,3-d]pyrimidine derivatives along with their inhibitory activity against SFK enzymes such as Fyn, Lyn, Hck, and c-Src were reported. The results indicated that compounds were slightly active of tested SFK enzymes in comparison with PP2 for Fyn, A-419259 for Lyn and CGP77675 for c-Src. Compound N-((2-amino-4-oxo-4,7-dihydro-3H-pyrrolo[2,3-d]pyrimidin-5-yl)methyl)-4-(3,4-dimethoxyphenyl)butanamide (5) was identified as a non-selective slight inhibitor against Fyn, Lyn and c-Src. However, compounds did not show any inhibitory effects on Hck. Docking studies were performed to analyze the binding mode of compounds against SFKs. The best interaction was obtained between compound 5 and the active site of Fyn and c-Src enzymes in comparison with reference compounds PP2 and CGP77675, respectively.
Introduction
Src family kinases (SFKs) are involved in a broad spectrum of cellular processes, such as cell growth, differentiation, survival, adhesion, and migrationCitation1,Citation2. Many evidences showed that abnormal SFK signalling participates in the pathogenesis of many diseases including cancers, osteoporosis, inflammation-mediated bone loss, rheumatoid arthritis, Alzheimer’s and Parkinson’s diseaseCitation3–6. The activation of SFK in human cancers may occur through a variety of mechanisms that include domain interaction and structural remodelling in response to various activators or upstream kinases and phosphatasesCitation7. The Src family of cytoplasmic protein tyrosine kinases includes Src, Lck, Fyn, Lyn, Hck, Fgr, Blk, Yrk, and YesCitation8. The prototype member of SFKs, c-Src (Src) is frequently overexpressed and activated in a variety of epithelial and non-epithelial cancers such as colorectal, breast, ovarian, lung and pancreatic cancer and melanomasCitation9–11. Src also plays prominent roles in invasion, tumour progression and development of metastasis, therefore Src is a promising target for cancer theraphyCitation12–15. Lck is a positive activator in T-cell signalling (TCR), and its activation is involved in autoimmune diseases such as rheumatoid arthritis, organ transplant rejection and asthma. TCR signals initiated by Lck ultimately lead to gene regulation events triggering cytokine release, proliferation and survival of antigen-specific T cell thereby amplifying specific immune responsesCitation16. Hck plays an important role in signalling process of inflammationCitation17. Hck is a phagocyte-specific proto-oncogene of the Src family and is expressed as two isoforms, p59Hck and p61Hck. It plays a critical role in Bcr/Abl-chronic myeloid leukaemia and is able to transform fibroblasts in vitro. However, the tumourigenic activity of Hck and the respective oncogenic functions of Hck isoforms have not been examinedCitation18. Lyn is phosphorylated in a KIT-independent manner in neoplastic mast cells (MCs) and it was shown that inhibitors such as dasatinib and bosutinib disrupt Lyn-driven oncogenic signalling in neoplastic MCCitation19. Among the SFKs, excess Fyn activity in the brain is associated with conditions such as Alzheimer’s and Parkinson’s diseases. Therefore inhibition of Fyn kinase may help counteract these nervous system disordersCitation20. Elevated levels of Fyn in fibroblast cells exhibited a cancer-like phenotype with increased anchorage-independent growth and prominent morphologic changes. Fyn is overexpressed in various cancers, including glioblastoma, head, neck, and prostate cancer. It is also involved in melanoma induces morphogenic transformation, alteration of mitogenic signals and stimulation of cell growth and proliferationCitation21. Most of the kinase activity inhibitors possess at least two aromatic rings, which interfere with ATP bindingCitation22,Citation23. Several small molecule inhibitors of SFKs are currently investigated in clinical trials. These Src inhibitors include heterocyclic ATP analogs, pyrazolo[2,3-d]pyrimidines, pyrrolo[2,3-d]pyrimidines, pyrido[2,3-d]pyrimidines, quinolines, and olomucinesCitation24. Here, the information about SFK enzymes inhibitors pyrrolo and pyrazolo[3,4-d]pyrimidine were summarized, since both skeletons are bioisosteres. Pyrazolo[3,4-d]pyrimidine derivatives, PP1 and PP2 belong to traditional ATP analogues (), which could inhibit c-Src, Lck and Fyn enzymes of SFK, showing IC50 values in the range of 4–6 nMCitation22,Citation25–28. Amide derivatives of PP1 and PP2, which bearing a C3 un-substituted phenyl ring were found potent inhibitor of Fyn with similar potency, with IC50 values of 6.5 nMCitation28. Pyrrolopyrimidine compounds CGP76030 and CGP77675, were reported to possess significant inhibitory activities against c-Src, Lck and Yes at nanomolar levels in osteoporosis and cancer models, both in vitro and in vivo ()Citation16,Citation29. Other pyrrolo[2,3-d]pyrimidine compound PKI166Citation30, inhibits c-Src with an IC50 value of 130 nM and A-419259Citation31,Citation32 inhibits c-Src, Lck and Lyn with IC50 values of 9, <3 and <3 nM, respectively (). Compound (S)-6-(4-(2-(dimethylamino)ethoxy)phenyl)-5-phenyl-N-((tetrahydrofuran-2-yl)methyl)-7H-pyrrolo[2,3-d]pyrimidin-4-amineCitation16 was found as Lck inhibitor with IC50 of <10 nM ().
Figure 1. Pyrazolopyrimidine and pyrrolopyrimidine derivatives as SFK inhibitors.
In this work, we have prepared new Pyrrolo[2,3-d]pyrimidine derivatives and evaluated for their inhibitory activities and selectivities against SFKs, namely Fyn, Lyn, Hck and c-Src. (). For this aim, structurally different type of amide derivatives are randomly designed to find new promising candidate molecules for specific SFKs inhibition. Subsequently, docking analyses were performed to evaluate binding properties and establishing relationships between biological activity-binding affinities of compounds.
Scheme 1. Synthesis of pyrrolo[2,3-d]pyrimidine amide derivatives.
![Scheme 1. Synthesis of pyrrolo[2,3-d]pyrimidine amide derivatives.](/cms/asset/78a16d30-954c-429a-b145-0a379e0234a2/ienz_a_715288_f0002_b.gif)
Materials and methods
Chloroacetonitrile, methylformate, 2,6-diamino-4-hydroxy pyrimidine, sodium methoxide, sodium acetate, 10% Pd/C, 4-(3,4-dimethoxyphenyl)butyric acid, (4-methylthio)benzoic acid, 5-methylthiophene-2-carboxylic acid, (4-chlorophenoxy)acetic acid (from Sigma-Aldrich, St. Louis, MO, USA); ethanol, hydrochloric acid, isopropyl alcohol, sodium hydroxide, sulphuric acid, dimethylsulfoxide (from Merck, Darmstadt, Germany), Fyn, Lyn, and Hck kinases (Millipore, MA, USA), c-Src kinase (Invitrogen, NY, USA) and ProFluor Src-family kinase assay kit, 8 plate (Promega, Madison, USA) were purchased. Melting points were measured with a capillary melting point apparatus (Electrothermal 9100; Essex, UK). The Nuclear Magnetic Resonance (1H-NMR) spectra were recorded on Varian Mercury 400 NMR spectrometer for 400 MHz (Varian Inc., Palo Alto, CA, USA). The chemical shift values were expressed in parts per million (ppm) relative to tetramethylsilane as an internal standard and signals were reported as s (singlet), d (doublet), t (triplet), q (quartet), m (multiplet). Mass spectra were recorded on a Waters ZQ Micromass LC-MS spectrometer (Waters Corporation, Milford, MA, USA). Perkin Elmer S-55 Spectrofluorimetry (from Perkin Elmer, Shelton, CT, USA) was used to measure fluorescence of phosphorylation reaction for enzyme activity studies. Analytical TLC was carried out on Merck 0.2 mm precoated silica gel (60 F-254) aluminium sheets (from Merck, Darmstadt, Germany), with visualization by irradiation with a UV lamp. The flash column chromatography was accomplished on silica gel 60 (230–400 mesh, Merck, Darmstadt, Germany). Elemental analysis was taken on a Leco-932 CHNS-O analyzer (St. Joseph, MI, USA).
5-(Cyano)-2-aminopyrrolo[2,3-d]pyrimidin-4(3H)-one (PreQ0, compound 3)
Methyl formate was added to a stirred mixture of NaOCH3 in anhydrous toluene at 0°C and chloroacetonitrile was added as dropwise over 30 min at 0°C. The reaction mixture was stirred at 0°C for 3 h. Chloro(formyl) acetonitrile 1 was obtained as an oily compound (6.3 g), which was used without further purification for next step (). NaOAc (0.101 mol, 8.29 g) and 2,6-diamino-4-hydroxy pyrimidine 2 (0.051 mol, 6.37 g) was mixed and heated at 100°C. Compound 1 in water was added to this mixture and it was refluxed for 5 h. The yellow solid was collected and dried in vacuo oven at 50°C for 24 h. Compound 3Citation33 was obtained with 36%. M.p. >300°C; 1H-NMR (400 MHz, DMSO-d6, δ, ppm): 6.40 (s, 2-NH2, 2H), 7.60 (s, 7-CH, 1H), 10.70 (s, 8 NH, 1H), 11.98 (s, 3-NH, 1H); IR (KBr, cm−1): 3450, 3400, 3100 (N-H signals), 2240 (C≡N), 1620 (C=C); ESI-MS: m/z, 176.3 (M+1).
2-Amino-5-(aminomethyl)pyrrolo[2,3-d]pyrimidin-4(3H)-one (compound 4, PreQ1)
PreQo was suspended in 50 mL MeOH and 50 mL of 1 N HCl. Ten percentage of Pd/C was added. The compound was treated with a hydrogenation gas (3 atm) at room temperature and stirred for 24 h. The precipitate was purified by silica gel column chromatography. Compound 4 was obtained with Yield 95%. M.p. 220–225°C; 1H NMR (400 MHz; DMSO-d6, δ, ppm): 4.00 (2H, s, CH2-NH2), 6.46 (2H, s, 2-NH2), 6.78 (1H, s 7-CH), 10.58 (1H, s, 8-NH), 11.21 (1H, s, 3-NH); IR (KBr, cm−1): 3440, 3340, 3225 (N-H signals), 1610 (C=C); LC/MS, (ESI, m/z): 180.5 (M+1), 163.0 (-NH3, %100).
The general synthesis of 2-amino-pyrrolo[2,3-d]pyrimidin-4(3H)-one-5-(substituted) amide derivatives (compounds 5–8)
One millimolar appropriate acid was dissolved in DMF and 1.2 mmol CDI was added at 0°C. The mixture was stirred at room temperature for 1 h. One millimolar PreQ1 was added and the mixture was stirred at 50°C for 12–24 h. The solvent was removed in vacuo and the resulting residue was purified by column chromatography on silica gel (CH2Cl2/MeOH, 9.5:0.5) to provide compounds 5–8. Purity of compounds were determined by HPLC and RT (Retention Time) were given as below. Chromatographic conditions; column C18 (Isterra) 5 mm × 25 cm, mobile phase 15 H2O: 65 CH3CN: 10 MeOH: 10 formic acid 0.1% in CH3CN, flow rate: 0.55 mL/min, temperature: 25°C.
N-((2-Amino-4-oxo-4,7-dihydro-3H-pyrrolo[2,3-d]pyrimidin-5-yl)methyl)-4-(3,4-dimethoxy phenyl)butanamide (5)
Yield 0.086 g, 40%; m.p. 256–258°C; 1H NMR (400 MHz, DMSO-d6, δ, ppm): 1.77 (2H, m, J = 7.6 Hz, CH2-CH2-CH2), 2.09 (2H, t, J = 7.6 Hz, O=C-CH2-CH2), 2.47 (2H, t, J = 7.6 Hz, -CH2-CH2-Ar), 3.70 (3H, s, Ar-OCH3), 3.72 (3H, s, Ar-OCH3), 4.28 (2H, d, J = 5.2 Hz, -CH2-NH-C=O), 6.09 (2H, s, 2-NH2), 6.48 (1H, d, J = 2.4 Hz, CH-7), 6.64 (1H, dd, J = 8.0, 2.0, Ar-H2′), 6.75 (1H, d, J = 2.0 Hz, Ar-H6′), 6.82 (1H, d, J = 8.0 Hz, Ar-H3′), 8.19 (1H, t, J = 5.2 Hz, -CH2-NH-C=O), 10.35 (1H, s, 8-NH), 10.85 (1H, s, 3-NH); 13C NMR (δ, ppm): 27.9 (-C-C-C), 34.9 (O=C-C-C-C), 35.7 (-C-C-C-Ar), 35.7 (-C-NH-), 56.0 (Ar-OCH3), 56.2 (Ar-OCH3), 99.0 (C-6′), 112.5 (C-5), 112.8 (C-3′), 114.4 (C-7), 116.2 (C-6), 120.7 (C-2′), 134.9 (C-1′), 147.6 (C-4′), 149.3 (C-5′), 152.2 (C-9), 152.9 (C-2), 160.2 (C-4), 171.9 (NH-C=O); IR (KBr, cm−1): 3450, 3375, 3320, 3125 (N-H signals), 1630 (C=O Amide), 1600 (C=C); LC/MS (ESI, m/z): 386.8 (M+1, 100%); HPLC-RT: 3.54 min (pure single peak); C19H23N5O4, calcd. C 59.21, H 6.02, N 18.17; found C 59.57, H 6.28, N 18.00.
N-[(2-Amino-4-oxo-4,7-dihydro-3H-pyrrolo[2,3-d]pyrimidin-5-yl)methyl]-4-(methylthio) benzamide (6)
Yield 0.030 g, 16%; m.p. 238–240°C; 1H NMR (400 MHz, DMSO-d6, δ, ppm): 4.25 (3H, s, SCH3), 4.46 (2H, d, J = 5.6 Hz, -CH2-NH-C=O), 6.11 (2H, s, 2-NH2), 6.55 (1H, d, J = 2.0 Hz, CH-7), 7.28 (2H, d, J = 8.4 Hz, Ar-H3′,5′), 7.77 (2H, d, J = 8.4 Hz, Ar-H2′,6′), 9.27 (1H, t, J = 5.2 Hz, -CH2-NH-C=O), 10.51 (1H, s, 8-NH), 10.88 (1H, s, 3-NH); 13C NMR (δ, ppm): 14.8 (SCH3), 36.4 (-C-NH-), 99.2 (C-5), 114.6 (C-7), 115.8 (C-6), 125.7 (C-3′), 125.9 (C-5′), 128.1 (C-2′), 128.2 (C-6′), 131.4 (C-1′), 142.9 (C-9), 152.5 (C-2), 152.9 (C-4′), 160.7 (C-4), 165.5 (NH-C=O); IR (KBr, cm−1): 3450, 3400, 3190, 3145 (N-H signals), 1700 (C=O Amide), 1600 (C=C); LC/MS (ESI, m/z): 330.9 (M+1, 100%); HPLC-RT: 4.75 min (pure single peak); C15H15N5O2S calcd. C 54.70, H 4.59, N 21.26; found C 54.98, H 4.33, N 21.45.
N-[(2-Amino-4-oxo-4,7-dihydro-3H-pyrrolo[2,3-d]pyrimidin-5-yl)methyl]-5-methylthiophene-2-carboxamide (7)
Yield 0.118 g, 35%; m.p. 260–262°C; 1H NMR (400 MHz, DMSO-d6, δ, ppm): 2.46 (3H, s, thiophene-CH3), 4.44 (2H, d, J = 5.2 Hz, -CH2-NH-C=O), 6.15 (2H, s, 2-NH2), 6.57 (1H, d, J = 2.0 Hz, CH-7), 6.83 (1H, dd, J = 4.0, 1.2 Hz, thiophene-H4′), 7.49 (1H, d, J = 4.0 Hz, thiophene-H5′), 9.19 (1H, t, J = 5.2 Hz, -CH2-NH-C=O), 10.52 (1H, s, 8-NH), 10.91(1H, s, 3-NH); 13C NMR (δ, ppm): 14.8 (CH3), 36.2 (-C-NH-), 99.2 (C-5), 114.6 (C-7), 115.8 (C-6), 127.0 (C-4′), 128.4 (C-5′), 138.4 (C-1′), 144.9 (C-9), 152.5 (C-2), 152.9 (C-3′), 160.7 (C-4), 161.2 (NH-C=O); IR (KBr, cm−1): 3400, 3340, 3130 (N-H signals), 1640 (C=O Amide), 1600 (C=C); LC/MS (ESI, m/z): 304.7 (M+1, 100%); HPLC-RT: 4.15 min (pure single peak); C13H13N5O2S calcd. C 55.03, H 4.01, N 21.39; found C 54.76, H 3.69, N 21.50.
N-[(2-Amino-4-oxo-4,7-dihydro-3H-pyrrolo[2,3-d]pyrimidin-5-yl)methyl]-2-(4-chlorophenoxy) acetamide (8)
Yield 0.065 g, 33%; m.p. 200–202°C; 1H NMR (400 MHz, DMSO-d6, δ, ppm): 4.36 (2H, d, J = 6.0 Hz, CH2-NH-C=O), 4.49 (2H, s, -O-CH2-C=O), 6.10 (2H, s, 2-NH2), 6.53 (1H, d, J = 2.0 Hz, CH-7), 7.00 (2H, d, J = 9.2 Hz, Ar-H3′,5′), 7.35 (2H, d, J = 9.6 Hz, Ar-H2′,6′), 8.88 (1H, t, J = 5.6 Hz, -CH2-NH-C=O), 10.52 (1H, s, 8-NH), 10.87 (1H, s, 3-NH); 13C NMR (δ, ppm): 35.3 (-C-NH-), 67.1 (CH2OPh), 99.3 (C-5), 114.5 (C-7), 117.4 (C-6), 115.5 (C-2′), 115.9 (C-6′), 129.8 (C-5′), 129.9 (C-3′), 135.8 (C-9), 153.0 (C-2), 153.2 (C-4′), 157.1 (C-1′), 160.5 (C-4), 167.3 (NH-C=O); IR (KBr, cm−1): 3420, 3310, 3150 (N-H signals), 1640 (C=O Amide), 1600 (C=C); LC/MS (ESI, m/z): 348.8 (M+, 100%); HPLC-RT: 4.79 min (pure single peak); C15H14N5O3Cl.H2O calcd. C 49.26, H 4.41, N 19.15; found C 49.13, H 4.68, N 19.27.
In vitro assay for Src tyrosine kinase activity
Kinase activity (Src-family kinases, Hck, Lyn, Fyn, and c-Src) and the effect of the molecules were determined by ProFluor Src-Family Kinase Assay Kit (Promega). A titration assay was performed for each kinase to determine the amount of the enzyme that results in approximately 80% phosphorylation as suggested by the manufacturerCitation34. The compounds were dissolved in 10% DMSO and tested at 1, 10 and 100 µM concentrations. Briefly, the molecules were mixed with a reaction buffer that included a specific substrate for Src-family kinases (R110), a control substrate and the kinase. The reaction was initiated by the addition of ATP. After incubating the 96-well reaction plate at 22°C for 1 h, protease solution was added to each well and the plate was incubated for 1 h. The fluorescence of the liberated R110 was read at an excitation wavelength of 485 nm and emission wavelength of 530 nm. The strength of fluorescence is inversely correlated with the kinase activity. The Src kinase activity is measured as the difference between the total activity of no vehicle (DMSO) and the activity of enzyme in the presence of DMSO. Percentage inhibition value of compounds was determined as the mean of triplicate measurements.
Molecular modelling
For this study, X-ray crystal structure of Fyn-saturosporine complex (PDB entry 2DQ7), Lyn-PP2 complex (PDB entry 2ZV9), Hck-PP1 complex (PDB entry 1QCF), and c-Src-CGP77675 complex (PDB entry 1YOL) were downloaded from Protein Data Bank (PDB). Solvents of molecules were deleted and hydrogen atoms were added to obtain the docking grid the active site was defined using Autogrid. The grid size was set to 40 × 40 × 40 points with grid spacing of 0.375 Å. The grid box was placed on the centre of the ligand from the corresponding crystal structure complexes. 2D structures of inhibitors were established by using ChemBioDrawUltra 11.0 and minimized with ChemBio3DUltra 11.0. To be exported to Autodock, the minimized structures were converted into the mol2 file format. The structures of proteins and ligands were combined using the Autodock 4.2.1 and Vina 1.0.2 software package (The Scripps Research Institute, La Jolla CA, USA). The rigid root and rotatable bonds were defined using ADT. The reliability of the docking protocol was first tested by simulations of the binding mode of known SFK inhibitors (PP1, PP2, CGP77675, and A419259). The results were ranked by the scoring functions of ligand-fit, and analyzed for relatively lower binding energy, and potential in forming hydrogen bonds with SFK kinases. The ligand was fully optimized inside the binding site during the docking simulations. The conformation with the lowest predicted binding free energy of the most occurring binding modes in SFKs active pockets were evaluated to binding properties and relationships between biological activity-binding affinities of compounds. Representation binding modes of compounds were shown by using PyMOL Molecular Graphics System (DeLano Scientific LLC, 2008).
Results and discussion
The starting compound 1 was synthesized with the reaction of methyl formate and chloroacetonitrile in anhydrous toluene (). To obtain compound 3 (PreQ0), 2,6-diamino-4-hydroxy pyrimidine 2 was reacted with compound 1 in presence of sodium acetate. PreQ1 (compound 4) was prepared by reduction of cyano group of PreQ0 to amine using H2/Pd in 1 M HClCitation33. The synthesis of amide derivatives of pyrrolo[2,3-d]pyrimidine (compounds 5–8) were achieved by the reaction of PreQ1 and corresponding acid derivatives in presence of 1,1′-carbonyldiimidazole (CDI) in DMF. The structural elucidations of compounds were established by 1H-NMR, IR, HPLC and MS. The proton numbering for NMR interpretation is indicated on the formula in .
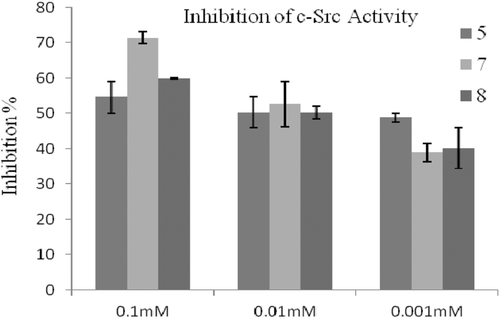
The inhibitory activities of compounds were evaluated via measurement the strength of fluorescence, which are inversely correlated with the kinase activity. For the primary assay, compounds were tested against Lyn, Fyn, Hck and c-Src of SFK enzymes at 0.1, 0.01 and 0.001 mM concentrations. In these concentrations range, compounds inhibited the enzymes Fyn (), Lyn () and c-Src () by 18–46, 12–42, and 14–71%, respectively. These results indicated that compounds are slightly active inhibitors of the tested SFK enzymes in comparison with PP2 (IC50 = 0.5 nM) for Fyn, A-419259 (IC50 = 0.3 nM) for Lyn and CGP77675 (IC50 = 0.4 nM) for c-Src. Compounds did not show any inhibitory properties against Hck. The best Fyn and Lyn inhibition () was obtained with compound 6 as shown by an inhibition rate of 46 and 42% at 0.1 mM concentration, respectively. Moreover, compound 5 showed Fyn inhibitions rates of 35 and 32% at 0.01 and 0.001 mM concentrations, respectively. On the other hand, compound 5 possessed weaker inhibitory effect on Lyn as shown by 25 and 17% inhibitions at 0.01 and 0.001 mM concentrations, respectively. To evaluate the inhibitory activities at lower concentrations, the data confirmed that compound 5 had higher inhibition profiles against Fyn and Lyn. This result revealed that compound 5 was more potent than the other compounds. As seen in , compound 7 showed 53% c-Src inhibition 0.01 mM concentration (IC50 was determined as 16 µM), whereas compound 5 showed 50% c-Src inhibition at much lower 0.001 mM concentration (IC50 was determined as 21 µM). The results indicate that compound 5 is slightly inhibitor of all three kinases, which suggest compound 5 has a non-selective inhibitory properties. While sulphur containing compounds 6 and 7 have similar inhibitory potencies against Fyn and Lyn, compound 6 has not any inhibition against c-Src. However, compound 7 and 8 are better selective inhibitors of c-Src. Overall, these findings suggest that the features of substituents on fifth position might have significant influence on the inhibitory potencies and selectivity profiles of compounds. It is noteworthy to insert new substituents on this position to design more potent and selective pyrrolopyrimidine compounds against SFKs.
Table 1. Inhibition of SFKs (c-Src, Fyn and Lyn) by compounds 5–8 and reference compounds.
Figure 2. The inhibitory effect of the molecules at 0.1, 0.01 and 0.001 mM concentrations on Fyn activity.
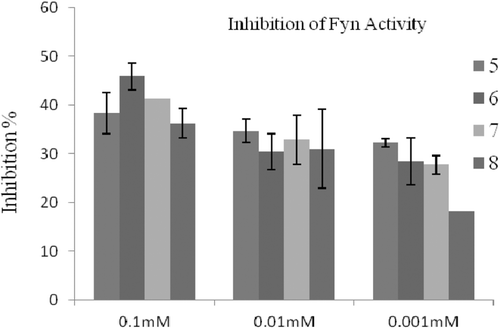
Figure 3. The inhibitory effect of the molecules at 0.1, 0.01 and 0.001 mM concentrations on Lyn activity.
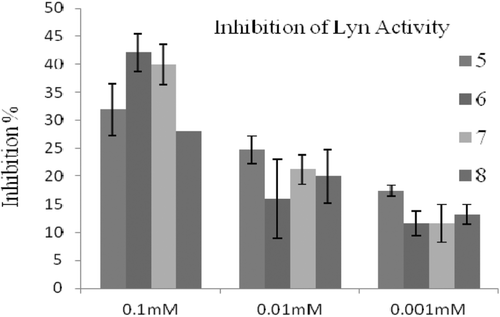
Figure 4. The inhibitory effect of the molecules at 0.1, 0.01 and 0.001 mM concentrations on c-Src activity.
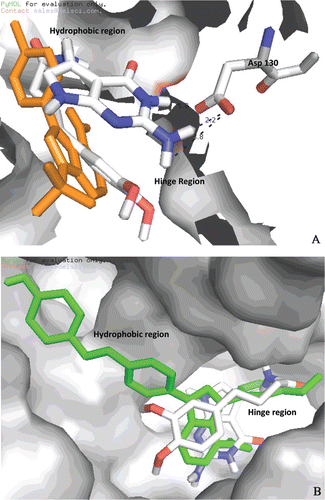
Binding conformation and the degree of fit into the SFKs (c-Src, Fyn, Lyn and Hck) of the compounds were evaluated by using Autodock 4.2.1 and Vina 1.0.2. The program successfully reproduced the X-ray coordinates of the inhibitor binding with RMSD values ranging from 1–3 Å and exhibited a similar accuracy with the same compounds when docked into the SFK binding sites. Each docked compound was assigned a score according to its binding mode onto the binding site of enzymes. The results suggest that ligands with highest binding affinities have a better chance of interaction with enzymes and have also a good probability of acting as inhibitors. From the interaction mode of the compounds with the predicated active site, it has been found that only compound 5 makes three hydrogen bonds with Fyn. One of the hydrogen bound was observed between the H (NH-3 of pyrolopyrimidine ring) and the O (OH) backbone of ASP130 with the distance of 1.8 Å. A couple of hydrogen bonds formed between the amino group at 2nd position of pyrrolopyrimidine ring and C=O of ASP130 with the distances of 2.2 Å and 3.8 Å (). All compounds were found to bind into the active site of Fyn similar to PP2, which is known as most potent inhibitor of Fyn but none of them are overlapped well with PP2, including compound 5 (). depicted the interactions of compound 5 and CGP77675 with the active site of c-Src. Compound 5 was found to bind into the ATP binding site of c-Src similar to the crystallized inhibitor CGP77675 but none of hydrogen bonds involving the pyrrolopyrimidine nucleus and the hinge region were determined. Compounds were not docked well onto the Lyn and Hck in comparison with potent known inhibitors A419259 and PP1, respectively. The docking studies suggested that the flexible or longer side chains of our compounds at 5th position might be the reason for weak binding property and activity in comparison with the reference compounds. As conclusion of above results, it was impossible to identify for each inhibitor a molecular portion that interacts with the same pocket of the SFKs binding site, since inhibitors showed quite different binding modes.
Figure 5. (A) Comparison of compound 5 and PP2 (orange) with Fyn binding site; (B) Comparison of compound 5 and CGP77675 (green) with c-Src binding site.
Declaration of interest
This work was partially supported by grants from the University of Ankara Research Fund (BAP-09B3336002).
References
- Ingley E. Src family kinases: regulation of their activities, levels and identification of new pathways. Biochim Biophys Acta 2008;1784:56–65.
- Parang K, Sun G. Recent advances in the discovery of Src kinase inhibitors. Expert Opin Ther Patents 2005;15:1183–1207.
- Gupta S, El-Rayes BF. Small molecule tyrosine kinase inhibitors in pancreatic cancer. Biologics 2008;2:707–715.
- Chong YP, Ia KK, Mulhern TD, Cheng HC. Endogenous and synthetic inhibitors of the Src-family protein tyrosine kinases. Biochim Biophys Acta 2005;1754:210–220.
- Sen B, Johnson FM. Regulation of SRC family kinases in human cancers. J Signal Transduct 2011;2011:865819.
- Cao X, You QD, Li ZY, Wang XJ, Lu XY, Liu XR et al. Recent progress of SRC family kinase inhibitors as anticancer agents. Mini Rev Med Chem 2008;8:1053–1063.
- Roskoksi R. Src kinase regulation by phosphorylation and dephosphorylation. Biochem Biophysical Res Commun 2005;331:1–14.
- Hu G, Place AT, Minshall RD. Regulation of endothelial permeability by Src kinase signaling: vascular leakage versus transcellular transport of drugs and macromolecules. Chem Biol Interact 2008;171:177–189.
- Finn RS. Targeting Src in breast cancer. Ann Oncol 2008;19:1379–1386.
- Edwards J. Src kinase inhibitors: an emerging therapeutic treatment option for prostate cancer. Expert Opin Investig Drugs 2010;19:605–614.
- Rothschild SI, Gautschi O, Haura EB, Johnson FM. Src inhibitors in lung cancer: current status and future directions. Clin Lung Cancer 2010;11:238–242.
- Sakamoto M, Takamura M, Ino Y, Miura A, Genda T, Hirohashi S. Involvement of c-Src in carcinoma cell motility and metastasis. Jpn J Cancer Res 2001;92:941–946.
- Sawyer TK. Cancer metastasis therapeutic targets and drug discovery: emerging small-molecule protein kinase inhibitors. Expert Opin Investig Drugs 2004;13:1–19.
- Rucci N, Susa M, Teti A. Inhibition of protein kinase c-Src as a therapeutic approach for cancer and bone metastases. Anticancer Agents Med Chem 2008;8:342–349.
- Saad F, Lipton A. SRC kinase inhibition: targeting bone metastases and tumor growth in prostate and breast cancer. Cancer Treat Rev 2010;36:177–184.
- Martin MW, Machacek MR. Update on lymphocyte specific kinase inhibitors: a patent survey. Expert Opin Ther Pat 2010;20:1573–1593.
- Smolinska MJ, Page TH, Urbaniak AM, Mutch BE, Horwood NJ. Hck tyrosine kinase regulates TLR4-induced TNF and IL-6 production via AP-1. J Immunol 2011;187:6043–6051.
- Poincloux R, Al Saati T, Maridonneau-Parini I, Le Cabec V. The oncogenic activity of the Src family kinase Hck requires the cooperative action of the plasma membrane- and lysosome-associated isoforms. Eur J Cancer 2009;45:321–327.
- Gleixner KV, Mayerhofer M, Cerny-Reiterer S, Hörmann G, Rix U, Bennett KL et al. KIT-D816V-independent oncogenic signaling in neoplastic cells in systemic mastocytosis: role of Lyn and Btk activation and disruption by dasatinib and bosutinib. Blood 2011;118:1885–1898.
- Resh MD. Fyn, a Src family tyrosine kinase. Int J Biochem Cell Biol 1998;30:1159–1162.
- Saito YD, Jensen AR, Salgia R, Posadas EM. Fyn: a novel molecular target in cancer. Cancer 2010;116:1629–1637.
- Belsches-Jablonski AP, Demory ML, Parsons JT, Parsons SJ. The src pathway as a therapeutic strategy. Drug Discov. Today: Ther. Strate. 2005;2:313–321.
- Noble ME, Endicott JA, Johnson LN. Protein kinase inhibitors: insights into drug design from structure. Science 2004;303:1800–1805.
- Ye G, Tiwari R, Parang K. Development of Src tyrosine kinase substrate binding site inhibitors. Curr Opin Investig Drugs 2008;9:605–613.
- Zhu X, Kim JL, Newcomb JR, Rose PE, Stover DR, Toledo LM et al. Structural analysis of the lymphocyte-specific kinase Lck in complex with non-selective and Src family selective kinase inhibitors. Structure 1999;7:651–661.
- Nam JS, Ino Y, Sakamoto M, Hirohashi S. Src family kinase inhibitor PP2 restores the E-cadherin/catenin cell adhesion system in human cancer cells and reduces cancer metastasis. Clin Cancer Res 2002;8:2430–2436.
- Meyn MA 3rd, Smithgall TE. Small molecule inhibitors of Lck: the search for specificity within a kinase family. Mini Rev Med Chem 2008;8:628–637.
- Schenone S, Brullo C, Musumeci F, Biava M, Falchi F, Botta M. Fyn kinase in brain diseases and cancer: the search for inhibitors. Curr Med Chem 2011;18:2921–2942.
- Recchia I, Rucci N, Festuccia C, Bologna M, MacKay AR, Migliaccio S et al. Pyrrolopyrimidine c-Src inhibitors reduce growth, adhesion, motility and invasion of prostate cancer cells in vitro. Eur J Cancer 2003;39:1927–1935.
- Traxler P, Bold G, Buchdunger E, Caravatti G, Furet P, Manley P et al. Tyrosine kinase inhibitors: from rational design to clinical trials. Med Res Rev 2001;21:499–512.
- Wilson MB, Schreiner SJ, Choi HJ, Kamens J, Smithgall TE. Selective pyrrolo-pyrimidine inhibitors reveal a necessary role for Src family kinases in Bcr-Abl signal transduction and oncogenesis. Oncogene 2002;21:8075–8088.
- Pene-Dumitrescu T, Peterson LF, Donato NJ, Smithgall TE. An inhibitor-resistant mutant of Hck protects CML cells against the antiproliferative and apoptotic effects of the broad-spectrum Src family kinase inhibitor A-419259. Oncogene 2008;27:7055–7069.
- Gangjee A, Vidwans A, Elzein E, McGuire JJ, Queener SF, Kisliuk RL. Synthesis, antifolate, and antitumor activities of classical and nonclassical 2-amino-4-oxo-5-substituted-pyrrolo[2,3-d]pyrimidines. J Med Chem 2001;44:1993–2003.
- Technical Bulletin. ProFluor Src-family kinase assay, Revised 6/09, Promega Corporation, USA.