
Abstract
Since the first histone deacetylase (HDAC) inhibitor (Zolinza®, widely known as suberoylanilide hydroxamic acid; SAHA) was approved by the Food and Drug Administration for the treatment of T-cell lymphoma in 2006, the search for newer HDAC inhibitors has attracted a great deal of interest of medicinal chemists worldwide. As a continuity of our ongoing research in this area, we designed and synthesized a series of 5-substitutedphenyl-1,3,4-thiadiazole-based hydroxamic acids as analogues of SAHA and evaluated their biological activities. A number of compounds in this series, for example, N1-hydroxy-N8-(5-(2-chlorophenyl)-1,3,4-thiadiazol-2-yl)octandiamide (5b), N1-hydroxy-N8-(5-(3-chlorophenyl-1,3,4-thiadiazol-2-yl)octandiamide (5c) and N1-hydroxy-N8-(5-(4-chlorophenyl)-1,3,4-thiadiazol-2-yl)octandiamide (5d), were found to possess potent anticancer cytotoxicity and HDAC inhibition effects. Compounds 5b–d were generally two- to five-fold more potent in terms of cytotoxicity compared to SAHA against five cancer cell lines tested. Docking studies revealed that these hydroxamic acid displayed higher affinities than SAHA toward HDAC8.
Introduction
Histone deacetylases (HDACs) constitute a group of enzymes that have important roles in gene transcription because acetylation or deacetylation is associated with an open chromatin configuration and a permissive gene transcription stateCitation1. To date, 18 HDAC enzymes have been identified in human and they are subdivided into four classes based on their homologies to yeast HDACs. Class I includes HDAC 1–3 and 8; class II includes HDACs 4–7, 9 and 10. Class III HDACs, known as sirtuins, include Sirt1–7, which are NAD+-dependent enzymes; class IV has only one member, HDAC11, exhibits properties of both classes I and II HDACsCitation1.
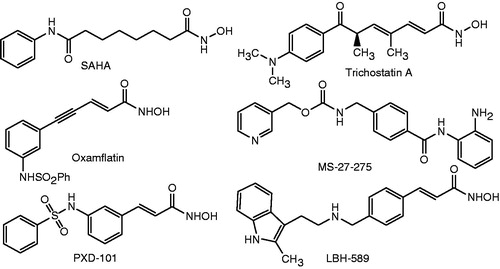
A number of evidences have demonstrated that these enzymes are not only involved in the regulation of chromatin structure and gene expression but also they can also regulate cell-cycle progression and carcinogenic process, which in turn involves in the formation of malignant tumorsCitation2,Citation3. HDACs have, therefore, become an attractive target for anticancer drug discoveryCitation4–6. Intensive efforts of medicinal chemists have resulted in the finding of a variety of HDAC inhibitors, such as trichostatin A, suberoylanilide hydroxamic acid (SAHA; vorinostat), MS-27-275 (entinostat), LBH-589 (panobinostat), PXD-101 and oxamflatin, among others ()Citation7–9. Of these, SAHA has been approved by the Food and Drug Administration in 2006 to treat several types of lymphoma, including cutaneous T-cell lymphoma. To date, at least 12 other HDAC inhibitors are currently in some phase of clinical trials, either as monotherapy or in combination with other chemotherapeutic agents or radiation, in patients with hematologic and solid tumors, including lung, breast, pancreas, renal, bladder cancers, melanoma, glioblastoma, leukemias, lymphomas and multiple myeloma.
Figure 1. Structures of some HDAC inhibitors.
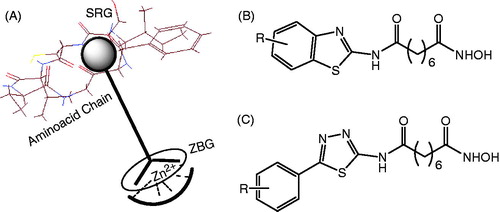
The common pharmacophore motif of a majority of HDAC inhibitors consists of three distinct domains: a metal binding head group, which interacts with the Zn2+ ion at the bottom of the active binding site of the enzyme; a long aliphatic linker, which occupies the narrow hydrophobic tubular channel; and a surface recognition group (SRG) (a cap group) ()Citation9. The surface recognition domain is essential for recognizing and binding to aminoacid chains at the entrance of the active pocket of enzymes. Based on this pharmacophore, a number of SRGs have been studied. In our previous report, we have also demonstrated that a thiazole and benzothiazole system could be excellent substitution for a phenyl ring in SAHACitation10. As a continuity of our search for new HDAC inhibitors, we have designed and synthesized a series of 5-substitutedphenyl-1,3,4-thiadiazole-based hydroxamic acids (). In this article, the results from this study are reported.
Figure 2. A pharmacophore motif of HDACIs (A) and benzothiazole/5-substitutedphenyl-1,3,4-thiadiazole-based hydroxamic acids as HDACIs (B and C).
Materials and methods
Chemistry
All products were homogenous, as examined by thin-layer chromatography, performed on Whatman® 250 µm Silica Gel GF Uniplates and visualized under UV light at λ 254 nm. Melting points were determined by a Gallenkamp melting point apparatus (LabMerchant, London, United Kingdom) and are uncorrected. Nuclear magnetic resonance spectra (1H NMR) were recorded on a Bruker DPX 500 MHz FT NMR spectrometer using tetramethylsilane as an internal standard and dimethyl sulfoxide-d6 (DMSO-d6) as solvent unless otherwise indicated. Chemical shifts were reported in parts per million (ppm) downfield from tetramethylsilane as an internal standard. Splitting patterns were designated as follows: s, singlet; d, doublet; t, triplet; q, quartet; and m, multiplet. Electron ionization, electrospray ionization (ESI) and high-resolution mass spectra were obtained using PE Biosystems API 2000 (Perkin Elmer, Palo Alto, CA) and Mariner® mass spectrometer (Azco Biotech, Inc., Oceanside, CA), respectively. Reagents and solvents were purchased from Aldrich or Fluka Chemical Corp. (Milwaukee, WI) and were used directly without additional steps of purification. Media, sera and other reagents used for cell culture were purchased from GIBCO Co. Ltd. (Grand Island, New York, NY).
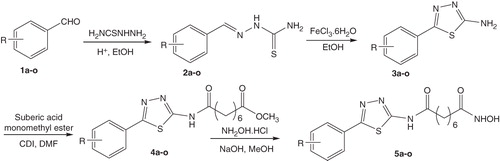
The synthesis of the series of N1-hydroxy-N8-(5-substitutedphenyl-1,3,4-thiadiazol-2-yl)octandiamides (5a–o) was carried out as illustrated in . Details are described as follows.
Scheme 1. Synthetic pathway for N1-hydroxy-N8-(5-substitutedphenyl-1,3,4-thiadiazol-2-yl)octandiamides (5a–o).
Synthesis of N1-hydroxy-N8-(5-phenyl-1,3,4-thiadiazol-2-yl)octanediamide (5a)
2-Phenylidenehydrazinecarbothioamide (2a)
The synthesis of compound 5a used benzaldehyde (1a) as the starting material. About two drops of acetic acid was added to the mixture of 1a (0.2 mL; 2 mmoL) and thiosemicarbazide (0.218 g; 2.4 mmoL) in ethanol (15 mL). The mixture was refluxed for about 4 h. Upon completion, the reaction mixture was cooled to room temperature. Water (15 mL) was added to induce precipitation. The precipitate was filtered and washed with water (two times), dried at 60 °C to yield a white–yellow solid (0.33 g, 91.2%) of 2a. mp: 165–163 °C; Rf = 0.54 (dichloromethane/methanol (DCM/MeOH) = 9/1).
5-Phenyl-1,3,4-thiadiazol-2-amine (3a)
Compound 3a was obtained from 2a, using Fe3+ reagent. The synthesis was carried out as follows: a suspension of FeCl3.12H2O in ethanol was slowly added to the solution of 2a in ethanol. The mixture was refluxed for 15 min, then, it was allowed to cool to room temperature, diluted by water (15 mL), alkalized by a NaOH 10% solution (tested by litmus paper) and extracted with methylene chloride (40 mL). The extracts were pooled, and methylene chloride was evaporated under reduced pressure. The residue was recrystallized from ethanol to give compound 3a as a yellow solid. Yield: 72.5%; mp: 225–227 °C; Rf = 0.66 (DCM/MeOH = 9/1). IR (KBr, cm−1): 3277 (NH2), 1514, 1468 (C=C).
Methyl 8-oxo-8-[(5-phenyl-1,3,4-thiadiazol-2-yl)amino]octanoate (4a)
Compound 4a was obtained as follows: 1,1′-carbodiimidazole (CDI) (162 mg; 1 mmoL) was dissolved in DCM and suberic monomethyl ester acid (1.9 mL; 1 mmoL) was added. The mixture was stirred for 10 min, and a solution of 3a (180 mg) in DMF (2 mL) was added. The reaction mixture was stirred at 60 °C for 24 h. DCM was evaporated under reduced pressure, then the mixture was poured into 20 mL of cold water. The precipitate appeared was filtered and washed, dried at 70 °C to give a white–pink solid. Yield: 58.5%; mp: 162.0–164.0 °C; Rf = 0.80 (DCM/MeOH = 9/1). IR (KBr, cm−1): 3162 (NH), 2853 (CH2), 1737 (C=O), 1562, 1436 (C=C); CI-MS (m/z): 347.0 [M+].
N1-Hydroxy-N8-(5-phenyl-1,3,4-thiadiazol-2-yl)octanediamide (5a)
Compound 5a was obtained from 4a. Synthesis of 5a was carried out as follows: To a solution of 4a (173 mg, 0.5 mmoL) in a mixture of MeOH (5 mL) and DMF (3 mL) was added NH2OH.HCl (490 mg, 7 mmoL). Ultrasound was used to dissolve the mixture. The mixture was cooled in a mixture of salt and crushed ice, and NaOH (17 mmoL) in H2O (1 mL) was added. The reaction mixture was stirred for 1 h and 30 min at 0 °C and poured slowly into 30 mL of cold water, acidified with HCl 5% to pH 5. The precipitate appeared was filtered and washed. Recrystallization from EtOH gave white crystals. The product was dried at 40 °C for 24 h in a vacuum oven. Yield: 52.0%; mp: 202.5–204.0 °C; Rf = 0.62 (DCM/MeOH = 9/1). IR (KBr, cm−1): 3251 (OH, acid), 3050 (NH), 2927, 2865 (CH, CH2), 1650, 1629 (C=O) and 1558 (C=C). CI-MS (m/z): 347.1 [M − H]−. 1H-NMR (500 MHz, DMSO-d6, ppm): δ 12.61 (1 H, s, NH), 10.36 (1 H, s, NH), 8.65 (1 H, brs, OH), 7.92–7.93 (2 H, m), 7.51–7.52 (3 H, m), 2.39–2.42 (2 H, m, CH2), 1.91–1.94 (2 H, m, CH2), 1.56–1.59 (2 H, m, CH2), 1.46–1.49 (2 H, m, CH2) and 1.25–1.26 (4 H, m, CH2). 13C NMR (125 MHz, DMSO-d6, ppm): δ 171.6 (C-5′′), 169.2 (C-8), 161.7 (C-1), 158.4 (C-2′), 130.5 (C-1′′), 130.3 (C-2′′,C-6′′), 129.4 (C-3′′, C-5′′), 126.9 (C-4′′), 32.3 (C-7), 34.8 (C-2), 28.3 (C-3, C-6), 25.0, 24.5 (C-4, C-5). Anal. Calcd. for C16H20N4O3S (348.13): C, 55.16; H, 5.79; N, 16.08. Found: C, 55.21; H, 5.75; N, 16.14.
Compounds 5b–o were synthesized via intermediates 2b–o, 3b–o and 4b–o by similar procedures as described above for 5a. Only data for the final compound series are described in this study.
N1-Hydroxy-N8-(5-(2-chlorophenyl)-1,3,4-thiadiazol-2-yl)octanediamide (5b)
White crystals; yield: 62.1%; mp: 163.5–165.0 °C; Rf = 0.52 (DCM/MeOH = 9/1). IR (KBr, cm−1): 3288 (OH, acid), 3030 (NH), 2927 (CH, CH2), 2857 (CH, CH2), 1650, 1630 (C=O) and 1584 (C=C). CI-MS (m/z): 381.5 [M − H]−. 1H-NMR (500 MHz, DMSO-d6, ppm): δ 12.67 (1 H, s, NH), 10.34 (1 H, s, NH), 8.68 (1 H, brs, OH), 8.09 (1 H, d, J = 7.50 Hz), 7.68 (1 H, d, J = 7.75 Hz), 7.50–7.58 (2 H, m), 2.18–2.21 (2 H, m, CH2), 1.92–1.95 (2 H, m, CH2), 1.58–1.62 (2 H, m, CH2), 1.46–1.51 (2 H, m, CH2) and 1.25–1.30 (4 H, m, CH2). 13C NMR (125 MHz, DMSO-d6, ppm): δ 171.7 (C-5′′), 169.1 (C-8), 159.9 (C-1), 157.6 (C-2′), 131.7 (C-1′′), 131,0 (C-2′′), 130.8 (C-4′′), 130.5 (C-6′′), 128.9 (C-3′′), 127.8 (C-5′′), 34.7 (C-7), 33.6 (C-2), 28.2 (C-3, C-6), 24.9 and 24.3 (C-4, C-5). Anal. Calcd. for C16H19ClN4O3S (382.87): C, 50.19; H, 5.00; and N, 14.63. Found: C, 50.15; H, 5.11; and N, 14.45.
N1-hydroxy-N8-(5-(3-chlorophenyl)-1,3,4-thiadiazol-2-yl)octanediamide (5c)
White crystals; yield: 62.1%; mp: 163.5–165.0 °C; Rf = 0.52 (DCM/MeOH = 9/1). IR (KBr, cm−1): 3430 (OH), 3040 (NH), 2927, 2862 (CH, CH2), 1645, 1635 (C=O) and 1567 (C=C). CI-MS (m/z): 381.6 [M − H]−, 340.5 [M − C3H5]−. 1H-NMR (500 MHz, DMSO-d6, ppm): δ 12.69 (1 H, s, NH), 10.36 (1 H, s, NH), 8.68 (1 H, brs, OH), 7.97 (1 H, s), 7.88 (1 H, d, J = 6.0 Hz), 7.55–7.57 (2 H, m), 1.92–1.95 (2 H, m, CH2), 1.57–1.60 (2 H, m, CH2), 1.48–1.51 (2 H, m, CH2) and 1.23–1.26 (6 H, m, CH2). Citation13C NMR (125 MHz, DMSO-d6, ppm): δ 171.7 (C-5′), 169.2 (C-8), 160.2 (C-1), 158.8 (C-2′), 134.0 (C-1″), 132.2 (C-3″), 131.3 (C-2″), 130.3 (C-6″), 126.1 (C-5″), 125.8 (C-4″), 34.9 (C-7), 32.2 (C-2), 28.2 (C-3, C-6), 25.0 and 24.5 (C-4, C-5). Anal. Calcd. for C16H19ClN4O3S (382.87): C, 50.19; H, 5.00; and N, 14.63. Found: C, 50.21; H, 5.04; and N, 14.59.
N1-hydroxy-N8-(5-(4-chlorophenyl)-1,3,4-thiadiazol-2-yl)octanediamide (5d)
White powder; yield: 58.4%; mp: 231.5–233.0 °C; and Rf = 0.53 (DCM/MeOH = 9/1). IR (KBr, cm−1): 3340 (OH acid), 3171 (NH), 2936, 2853 (CH, CH2), 1630, 1620 (C=O) and 1581 (C=C). CI-MS (m/z): 381.2 [M − H]− and 340.1 [M − C3H5]−. 1H-NMR (500 MHz, DMSO-d6, ppm): δ 12.66 (1 H, s, NH), 10.36 (1 H, s, NH), 8.69 (1 H, brs, OH), 7.94 (2 H, d, J = 8.0 Hz); 7.58 (2 H, d, J = 8.0 Hz), 1.92–1.95 (2 H, m, CH2), 1.57–1.61 (2 H, m, CH2), 1.45–1.50 (2 H, m, CH2) and 1.26–1.27 (6 H, m, CH2). Citation13C NMR (125 MHz, DMSO-d6, ppm): δ 171.7 (C-5′), 169.2 (C-8), 160.6 (C-1), 158.6 (C-2′), 135.1 (C-4″), 129.4 (C-1″), 129.1 (C-3″, C-5″), 128.6 (C-2″, C-6″), 34.9 (C-7), 33.2 (C-2), 28.3 (C-3, C-6), 25.0 and 24.4 (C-4, C-5). Anal. Calcd. for C16H19ClN4O3S (382.87): C, 50.19; H, 5.00; and N, 14.63. Found: C, 50.25; H, 5.10; and N, 14.48.
N1-hydroxy-N8-(5-(4-fluorophenyl)-1,3,4-thiadiazol-2-yl)octanediamide (5e)
White crystals; yield: 60.0%; mp: 212.0–213.0 °C; Rf = 0.55 (DCM/MeOH = 9/1). IR (KBr, cm−1): 3400 (OH), 3239, 3054 (NH), 2939, 2853 (CH2), 1687, 1628 (C=O), 1597, 1557 and 1515 (C=C). CI-MS (m/z): 756.0 [2M + Na]+, 363.8 [M − 2H]−. 1H-NMR (500 MHz, DMSO-d6, ppm): δ 12.63 (1 H, s, NH), 10.42 (1 H, s, NH), 7.97 (2 H, d, J = 8.0 Hz), 7.34 (2 H, d, J = 8.0 Hz), 2.47–2.50 (2 H, m, CH2), 1.93–1.96 (2 H, m, CH2), 1.56–1.60 (2 H, m, CH2), 1.46–1.50 (2 H, m, CH2) and 1.25–1.26 (4 H, m, CH2). Citation13C NMR (125 MHz, DMSO-d6, ppm): δ 171.6 (C-5′), 169.2 (C-8), 162.3 (C-1), 160.6 (C-4″), 158.3 (C-2′), 129.2 (C-3″, C-5″), 126.2 (C-1″), 116.4 (C-2″, C-6″), 34.8 (C-7), 33.6 (C-2), 28.3 (C-3, C-6) and 24.3 (C-4, C-5, overlap). Anal. Calcd. for C16H19FN4O3S (366.41): C, 52.45; H, 5.23; and N, 15.29. Found: C, 52.50; H, 5.27; and N, 15.32.
N1-hydroxy-N8-(5-(4-bromophenyl)-1,3,4-thiadiazol-2-yl)octanediamide (5f)
White powder; yield: 60.5%; mp: 202.5–204.0 °C; Rf = 0.57 (DCM/MeOH = 9/1). IR (KBr, cm−1): 3400 (OH), 3163, 3049 (NH), 2923, 2850 (CH2), 1691, 1616 (C=O), 1571, 1557 and 1469 (C=C). CI-MS (m/z): 425.9 [M − H]−. 1H-NMR (500 MHz, DMSO-d6, ppm): δ 12.68 (1 H, s, NH), 10.37 (1 H, s, NH), 7.86–7.88 (2 H, d, J = 8.00 Hz), 7.71 (2 H, d, J = 8.00 Hz), 2.48–2.50 (2 H, m, CH2), 1.94–2.16 (2 H, m, CH2), 1.59–1.61 (2 H, m, CH2), 1.45–1.49 (2 H, m, CH2) and 1.27–1.29 (4 H, m, CH2). Citation13C NMR (125 MHz, DMSO-d6, ppm): δ 174.3 (C-5′), 171.6 (C-8), 160.6 (C-1), 158.6 (C-2′), 132.3 (C-3″, C-5″), 129.4 (C-1″), 128.7 (C-2″, C-6″), 128.6 (C-3″, C-5″), 123.8 (C-4″), 34.7 (C-7), 33.6 (C-2), 32.1, 28.2 (C-3, C-6), 25.0 and 24.3 (C-4, C-5). Anal. Calcd. for C16H19BrN4O3S (427.32): C, 44.97; H, 4.48; and N, 13.11. Found: C, 45.03; H, 4.51; and N, 13.07.
N1-hydroxy-N8-(5-(4-methylphenyl)-1,3,4-thiadiazol-2-yl)octanediamide (5g)
White solid; yield: 65.0%; mp: 170.0–172.0 °C; Rf = 0.52 (DCM/MeOH = 9/1). IR (KBr, cm−1): 3500 (OH), 3162, 3023 (NH), 2929, 2862 (CH2), 1702, 1650, 1615 (C=O), 1560 and 1463 (C=C). CI-MS (m/z): 359.8 [M − 2H]− and 344.9 [M − OH]−. 1H-NMR (500 MHz, DMSO-d6, ppm): δ 12.59 (1 H, s, NH), 10.36 (1 H, s, NH), 7.80 (2 H, d, J = 8.50 Hz), 7.32 (2 H, d, J = 8.50 Hz), 2.47–2.50 (2 H, m, CH2), 2.19–2.16 (1 H, t, CH2b), 2.35 (3 H, s, -CH3), 1.90–1.94 (1 H, t, CH2a), 1.58–1.61 (2 H, m, CH2), 1.47–1.50 (2 H, m, CH2) and 1.27–1.29 (4 H, m, CH2). Citation13C NMR (125 MHz, DMSO-d6, ppm): δ 174.4 (C-5′), 169.1 (C-8), 161.7 (C-1), 158.0 (C-2′), 140.1 (C-1″), 129.9 (C-3″, C5″), 127.5 (C-4″), 126.8 (C-2″, C-6″), 34.8 (C-7), 33.6 (C-2), 32.2 (CH3), 28.3, 28.2 (C-3, C-6), 24.3 and 24.3 (C-4, C-5). Anal. Calcd. for C17H22N4O3S (362.14): C, 56.33; H, 6.12; and N, 15.46. Found: C, 56.36; H, 6.13; and N, 15.43.
N1-hydroxy-N8-(5-(4-methoxyphenyl)-1,3,4-thiadiazol-2-yl)octanediamide (5h)
White crystals; yield: 62.5%; mp: 178.0–179.0 °C; Rf = 0.58 (DCM/MeOH = 9/1). IR (KBr, cm−1): 3400 (OH), 3183, 3071 (NH), 2938, 2856 (CH2), 1693, 1611 (C=O), 1580, 1563 and 1519 (C=C). ESI-MS (m/z): 401.3369 [M + Na]+ and 377.3847 [M + H]+. 1H-NMR (500 MHz, DMSO-d6 + CDOD3, ppm): δ 7.85 (2H, d, J = 8.00 Hz), 7.05 (2 H, d, J = 8.00 Hz), 3.81 (3 H, s, OCH3); 2.46–2.50 (2 H, m, CH2), 1.95 (2 H, t, CH2), 1.56–1.60 (2 H, m, CH2), 1.46–1.50 (2 H, m, CH2) and 1.26–1.30 (4 H, m, CH2). Citation13C NMR (125 MHz, DMSO-d6 + CDOD3, ppm): δ 171.7 (C-5″), 169.0 (C-8), 161.3 (C-1), 160.9 (C-4″), 158.0 (C-2′), 128.4 (C-2″, C-6″), 122.9 (C-1″), 114.7 (C-3″, C-5″), 55.4 (OCH3), 35.0 (C-7), 32.2 (C-2), 28.2 (C-3, C-6), 25.0 and 24.3 (C-4, C-5). Anal. Calcd. for C17H22N4O4S (378.45): C, 53.95; H, 5.86; and N, 14.80. Found: C, 53.90; H, 5.84; and N, 14.83.
N1-hydroxy-N8-(5-(4-(dimethylamino)phenyl)-1,3,4-thiadiazol-2-yl)octanediamide (5i)
White powder; yield: 61.0%; mp: 180.0–181.0 °C; and Rf = 0.45 (DCM/MeOH = 9/1). IR (KBr, cm−1): 3350 (OH), 3227 (NH), 2926, 2853 (CH2), 1652 (C=O), 1609, 1570 and 1539 (C=C). CI-MS (m/z): 389.0 [M − 2H]−. 1H-NMR (500 MHz, DMSO-d6, ppm): δ 12.41 (1 H, s, NH), 10.37 (1 H, brs, NH), 7.71 (2 H, d, J = 7.80 Hz), 6.79 (2 H, d, J = 7.80 Hz), 2.98 (6 H, s, 2CH3), 2.46–2.50 (2 H, m, CH2), 1.95 (2 H, t, CH2), 1.58–1.61 (2 H, m, CH2), 1.47–1.50 (2 H, m, CH2), 1.27–1.31 (4 H, m, CH2). Citation13C NMR (125 MHz, DMSO-d6, ppm): δ 171.3 (C-5′), 169.1 (C-8), 162.2 (C-1), 156.6 (C-2′), 151.5 (C-4″), 127.8 (C-2″, C-6″), 114.4 (C-3″, C-5″), 40.2 (2CH3), 34.8 (C-7), 32.2 (C-2), 28.2 (C-3, C-6) and 24.3 (C-4, C-5, overlap). Anal. Calcd. for C18H25N5O3S (391.17): C, 55.22; H, 6.44; and N, 17.89. Found: C, 55.25; H, 6.41; and N, 17.85.
N1-hydroxy-N8-(5-(2-nitrophenyl)-1,3,4-thiadiazol-2-yl)octanediamide (5j)
Yellowish solid; yield: 52.0%; mp: 175.0–176.5 °C; and Rf = 0.50 (DCM/MeOH = 9/1). IR (KBr, cm−1): 3300 (OH), 3097, 3012 (NH), 2935, 2866 (CH2), 1709, 1698 (C=O), 1537 and 1470 (C=C). CI-MS (m/z): 391.5 [M − H]−; 1H-NMR (500 MHz, DMSO-d6, ppm): δ 12.76 (1 H, s, NH), 10.34 (1 H, s, NH), 8.06 (1 H, d, J = 7.50 Hz), 7.88 (1 H, d, J = 7.5 Hz), 7.84 (1 H, t, J = 7.50 Hz), 7.78 (1 H, t, J = 7.50 Hz), 2.50–2.53 (2 H, m, CH2), 1.94–1.91 (2 H, m, CH2), 1.58–1.63 (2 H, m, CH2), 1.46–1.52 (2 H, m, CH2) and 1.25–1.29 (4 H, m, CH2). Citation13C NMR (125 MHz, DMSO-d6, ppm): δ 171.9 (C-5′), 169.1 (C-8), 159.7 (C-1), 156.8 (C-2′), 148.5 (C-2″), 133.1 (C-5″), 131.9 (C-1″), 131.5 (C-4″), 124.5 (C-6″), 123.4 (C-3″), 34.8 (C-7), 32.2 (C-2), 28.2 (C-3, C-6), 25.0 and 24.3 (C-4, C-5). Anal. Calcd. for C16H19N5O5S (393.11): C, 48.85; H, 4.87; and N, 17.80. Found: C, 48.88; H, 4.91; and N, 17.77.
N1-hydroxy-N8-(5-(4-nitrophenyl)-1,3,4-thiadiazol-2-yl)octanediamide (5k)
Yellowish powder; yield: 53.5%; mp: 170.0–172.0 °C; and Rf = 0.52 (DCM/MeOH = 9/1). IR (KBr, cm−1): 3410 (OH), 2925, 2853 (CH2), 1691, 1624 (C=O), 1599, 1547 and 1519 (C=C). CI-MS (m/z): 391.5 [M − H]−; 1H-NMR (500 MHz, DMSO-d6, ppm): δ 10.42 (1 H, s, NH), 9.70 (1 H, brs, OH), 8.32 (2 H, d, J = 8.30 Hz), 8.16 (2 H, d, J = 8.30 Hz), 2.45 (2 H, t, CH2), 2.06 (1 H, t, CH2b), 1.95 (1 H, t, CH2a), 1.58–1.59 (2 H, m, CH2), 1.47 (2 H, m, CH2) and 1.21–1.26 (4 H, m, CH2). Citation13C NMR (125 MHz, DMSO-d6, ppm): δ 173.9 (C-5′), 169.7 (C-8), 162.9 (C-1), 152.2 (C-2′), 147.7 (C-4″), 137.6 (C-1″), 127.1 (C-2″, C-6″), 124.2 (C-3″, C-5″), 40.0 (C-7), 34.9 (C-2), 28.2 (C-3, C-6) and 24.3 (C-4, C-5, overlap). Anal. Calcd. for C16H19N5O5S (393.11): C, 48.85; H, 4.87; and N, 17.80. Found: C, 48.91; H, 4.86; and N, 17.82. HRMS (ESI) calcd for [M–H] C16H19N5O5S, m/z: 392.1029, observed: 392.1021.
N1-hydroxy-N8-(5-(2,6-dichlorophenyl)-1,3,4-thiadiazol-2-yl)octanediamide (5l)
White solid; yield: 50.0%; mp: 175.0–176.0 °C; and Rf = 0.56 (DCM/MeOH = 9/1). IR (KBr, cm−1): 3350 (OH), 3155 (NH), 2927, 2856 (CH2), 1701, 1629 (C=O) and 1534 (C=C). CI-MS (m/z): 416.3 [M − H]−, 499.3 [M − OH]−; 1H-NMR (500 MHz, DMSO-d6, ppm): δ 10.46 (1 H, s, NH), 9.71 (1 H, s, br, NH), 8.75 (1 H, brs, OH), 7.67 (2 H, d, J = 7.30 Hz), 7.61 (1 H, t, J = 7.30 Hz), 2.50–2.54 (2 H, m, CH2), 1.93–1.95 (2 H, m, CH2), 1.59–1.61 (2 H, m, CH2), 1.47–1.49 (2 H, m, CH2) and 1.22–1.29 (4 H, m, CH2). Citation13C NMR (125 MHz, DMSO-d6, ppm): δ 172.0 (C-5′), 169.0 (C-8), 160.5 (C-1), 155.3 (C-2′), 135.1 (C-2″, C-6″), 132.9 (C-4″), 128.7 (C-3″, C-5″), 34.9 (C-7), 32.2 (C-2), 28.2 (C-3, C-6), 25.0 and 24.3 (C-4, C-5). Anal. Calcd. for C16H18Cl2N4O3S (416.05): C, 46.05; H, 4.35; and N, 13.43. Found: C, 46.21; H, 4.39; and N, 13.40.
N1-hydroxy-N8-(5-(benzo[d][1,3]dioxol-5-yl)-1,3,4-thiadiazol-2-yl)octanediamide (5m)
White solid; yield: 55.0%; mp: 190.5–192.0 °C; Rf = 0.54 (DCM/MeOH = 9/1). IR (KBr, cm−1): 3340–3300 (OH), 3161, 3032 (NH), 2923, 2854 (CH2), 1692, 1637 (C=O), 1574, 1505 and 1458 (C=C). CI-MS (m/z): 393.12 [M]+, 391.12 [M − H]− and 376.13 [M − OH]+; 1H-NMR (500 MHz, DMSO-d6 + CDOD3, ppm): δ 7.46 (1 H, s), 7.39 (1 H, d, J = 7.50 Hz), 7.01 (1 H, d, J = 7.50 Hz), 6.10 (2 H, s, CH2), 2.43–2.47 (2 H, m, CH2), 2.15–2.18 (1 H, m, CH2), 1.90–1.94 (1 H, m, CH2), 1.59–1.61 (2 H, m, CH2), 1.48–1.49 (2 H, m, CH2) and 1.18–1.27 (4 H, m, CH2). Citation13C NMR (125 MHz, DMSO-d6 + CDOD3, ppm): δ 171.4 (C-5′), 169.1 (C-8), 161.7 (C-1), 157.8 (C-2′), 149.2 (C-3″), 148.0 (C-4″), 124.3 (C-1″), 121.8 (C-6″), 108.9 (C-5″), 106.4 (C-2″), 101.8 (-OCH2O-), 34.8 (C-7), 32.2 (C-2), 28.3, 28.2 (C-3, C-6), 25.0 and 24.4 (C-4, C-5). Anal. Calcd. for C17H20N4O5S (392.12): C, 52.03; H, 5.14; and N, 14.28. Found: C, 52.09; H, 5.11; and N, 14.25.
N1-hydroxy-N8-(5-(2,3,4-trimethoxyphenyl)-1,3,4-thiadiazol-2-yl)octanediamide (5n)
White solid; yield: 50.0%; mp: 175.0–176.0 °C; and Rf = 0.51 (DCM/MeOH = 9/1). IR (KBr, cm−1): 3424 (OH), 3171, 3044 (NH), 2940, 2860 (CH2), 1688, 1659 (C=O), 1594, 1576 and 1552 (C=C). CI-MS (m/z): 437.3 [M − H]−; 1H-NMR (500 MHz, DMSO-d6, ppm): δ 12.30 (1 H, s, NH), 10.32 (1 H, s, NH), 8.60 (1 H, brs, OH), 7.94 (1 H, d, J = 7.50 Hz), 6.99 (1 H, d, J = 7.50 Hz), 3.93 (3 H, s, CH3), 3.87 (3 H, s, CH3), 3.82 (3 H, s, CH3), 2.48–2.50 (2 H, m, CH2), 1.95–1.98 (2 H, m, CH2), 1.58–1.61 (2 H, m, CH2), 1.49–1.51 (2 H, m, CH2) and 1.25–1.28 (4 H, m, CH2). Citation13C NMR (125 MHz, DMSO-d6, ppm): δ 174.5 (C-5′), 169.1 (C-8), 159.6 (C-1), 156.2 (C-2″), 155.1 (C-4″), 150.1 (C-2″), 141.6 (C-3″), 121.9 (C-6″), 116.8 (C-1″), 108.9 (C-5″), 60.8 (-OCH3), 60.5 (-OCH3), 56.1 (-OCH3), 34.8 (C-7), 32.2 (C-2), 28.3 (C-3, C-6), 24.5 and 24.3 (C-4, C-5). Anal. Calcd. for C16H20N4O3S (438.16): C, 52.04; H, 5.98; and N, 12.78. Found: C, 52.09; H, 5.95; and N, 12.74.
N1-hydroxy-N8-(5-(3,4,5-trimethoxyphenyl)-1,3,4-thiadiazol-2-yl)octanediamide (5o)
White solid; yield: 53.5%; mp: 177.0–178.0 °C; and Rf = 0.54 (DCM/MeOH = 9/1). IR (KBr, cm−1): 3480 (OH), 3210 (NH), 2939, 2860 (CH2), 1708, 1651 (C=O), 1572, 1515 and 1467 (C=C). CI-MS (m/z): 437.41 [M − H]−; 1H-NMR (500 MHz, DMSO-d6, ppm): δ 12.59 (1 H, s, NH), 10.34 (1 H, s, NH), 8.67 (1 H, brs, OH), 7.18 (2 H, s), 3.87 (6 H, s, OCH3), 3.72 (3 H, s, CH3), 2.47–2.50 (2 H, m, CH2), 1.91–1.94 (2 H, m, CH2), 1.59–1.60 (2 H, m, CH2), 1.47–1.49 (2 H, m, CH2), 1.27–1.28 (4 H, m, CH2). Citation13C NMR (125 MHz, DMSO-d6, ppm): δ 171.5 (C-5′), 169.1 (C-8), 161.6 (C-1), 158.3 (C-2″), 153.4 (C-3″, C-5″), 139.4 (C-4″), 125.7 (C-1″), 104.2 (C-2″, C-6″), 116.8 (C-1″), 108.9 (C-5″), 60.1 (-OCH3), 56.1 (-2OCH3), 34.8 (C-7), 32.2 (C-2), 28.3 (C-3, C-6), 24.5 and 24.3 (C-4, C-5). Anal. Calcd. for C16H20N4O3S (438.16): C, 52.04; H, 5.98; and N, 12.78. Found: C, 52.05; H, 5.97; and N, 12.76.
Cytotoxicity assays
Human cancer cell lines, NCI-H460 (lung cancer), PC3 (prostate cancer), SW620 (colon cancer), MCF-7 (breast adenocarcinoma) and AsPC-1 (pancreatic cancer) cell lines were obtained from the American Type Culture Collection (Manassas, VA). Cells were plated at 9 × 103 cells/well in 96-well plates, incubated overnight and treated with samples for 48 h. Compounds were dissolved in DMSO. Cytotoxicity was measured by the method as described in literatureCitation11 with slight modificationsCitation12. The final concentration of DMSO in the wells containing cells was 0.01%, and this concentration did not affect the cell growthCitation11,Citation12. The IC50 values were calculated according to the Probit’s methodCitation13. The values reported for these compounds are averages of three separate determinations.
Western blot assay
Total protein extracts were prepared by lysing cells in radioimmunoprecipitation buffer (50 mM Tris-Cl [pH 8.0], 5 mM EDTA, 150 mM NaCl, 1% NP-40, 0.1% sodium dodecyl sulfate (SDS) and 1 mM phenylmethylsulfonyl fluoride). Protein concentrations in the lysates were determined using a Bio-Rad protein assay kit (Bio-Rad Laboratories Inc., Hercules, CA) according to the manufacturer’s instructions. Samples were separated on SDS polyacrylamide gels and transferred to nitrocellulose membranes. The membranes were incubated with blocking buffer (Tris-buffered saline containing 0.2% Tween-20 and 3% nonfat dried milk) and probed with the primary antibodies against acetyl histone-H3, -H4 and glyceraldehyde 3-phosphate dehydrogenase (GAPDH). After washing, membranes were probed with horseradish peroxidase-conjugated secondary antibodies. Detection was performed using an enhanced chemiluminescent protein detection system (Amersham Biosciences, Little Chalfont, UK).
Docking studies
AutoDock Vina programCitation14 was used in the docking studies. Initial structure of HDAC8 was obtained from the Protein Data Bank (PDB) (PDB ID: 1T69)Citation15, and coordinates for the compounds were generated using the GlycoBioChem PRODRG2 Server (http://davapc1.bioch.dundee.ac.uk/prodrg/)Citation16. The grid maps for docking studies were centered on the SAHA binding site and comprised 26 × 26 × 22 points with 1.0 Å spacing after SAHA was removed from the complex structure, as described previouslyCitation10. AutoDock Vina program was run with four-way multithreading, and the other parameters were default settings in Autodock Vina Program (The Scripps Research Institute, La Jolla, CA).
Results and discussion
Chemistry
A series of 5-substitutedphenyl-1,3,4-thiadiazole-based hydroxamic acids (5a–o) were synthesized via a four-step pathway (). In the first step, the thiosemicarbazones 2a–o were obtained by condensation of simple benzaldehydes 1a–o with thiosemicarbazide. Intramolecular cyclization of 2a–o proceeded smoothly using ferric chloride in ethanol to give 2-amino-5-phenyl-1,3,4-thiadiazole derivatives 3a–o. Coupling of 3a–o with suberic acid monomethyl ester using CDI as a carboxylic activating reagent generated the ester intermediates 4a–o. Reaction of the esters 4a–o with hydroxylamine in alkaline conditions gave the final products 5a–o in generally good yields. The presence of a nitro substituent on the phenyl ring was found to somewhat lower the yield of overall process.
The structures of the obtained compounds were straightforwardly and unambiguously confirmed by spectral studies, including IR, MS, 1H NMR and Citation13C NMR and elemental analysis.
Bioactivity
The sulforhodamine B cell proliferation assay was used to evaluate the antiproliferative activity of the synthesized compounds. The compounds 5a–o were first screened at 30 µM for cell growth inhibition against SW620 (human colon cancer) cell line. It was found that all compounds inhibited the growth of SW620 cells by more than 50% at this concentration. Therefore, the compounds were subjected to further evaluation at five concentrations (30, 10, 3, 1, 0.3 µM) in SW620 and four human cancer cell lines more, including MCF-7 (breast adenocarcinoma), PC-3 (prostate cancer), AsPC-1 (pancreas cancer) and NCI-H460 (lung cancer) cell lines. The IC50 (the concentration that causes 50% of cell proliferation inhibition) values of each compounds were determined and summarized in .
Table 1. HDAC inhibition and cytotoxicity of the compounds synthesized.
Data in demonstrated that compound 5a showed strong cytotoxicity against all five cell lines tested. Especially on SW620 and PC3 cell lines, this compound was approximately five-fold more potent than SAHA. Thus, the introduction of the 1,3,4-thiadiazole heterocycle between the phenyl ring and the amide moiety led to a better analogue than SAHA in term of cytotoxicity. It should be mentioned that in the course of our study, we noticed an article by Guan et al.Citation17 in which compound 5a was reported. In this article, compound 5a was reported to inhibit HDAC (came from HeLa cell nucleus extracts, containing mainly HDAC1 and HDAC2) with IC50 value of 0.27 µM). However, no cytotoxic data for this compound were revealed.
The presence of one halogen atom on the phenyl ring, either at position 2, 3 or 4, retained the cytotoxicity or even slightly enhanced it (compounds 5b–d, 5f). It seemed that substitution at position 2 was the most favorable for cytotoxicity. For example, compound 5b and 5c with a chlorine substituent at position 2 and 3, respectively, were more cytotoxic than compound 5a in SW620 cell line (IC50 values of 0.34 and 0.45 μM versus 0.70 μM). Compound 5b was even 10-fold more active than 5a in NCI-460 cell line (IC50 value of 0.11 μM versus 1.07 μM). Especially, compound 5f was about 30-times more potent than compound 5a in AsPC-1 cells (IC50 value of 0.08 μM versus 2.71 μM). Of exception was compound 5e, bearing a 4-fluorine substituent, which showed decreased cytotoxicity compared to compound 5a. Nevertheless, the cytotoxicity of these compounds were still comparable to that of SAHA in five cell lines tested.
The introduction of an additional chlorine at position 6 (compound 5l), however, led to a dramatic decrease in cytotoxicity. Within the series, two compounds bearing nitro substituents (compounds 5j and 5 k) were the least cytotoxic ones. Compound 5 g, with a 4-methyl substituent, and compound 5i, with a bulkier 4-dimethylamino group on the phenyl ring, showed almost 10-fold decrease of cytotoxicity compared to compound 5a. However, it was observed that the 3,4-methylenedioxy, 4-methoxy or polymethoxy substitutions were relatively tolerable for cytotoxic activity of this compound series, as evidenced that compounds 5h and 5m–o were equally active or even slightly more cytotoxic than SAHA against all five cancer cell lines assayed.
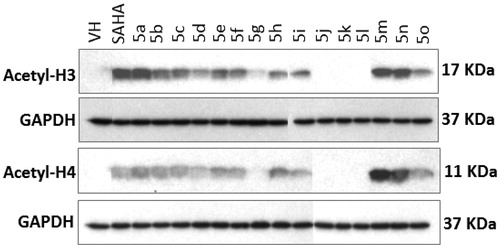
Next, we examined the effects of the compounds on the HDAC activity using the Western blot assay. Evaluation of HDAC inhibition in a whole cell system with primary antibodies against acetyl histone-H3, -H4, and GAPDH revealed that the acetylation of histone-H3 and histone-H4 increased in the presence of compounds 5a–f, 5h, 5i and 5m–o (), indicating that the HDAC activity had been inhibited. Meanwhile, in the presence of compounds 5g, 5j–l, the acetyl-H3 and acetyl-H3 were not observed, indicating that HDAC activity was not inhibited, leading to a complete deacetylation of histones H3 and H4. These results were found to be well correlated with the cytotoxicity, e.g. four compounds 5g, 5j–l were the least cytotoxic ones with the IC50 values against all five cancer cell lines higher than 11.52 µM. In contrast, other compounds like 5a–f, 5h, 5i and 5m–o showed much stronger cytotoxicity with IC50 values observed in low to sub-micromolar range in all five cancer cell lines tested (). The results demonstrate that inhibition of HDAC could be a prominent mechanism of these compounds’ cytotoxicity.
Figure 3. Effect of the compounds synthesized on histone acetylation in SW620 cells. Cells were treated with compounds (10 µM) for 24 h. Levels of acetylated histone-H3 and -H4 in total cell lysates were determined by Western immunoblot analysis. VH denotes vehicle treatment (blank control). SAHA (suberoylanilide hydroxamic acid) is a positive control.
Regarding drug-like properties of the compounds, it could be seen that all compounds have a molecular weight of less than 500 KDa and logP values from 0.86 to 2.64. The numbers of hydrogen bond donors (3) and hydrogen bond acceptors (8) fall within Lipinsky’s rule of five (). However, no clear correlation between logP values and bioactivity of the compounds was observed.
Docking study
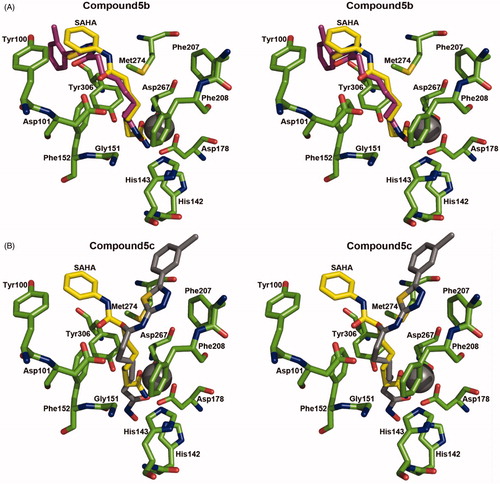
To speculate the interaction between these compounds and HDAC, we implemented docking experiments using the active site of HDAC. We selected the structure of HDAC8 in complex with SAHA as a docking template because its crystal structure is available (PDB ID: 1T69)Citation15, and it shares high structural similarity (DALI Z score = 40.4 and r.m.s.d. = 2.1 A)Citation18 and amino acid sequence similarity (46%) to HDAC4. We performed control docking experiments with SAHA to the crystal structure of HDAC8 using AutoDock Vina programCitation14 after SAHA was removed from the complex structure, as described previouslyCitation10. It was found from docking experiments that two compounds, 5b and 5c, were located in the active site () with more stable energy than that of SAHA and their aliphatic linkers seemed to interact with the residues in the active site tunnel of HDAC8 as the inhibition mode of SAHACitation15. Stabilization energies of predicted binding modes were −6.4 and −7.1 kcal/mol for compounds 5b and 5c, respectively, while the value for SAHA was −4.4 kcal/mol. Thus, based on the docking results, compounds 5b and 5c showed higher affinities than SAHA toward HDAC8.
Figure 4. Stereoview of the actual binding pose of SAHA and simulated docking pose of compound 5b (A) and 5c (B) to HDAC8. SAHA, compounds 5b, 5c, and the interaction parts of the HDAC8 were shown as a stick model.
Conclusion
In this study, fifteen 5-substitutedphenyl-1,3,4-thiadiazole-based hydroxamic acids analogous to SAHA have been designed, synthesized and evaluated for antitumor cytotoxicity and HDAC inhibitory activity. It was found that the presence of a 1,3,4-thiadiazole heterocycle between the phenyl ring and the amide moiety could be favorable for cytotoxicity. In general, halogen, methoxy or polymethoxy substituents on the phenyl ring were tolerated and, in some cases, led to increase in cytotoxicity. It was also found that the cytotoxicity of the compounds was well correlated with the inhibition of HDAC. Docking study performed with two representative compounds 5b (bearing 2-chlorophenyl) and 5c (bearing 3-chlorophenyl) revealed that these compounds had higher binding affinities to HDAC8 compared to SAHA. All of the compounds designed and synthesized possessed good drug-like properties. From this study, several compounds, such as 5b and 5c, were promising that warrant further investigation.
Declaration of interest
We acknowledge the principal financial supports from a National Foundation for Science and Technology of Vietnam (NAFOSTED), Grant number 104.01-2012.17 and the Medical Research Center program through the National Research Foundation of Korea, Grant number 2010-0029480. The docking study was supported by the National Research Foundation of Korea (NRF) grant for the Global Core Research Center (GCRC) funded by the Korea government (MSIP, Grant number 2011-0030001). The authors report no conflict of interest.
References
- De Ruijter AJM, Gennip AHV, Caron HN, et al. Histone deacetylases (HDACs): characterization of the classical HDAC family. Biochem J 2003;370:737–9
- Johnstone RW. Histone-deacetylase inhibitors: novel drugs for the treatment of cancer. Nature Rev Drug Disc 2002;1:287–99
- Marks PA, Rifkind RA, Richon VM, et al. Histone deacetylases and cancer: causes and therapies. Nature Rev Cancer 2001;1:194–202
- Witt O, Deubzer HE, Milde T, Oehme I. HDAC family: what are the cancer relevant targets? Cancer Lett 2009;277:8–21
- Dokmanovic M, Marks PA. Histone deacetylase inhibitors: discovery and development as anticancer agents. Expert Opin Investi Drug 2005;14:1497–511
- Glaser KB. HDAC inhibitors: clinical update and mechanism-based potential. Biochem Pharmacol 2007;74:659–871
- Dallavalle S, Cincinelli R, Nannei R, et al. Design, synthesis, and evaluation of biphenyl-4-yl-acrylohydroxamic acid derivatives as histone deacetylase (HDAC) inhibitors. Eur J Med Chem 2009;44:1900–12
- Bracker TU, Sommer A, Fichtner I, et al. Efficacy of MS-275, a selective inhibitor of class I histone deacetylases, in human colon cancer models. Int J Oncol 2009;35:909–20
- Finnin MS, Donigian JR, Cohen A, et al. Structures of a histone deacetylase homologue bound to the TSA and SAHA inhibitors. Nature 1999;401:188–93
- Oanh DTK, Hai HV, Hue VTM, et al. Benzothiazole-containing hydroxamic acids as histone deacetylase inhibitors and antitumor agents. Bioorg Med Chem Lett 2001;21:7509–12
- Skehan P, Storeng R, Scudiero D, et al. New colorimetric cytotoxicity assay for anticancer drug screening. J Natl Cancer Inst 1990;82:1107–12
- Nam NH, Kim Y, You YJ, et al. New constituents from Crinum latifolium with inhibitory activity against tube-like formation of human umbilical endothelial cells. Nat Prod Res 2004;18:485–91
- Wu L, Smythe AM, Stinson SF, et al. Multidrug-resistant phenotype of disease-oriented panels of human tumor cell lines used for anticancer drug screening. Cancer Res 1992;52:3029–34
- Trott O, Olson AJ. AutoDock Vina: improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J Comput Chem 2010;31:455–61
- Somoza JR, Skene RJ, Katz BA, et al. Structural snapshots of human HDAC8 provide insights into the class I histone deacetylases. Structure 2004;12:1325–34
- Schuttelkopf AW, van Aalten DM. PRODRG: a tool for high-throughput crystallography of protein-ligand complexes. Acta Crystallogr D Biol Crystallogr 2004;60:1355–63
- Guan P, Sun F, Hou X, et al. Design, synthesis and preliminary bioactivity studies of 1,3,4-thiadiazole hydroxamic acid derivatives as novel histone deacetylase inhibitors. Bioorg Med Chem 2012;20:3865–72
- Holm L, Rosenstrom P. Dali server: conservation mapping in 3D. Nucleic Acids Res 2010;38:W545–9