
Abstract
In order to find the new potent CK2 inhibitors the 60 derivatives of 2-aminopyrimidinone and their 6-aza-substituted analogs were synthesized and tested in vitro. Among them, the most efficient inhibitor 2-hydroxy-5-[4-(4-methoxyphehyl)-6-oxo-1,6-dihydropyrimidin-2-ylamino] benzoic acid was identified (IC50 = 1.1 μM). The structure--activity relationship study of newly synthesized derivatives was carried out and their binding mode with adenosine triphosphate-acceptor site of CK2 was proposed.
Introduction
Protein kinase CK2 (CK2) is a constitutively active pleiotropic kinase. It controls the key cellular activities and is involved in majority of signal transduction pathways. CK2 phosphorylates many substrates and participates in the complex series of cellular functions such as progression of the cell cycle, apoptosis, cell differentiation and transcription processesCitation1. CK2 overexpression and hyper-activation are observed in a large number of tumorsCitation2, including breast cancerCitation3, lungCitation4, prostateCitation5, colonCitation6, kidneyCitation7 and hematological malignanciesCitation8. Recently, it was reported that the decrease in CK2 regulation plays a critical role in inflammatoryCitation9 and infectious diseasesCitation10. Altogether, these facts raised the importance of finding pharmacologically useful inhibitors of this kinase. As a result, a large number of publications are devoted recently for the development of new CK2 inhibitorsCitation11.
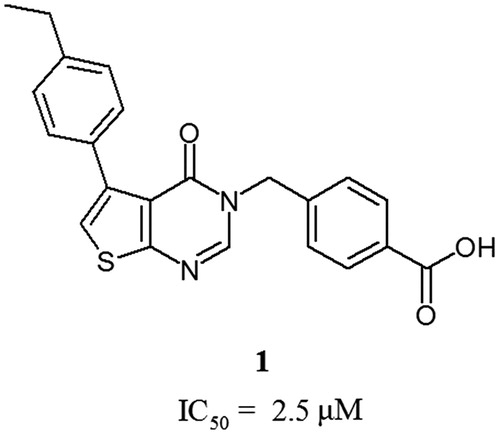
Accordingly to our previous report, the derivatives of fused pyrimidinone () are potent inhibitors of CK2Citation12.
Figure 1. Potent CK2 inhibitor representing fused pyrimidinone family.
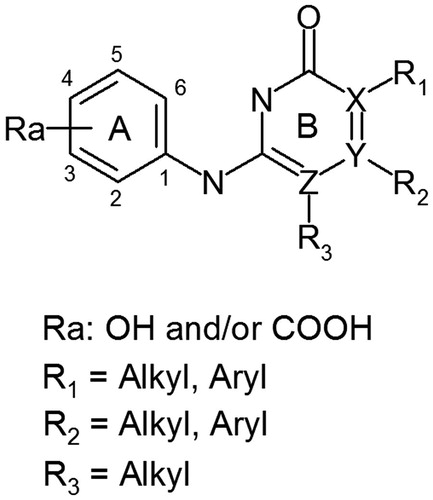
The aim of this study was the de novo design of protein kinase CK2 inhibitors based on 2-aminopyrimidinone and its 6-aza-analogues employing combinatorial chemistry techniques and molecular modeling methods. Considering the fact that most inhibitors of protein kinase CK2 published up to dateCitation12–16 have in their structure carboxyl and/or phenol groups, we focused our study only on derivatives containing these substituents ().
Figure 2. 2-Aminopyrimidinone scaffold.
Experimental
General
Starting materials and solvents were purchased from commercial suppliers and were used without further purification. 1HNMR spectra were recorded on a Varian VXR 400 instrument at 400 MHz (Agilent Technologies, Santa Clara, CA). 13C NMR spectra were recorded on a Varian VXR 400 instrument at 100 MHz. Chemical shifts are described as parts per million (δ) downfield from an internal standard of tetramethylsilane, and spin multiplicities are given as s (singlet), d (doublet), dd (double doublet), t (triplet), q (quartet) or m (multiplet). High-performance liquid chromatography-mass spectra (HPLC-MS) analysis was performed using the Agilent 1100 LC/MSD SL (Agilent Technologies) separations module and Mass Quad G1956B mass detector with electrospray ionization (Agilent Technologies) (+ve or −ve ion mode as indicated) and with HPLC performed using Zorbax SB-C18 (Agilent Technologies), Rapid Resolution HT Cartridge 4.6 × 30 mm 1.8-µ (Agilent Technologies P/N:823975-902) i.d. column, at a temperature of 40°C with gradient elution of 0–100% CH3CN (with 1 mL/L HCOOH): H2O (with 1 mL/L HCOOH) at a flow rate of 3 mL/min and a run time of 2.8 min. Compounds were detected at 215 nm using a Diode Array G1315B detector. All tested compounds had ≥90% purity as determined by this method. All purified synthetic intermediates had ≥95% purity as determined by this method.
Virtual screening
For virtual screening of a combinatorial library we used DOCK 4.0 package (Irwin “Tack” Kuntz's Group, San Francisco, CA)Citation17–20. Calculations of ligand geometry were performed using universal molecular mechanic force field (YFF) described previouslyCitation21,Citation22. Partial atomic charges of the ligands were calculated employing the Kirchhoff methodCitation23. Minimization of CK2 crystal structure (protein data bank (PDB) ID: 2ZJW) was performed with GROMACS (Biophysical Chemistry Department of University of Groningen, Netherlands)Citation24 package (steepest descent algorithm, GROMACS force field). Partial atomic charges of the receptor were calculated using assisted model building with energy refinement (http://en.wikipedia.org/wiki/AMBER) force field. Flexible docking and scoring were performed as reported elsewhereCitation25.
Biological testing
Selected compounds were tested using in vitro kinase assay. Each test was performed twice in a total reaction volume of 30 μL, containing 6 μg of peptide substrate RRRDDDSDDD (New England Biolabs Ltd., Herts, UK); 10 units of recombinant human CK2 holoenzyme (New England Biolabs); 50 μM adenosine triphosphate (ATP) and γ-labeled 32P ATP, diluted to specific activity 100 μCi/μM; CK2 buffer (20 mM Tris-HCl, pH 7.5; 50 mM KCl; 10 mM MgCl2) and inhibitor in varying concentrations. Incubation time was 20 min at 30°C. The reaction was stopped by adding an equal volume of 10% o-phosphoric acid and the reaction mixture was loaded onto 20-mm discs of phosphocellulose paper (Whatman plc, Kent, UK). Disks were washed three times with 1% o-phosphoric acid solution, air dried at room temperature and the level of radioactive signal was measured according to the Cherenkov method in a beta-counter (LKB Instruments; http://www.lkb.com.au/). As negative control an equal volume of dimethyl sulfoxide (DMSO) was added to the reaction mixture. Quercetin, a known CK2 inhibitorCitation26, was used as inhibition positive control. The final concentration of quercetin was 0.55 μM (which inhibited CK2 activity by 50%). Percent inhibition was calculated as ratio of substrate-incorporated radioactivity in the presence of inhibitor to the radioactivity incorporated in control reactions, i.e. in the absence of inhibitor. Serial dilutions of inhibitor stock solution were used to determine its IC50 concentration. The IC50 values represent means of triplicate experiments with standard error of the mean never exceeding 15%.
Chemistry
Accordingly to the synthetic proceduresCitation27, the series of building-blocks 2a–2u were synthesized. The structures of synthesized compounds are listed in . Their spectral properties are available online in Supplemental Information section.
Table 1. Chemical structures of building-blocks 2a–2u.
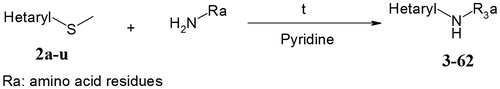
General procedure for the combinatorial synthesis of compounds 3–62
The solution of corresponding building-blocks 2a–2u (2.0 mmol) and corresponding aminophenol/amino acid (4.0 mmol) in pyridine was being heated for 8 h at 100°C. After that the reaction mixture was quenched by isopropyl alcohol (4 mL) and cooled down to room temperature. The precipitate was filtered, washed with isopropyl alcohol (2.0 mL) and air dried. The structures of synthesized compounds are summarized in . The spectra of the four selected compounds are presented below. The spectra of other compounds of the combinatorial library are available online in Supplemental Information section.
Table 2. Chemical structures and in vitro activities of compounds 3–15.
Table 3. Chemical structures and in vitro activities of compounds 16–34.
Table 4. Chemical structures and in vitro activities of compounds 35–62.
4-Methyl-3-[(4-methyl-6-oxo-1,6-dihydropyrimidin-2-yl)amino]benzoic acid (4)
Yield 58% (light gray solid). M.p. 227°C. LC-MS m/z 259 [M + H+], Rt = 0.96 min. 1H NMR (DMSO-d6) δ 2.06 (s, 3H, CH3), 2.30 (s, 3H, CH3), 5.51 (s, 1H, CH2), 7.22 (d, 1H), 7.60 (d, 1H), 8,32 (s, 1H).
3-[(4-Oxo-3,4-dihydroquinazolin-2-yl)amino]benzoic acid (7)
Yield 64% (light gray solid). M.p. >300° C dec. LC-MS m/z 285 [M + H+], Rt = 0.85 min. 1H NMR (DMSO-d6) δ 7.19 (t, 1H), 7.38–7.42 (m, 2H), 7.59–7.62 (m, 2H), 7.96 (dd, 1H), 8.08 (dd, 1H), 8.21 (s, 1H), 8.71(br. s., 1H, NH), 10.69 (br. s., 1H, NH), 12.54 (br. s., 1H, OH).
5-[(4-Butyl-6-oxo-1,6-dihydropyrimidin-2-yl)amino]-2-hydroxybenzoic acid (21)
Yield 51% (white solid). M.p. >260°C dec. LC-MS m/z 303 [M + H+], Rt = 0.76 min. 1H NMR (DMSO-d6) δ 0.93 (t, 3H, CH3), 1.30–1.40 (m, 2H, CH2), 1.59–1.66 (m, 2H, CH2), 2.34 (t, 2H, CH2), 5.51 (s, 1H), 6.82 (d, 1H), 7.65 (dd, 1H), 8.06 (d,1H), 11.01 (br. s., 1H, NH).
2-Hydroxy-5-{[4-(4-methoxyphenyl)-6-oxo-1,6-dihydropyrimidin-2-yl]amino}benzoic acid (25)
Yield 56% (light gray solid). M.p. >300°C dec. LC-MS m/z 353 [M + H+], Rt = 0.76 min. 1H NMR (DMSO-d6) δ 3.81 (s, 3H, OCH3), 6.33 (s, 1H), 6.95–7.00 (m, 3H), 7.58 (dd, 1H), 8.04 (d, 2H), 8.60 (s, 1H), 8.82 (br. s., 1H, NH), 10.97 (br. s., 1H, NH).
Results and discussion
To identify the potent inhibitors of CK2 among the 2-aminopyrimidinones and their 6-aza-analogues, the receptor-based virtual screening of the virtual library (1207 compounds) was carried out. The molecular docking of ligands was performed by DOCK 4.0 in ATP-binding siteCitation19,Citation20,Citation28 of CK2. Using the above-mentioned software, the 13 most promising compounds which had the best estimated binding energies were selected for further synthesis and biochemical evaluation.
The selected compounds were synthesized accordingly to the combinatorial chemistry procedures (). To synthesize the derivatives of 2-aminopyrimidinone and their 6-aza-analogs, the series of building-blocks 2a–2u () were synthesized accordingly to the previously reported proceduresCitation27.
Scheme 1. Combinatorial synthesis of 2-aminopyrimidinones and their 6-aza-analogs.
The newly synthesized compounds 3–15 were tested in vitro. Their chemical structures and results of the biochemical tests are shown in .
The biochemical testing revealed that compounds 4 and 7 were the most potent inhibitors of CK2 among 3–15 (IC50 is 28 and 24 μM, respectively).
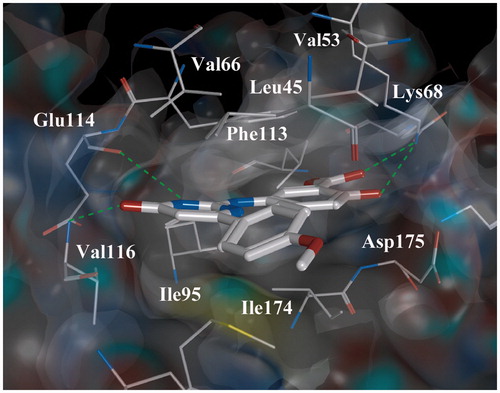
The analysis of a complex “Inhibitor 7-CK2” () obtained by the molecular docking method shows that stabilization of the ligand in ATP-binding site is achieved by the hydrophobic contacts with Leu45, Val53, Val66, Val116, Ile174 and stacking interaction with Phe113. Moreover, the hydrogen bond is present between ligand and Lys68 located deeply in the ATP-binding pocket. It is formed between the carboxyl oxygen of inhibitor and the side chain nitrogen of Lys68. It is worth to mention that this kind of interaction is observed in a number of small-molecule CK2 inhibitorsCitation29–33. Also, compound 7 forms additional hydrogen bond with Val116 in the hinge region of ATP-acceptor pocket. As it was previously published the bonding with that region is prerequisite to inhibitory activityCitation34. The H-bond is formed between the oxygen of pyrimidinone heterocycle and the backbone NH of Val116. It was found that the same interaction exists between inhibitor 4 and CK2.
Figure 3. Complex “compound 7-CK2” obtained by the molecular docking method. Intermolecular H-bonds are shown as dotted lines.
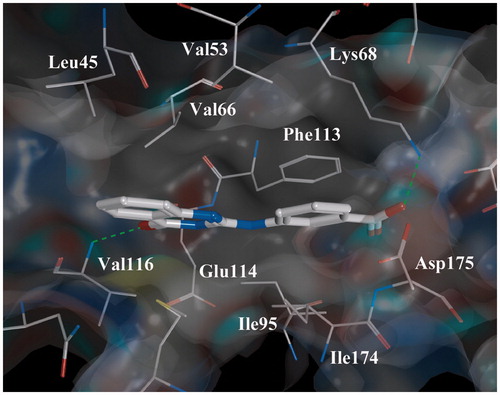
Thus, the analysis of obtained complexes showed that compounds 4 and 7 are complementary to the CK2 ATP-binding site. One of the key substituents that define the compounds activity is a carboxyl group at the third position of the ring A. For instance, compound 4 with a carboxyl group in this position has IC50 value 28 μM, while compound 3 with this substituent in the position 4 of the ring A is not active.
In order to increase the biological activity of the ligands, we carried out chemical optimization of 2-aminopirymidinons and their 6-aza-analogues according to the models obtained by molecular modeling methods. To perform the optimization of above-mentioned derivatives the following approaches were suggested:
to synthesize compounds with carboxyl residue at position 3 and hydroxyl residue at position 4 of the ring A (). It was expected that two acceptor groups (carboxyl and hydroxyl) in ortho-position will be favorable to formation stronger H-bonds with Lys68 of CK2 ATP-binding site;
to synthesize derivatives with the acceptor groups at ortho-, meta- or para-position of the ring A (). The aim of these modifications was to study influence of the acceptors and their position on hydrogen bonding between inhibitors and Lys68 of the ATP-binding site;
to synthesize ligands substituted with the nitrogen in a ring B at positions X, Y and Z (at the same time), X and Y (at the same time) (see ). This was suggested in order to evaluate the influence of number and position of nitrogen atoms on inhibitors activity and
to synthesize the derivatives containing hydrophobic aliphatic and aromatic substituents: R1 (methyl, butylene, t-butyl, benzyl, phenyl, phenylene, etc.), R2 (methyl, ethyl, n-propyl, iso-propyl, n-butyl, butylene, phenyl, phenylene, etc.) and R3 (methyl) in the ring B (). It was important to investigate the interaction between substituents and hydrophobic region II of CK2 ATP-acceptor pocket. It should be noted that these substituents may promote the formation of new hydrophobic contacts between the inhibitor and amino acid residues in the CK2-binding site (leading to increased inhibitory activity) and, in the same time, create steric hindrances destabilizing the ligand in CK2 ATP-acceptor pocket.
According to the chemical optimization plan, using building-blocks 2a–2u (), the 47 new compounds 16–62 were synthesized () and assayed in vitro ( and ).
The results of in vitro testing show that 10 optimized compounds inhibit the protein kinase CK2. 5-Aminosalicylic acid derivatives (17, 21, 22, 23, 25 and 29) with the carboxyl group at position 3 and hydroxyl group at position 4 of the ring A have the activities in the range of 1.1–20 μM (). Among aminophenol derivatives, the most active ligands were 35, 38, 43 and 44. These inhibitors show activity in the range of 15–27 μM and have a hydroxyl group at position 4 of the ring A ().
Activities of the six aminosalicylic acid derivatives depend on the size of substituents R1 and R2 in the ring B. Ligands with short substituents R2 such as ethyl, n-propyl and i-propyl were found inactive (compounds 18, 19 and 20) or showed low activity (compound 17, IC50 = 20 μM). At the same time, ligands with bigger substituents R2 made positive contributions to activity. For example, n-butyl-substituted compound 21 has the activity IC50 = 9.5 μM. Moreover, compound 25 () with 4-methoxyphenyl as R2 was the most potent among all tested ligands (IC50 = 1.1 μM). Thus, the large hydrophobic groups such as n-butyl, butylene, phenyl, phenylene and 4-methoxyphenyl are required at position Y of the ring B.
Figure 4. Structure of the most active CK2 inhibitor obtained as a result of chemical optimization.
On the contrary, increasing the size of the substituent R1 led to lower activities. For instance, compounds 26, 27 and 28 with relatively large groups such as benzyl, p-methylbenzyl and p-chlorobenzyl, respectively, were inactive. At the same time, compounds 17, 22, 23 and 29 with smaller R1 substituents (hydrogen, methyl, butylene and phenylene) have IC50 values 20, 17, 12 and 19 μM, respectively. Thus, the small groups such as hydrogen or methyl are required at position X of the ring B.
Among aminophenol derivatives of all four active compounds (35, 38, 43 and 44) have hydroxyl group at position 4 of the ring A (). Noteworthy, insertion of OH group at position 3 of the ring A clearly led to the complete loss of activity which is evident in compound pairs 7 and 45, 35 and 37, 44 and 45 (IC50 are 24 μM and >30, 23 μM and >30, 27 μM and >30 μM, respectively). Exactly as in the case of aminosalicylic acid derivatives, the inhibitory activity of aminophenol derivatives was influenced by the structure of substituents R1 and R2 of the ring B and the same trends were observed. For example, all active aminophenol derivatives (35, 38, 43 and 44) have small substituent R1 at position X of the ring B. Furthermore, the same as in the case of aminosalicylic acid derivatives, increase in the size of R2 at position Y of the ring B improved the activities (row 35, 38 and 43). The compound 43 with the phenyl group was more active (IC50 = 15 μM) than the compound 38 with the n-propyl group (IC50 = 20 μM) and compound 35 with the methyl group (IC50 = 23 μM).
The inhibitory properties of ligands were also affected by a substituent R4 at position 2 of the ring A. Compounds 35 and 38 substituted by hydrogen at this position showed the corresponding activities of 23 and 20 μM, whereas compounds 36 and 39 substituted by methyl groups were inactive.
In overall, the presence of carboxyl group in position 3 of the ring A and a hydroxyl group at position 4 had a mixed influence on inhibitory activities. In the one case, the presence of carboxyl groups led to the disappearance of activity (compounds 19 Ta 38, IC50 was >30 and 20 μM, respectively), in the other case the inhibitory properties of the ligands were almost the same (compounds 17 Ta 35, IC50 20 and 23 μM, respectively), in another instance it increased the activity (compounds 29 and 44, IC50 was 19 and 27 μM, respectively).
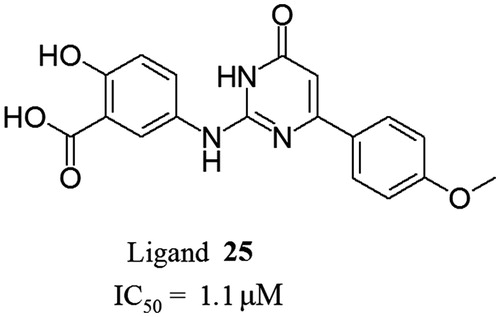
It is important to add that all aza-analogs of 2-aminopyrimidinone containing nitrogen at position X and/or Y were not active (ligands 10–15, 30–34 and 54–62). Accordingly to the calculations performed using Instant JChemCitation35, it was found that electronegative charge of oxygen atom in pyrimidinone heterocycle is decreased when additional atom of nitrogen is inserted into the heterocycle (). Thus, the following values were obtained for 2-aminopyrimidin-4-ones, 2-amino-6-azapyrimidin-4-ones and 4-amino-6-azapyrimidin-2-ones −0.51, −0.45 and −0.47, respectively. The oxygen of pyrimidinone cycle is forming important hydrogen bonds with amino acid residues of the kinase ( and ). Therefore, the decrease in negative charge on pyrimidinone’s oxygen a likely is reducing the stability of ligand-receptor complex. As a result, all the above-mentioned aza-analogs of 2-aminopyrimidine are binding with ATP-acceptor pocket weaker than 2-aminopyrimidines themselves and are not active.
Figure 5. Calculated electronegative charges at the oxygen atoms for (a) 2-aminopyrimidin-4-one, (b) 2-amino-6-azapyrimidin-4-one and (c) 4-amino-6-azapyrimidin-2-one.
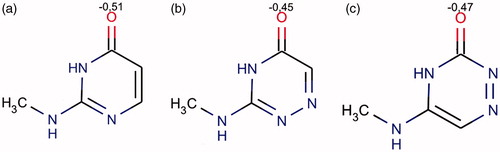
Figure 6. Complex of ligand 25 with CK2. Complex is obtained using a molecular docking method. Intermolecular H-bonds are shown as dotted lines.
To explain the obtained structure–activity relationships, the complexes of the ligands and CK2 were generated by molecular docking methods. It was shown that the ligands are complementary to the ATP-binding site of CK2 and located similarly to compounds 3–15 (see above). The compound 25 () which has the highest inhibitory activity among the other optimized ligands forms a number of additional intermolecular bonds with the CK2 catalytic subunit. The first additional bond is formed between hydroxyl group of the ligand and Lys68 while the bond between the ligand’s carboxyl group and this amino acid is remained. The second bond is formed with Glu114 in the hinge region of CK2. In addition, the 4-methoxyphenyl substituent of 25 is responsible for rigid ligand fixation in a hydrodrophobic pocket-2 of kinase ATP-binding site. Presumably, the summation of the above-mentioned additional interactions is responsible for higher inhibitory activity of 25 comparing to the other synthesized ligands.
As it was noticed before, the absence of activity in compounds 26, 27 and 28 is associated with presence of the large substituents R1 at position X of the ring A. The analysis of the obtained complexes showed that insertion of hydrophobic groups to this position leads to increased steric hindrances in the middle of the ATP-binding site which results in breaking of hydrogen bond with Val116. The substituent R4 of compounds 35–62 () is causing conformationally disfavored orientation of the ligands in the middle of the ATP-binding site. As a result, the hydrogen bond with Lys68 is broken. This causes the complete loss of inhibitory activity of compounds.
Thus, there are two key structural feature required for activity of 2-aminopyrimidinone derivatives. The presence of acceptor group in the ring A promotes stronger interaction with Lys68 which improves inhibitory properties of 2-aminopyrimidinone derivatives. And additional stabilization of the complex is provided by the large substituents oriented into hydrophobic pocket-II of the ATP-acceptor site of CK2 ().
Therefore, the 2-aminopyrimidinone derivatives are hopeful for further study and optimization which should be achieved by the formation of additional interactions with Glu114 and Asp175 as well as by enhancement of the complex stability via addition of hydrophobic groups into the pyrimidinone ring.
Conclusions
It was established that 2-aminopyrimidinone derivatives are promising class of compounds for the development of new potent inhibitors of protein kinase CK2. In order to find new and effective inhibitors of human protein kinase CK2, the new 2-aminopyrimidinone derivatives and their 6-aza-analogs were developed by methods of combinatorial chemistry and molecular docking. Among in vitro tested compounds, the most active ligand 2-hydroxy-5-[4-(4-methoxyphenyl)-6-oxo-1,6-dihydropyrimidin-2-ylamino] benzoic acid 25 was identified (IC50 = 1.1 μM). The relationship “chemical structure-biological activity” for 2-aminopyrimidinone derivatives and their 6-aza-analogs was investigated and the binding mode of the synthesized ligands with CK2 acceptor site was proposed. It was established that only 2-aminopyrimidinone derivatives inhibited protein kinase CK2 with the activities in the range of 1.1–30 μM while their 6-aza-analogs were not active. Probably, it is associated with the lower ability of carbonyl group of 6-aza-2-aminopyrimidinone ring to form hydrogen bonds with amino acid residues of CK2 ATP-binding site.
Declaration of interest
The authors report no conflicts of interest. The authors alone are responsible for the content and writing of this article.
Supplementary material available online Supplemental Information
Supplementary Material
Download PDF (159.7 KB)References
- Volynets GP, Golub AG, Bdzhola VG, et al. The role of protein kinase CK2 in the process of oncogenesis, regulation of apoptosis and cellular stress response. Ukr Bioorg Acta 2007;2:25–32
- Ruzzene M, Pinna LA. Addiction to protein kinase CK2: a common denominator of diverse cancer cells? Biochim Biophys Acta 2010;3:499–504
- Landesman-Bollag E, Romieu-Mourez R, Song DH, et al. Protein kinase CK2 in mammary gland tumorigenesis. Oncogene 2001;20:3247–57
- Lin YC, Hung MS, Lin CK, et al. CK2 inhibitors enhance the radio sensitivity of human non-small cell lung cancer cells through inhibition of stat3 activation. Cancer Biother Radiopharm 2011;26:381–8
- Laramas M, Pasquier D, Filhol O, et al. Nuclear localization of protein kinase CK2 catalytic subunit (CK2-alpha) is associated with poor prognostic factors in human prostate cancer. Eur J Cancer 2007;43:928–34
- Zou J, Luo H, Zeng Q, et al. Protein kinase CK2α is overexpressed in colorectal cancer and modulates cell proliferation and invasion via regulating EMT-related genes. J Transl Med 2011;9:97
- Stalter G, Siemer S, Becht E, et al. Asymmetric expression of protein kinase CK2 subunits in human kidney tumors. Biochem Biophys Res Commun 1994;202:141–7
- Piazza F, Manni S, Ruzzene M, et al. Protein kinase CK2 in hematologic malignancies: reliance on a pivotal cell survival regulator by oncogenic signaling pathways. Leukemia 2012;26:1174–9
- Singh NN, Ramji DP. Protein kinase CK2, an important regulator of the inflammatory response? J Mol Med (Berl) 2008;86:887–97
- Ivanov KI, Puustinen P, Gabrenaite R, et al. Phosphorylation of the potyvirus capsid protein by protein kinase CK2 and its relevance for virus infection. Plant Cell 2003;15:2124–39
- Cozza G, Pinna LA, Moro S. Protein kinase CK2 inhibitors: a patent review. Expert Opin Ther Pat 2012;22:1081–97
- Kotey IM, Balanda AO, Bdzhola VG, et al. Synthesis and biological evaluation of thienopyrimidinones as CK2 protein kinase inhibitors. Ukr Bioorg Acta 2008;2:3–9
- Pagano MA, Poletto G, Di Maira G, et al. Tetrabromocinnamic acid (TBCA) and related compounds represent a new class of specific protein kinase CK2 inhibitors. ChemBioChem 2007;8:129–39
- Sarno S, De Moliner E, Ruzzene M, et al. Biochemical and three-dimensional-structural study of the specific inhibition of protein kinase CK2 by [5-oxo-5,6-dihydroindolo-(1,2-a)quinazolin-7-yl] acetic acid (IQA). Biochem J 2003;374:639–46
- Golub AG, Yakovenko OY, Prykhod’ko AO, et al. Evaluation of 4,5,6,7-tetrahalogeno-1H-isoindole-1,3(2H)-diones as inhibitors of human protein kinase CK2. Biochim Biophys Acta 2008;1784:143–9
- Golub AG, Yakovenko OY, Bdzhola VG, et al. Evaluation of 3-carboxy-4(1H)-quinolones as inhibitors of human protein kinase CK2. J Med Chem 2006;49:6443–50
- Shoichet BK, Stroud RM, Santi DV, et al. Structure-based discovery of inhibitors of thymidylate synthase. Science 1993;259:1445–50
- Bodian DL, Yamasaki RB, Buswell RL, et al. Inhibition of the fusion-inducing conformational change of influenza hemagglutinin by benzoquinones and hydroquinones. Biochemistry 1993;32:2967–78
- Ring CS, Sun E, McKerrow JH, et al. Structure-based inhibitor design by using protein models for the development of antiparasitic agents. Proc Natl Acad Sci USA 1993;90:3583–7
- Ewing TJ, Makino S, Skillman AG, et al. DOCK 4.0: search strategies for automated molecular docking of flexible molecule databases. J Comput Aided Mol Des 2001;15:411–28
- Yakovenko OY, Oliferenko AA, Golub AG, et al. The new method of distribution integrals evaluations for high throughput virtual screening. Ukr Bioorg Acta 2007;1:52–62
- Yakovenko OY, Li YY, Oliferenko AA, et al. Ab initio parameterization of YFF1, a universal force field for drug-design applications. J Mol Model 2012;18:663–73
- Yakovenko OY, Oliferenko AA, Bdzhola VG, et al. Kirchhoff atomic charges fitted to multipole moments: implementation for a virtual screening system. J Comput Chem 2008;29:1332–43
- Lindahl E, Hess B, van der Spoel D. GROMACS 3.0: a package for molecular simulation and trajectory analysis. J Mol Mod 2001;7:306–17
- Prykhod’ko AO, Yakovenko OY, Golub AG, et al. Evaluation of 4H-4-chromenone derivatives as inhibitors of protein kinase CK2. Biopolym Cell 2005;21:1–6
- Meggio F, Pagano M, Moro S, et al. Inhibition of protein kinase CK2 by condensed polyphenolic derivatives. An in vitro and in vivo study. Biochemistry 2004;43:12931–6
- Chekanov MO, Tarnavskiy SS, Synyugin AR, et al. Solution phase combinatorial synthesis of a diverse library of 2-aminopyrimidinones and their 6-aza-analogs. Ukr Bioorg Acta 2011;10:29–40
- Ewing TJ, Kuntz ID. Critical evaluation of search algorithms for automated molecular docking and database screening. J Comput Chem 1996;18:1175–89
- Suzuki Ya, Oishi Sh, Takei Y, et al. Design and synthesis of a novel class of CK2 inhibitors: application of copper- and gold-catalysed cascade reactions for fused nitrogen heterocycles. Org Biomol Chem 2012;10:4907--15
- Fuchi N, Iura Y, Kaneko H, et al. Discovery and structure–activity relationship of 2,6-disubstituted pyrazines, potent and selective inhibitors of protein kinase CK2. Bioorg Med Chem Lett 2012;22:4358–61
- Golub AG, Bdzhola VG, Briukhovetska NV, et al. Synthesis and biological evaluation of substituted (thieno[2,3-d]pyrimidin-4-ylthio)carboxylic acids as inhibitors of human protein kinase CK2. Eur J Med Chem 2011;46:870–6
- Golub AG, Bdzhola VG, Kyshenia YV, et al. Structure-based discovery of novel flavonol inhibitors of human protein kinase CK2. Mol Cell Biochem 2011;356:107–15
- Ferguson AD, Sheth PR, Basso AD, et al. Structural basis of CX-4945 binding to human protein kinase CK2. FEBS Lett 2011;585:104–10
- Backes AC, Zech B, Felber B, et al. Small-molecule inhibitors binding to protein kinases. Part I: exceptions from the traditional pharmacophore approach of type I inhibition. Expert Opin Drug Discov 2008;3:1409–25
- Instant JChem, release version 5.5.1. ChemAxon. Available from: http://www.chemaxon.com