
Abstract
Herein, we report the design, syntheses and in vitro anti-microbial activity of two series of rhodanines with chalcone moiety. Anti-microbial tests showed that some of the synthesized compounds exhibited good inhibition (MIC = 1–8 µg/mL) against multi-drug-resistant Gram-positive organisms, including methicillin resistant and quinolone-resistant Staphylococcus aureus, in which the compound 4g was found to be the most potent with minimum inhibitory concentration (MIC) value of 1 µg/mL against two methicillin-resistant S. aureus.
Introduction
The severe infections caused by multi-drug-resistant microorganisms such as methicillin-resistant Staphylococcus aureus (MRSA) and glycopeptide-resistant Staphylococci (GRSA) have substantially increased over the last few years. However, the number of effective anti-bacterial agents has decreased gradually with the appearance of new resistance mechanisms of microorganisms, which is a major concern in medicine, especially for hospitals nowCitation1,Citation2.
Rhodanine scaffold is a powerful device for medicinal chemist and have a broad substrate scope for the synthesis of various heterocyclic moieties with wide range of pharmacological activities such as anti-bacterialCitation3–5, anti-fungalCitation6, anti-diabeticCitation7, anti-tubercularCitation8,Citation9, anti-HIVCitation10,Citation11 and anthelmintic agentsCitation12,Citation13. Recently, this scaffold showed important therapeutic targets against plasmodium fatty acid synthesis pathway (FAS-II) in the parasites which is distinct from type I fatty acid synthesis pathway (FAS-I) in humanCitation14. Hardej et al.Citation15 reported some rhodanine derivatives as anti-microbial agents against MRSA strains.
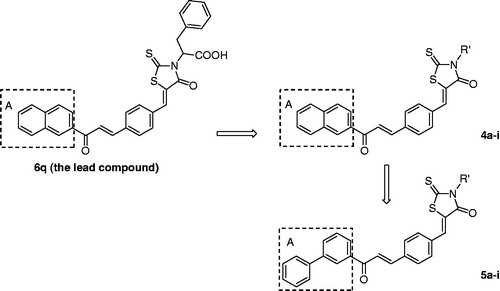
In our previous study, we designed and synthesized three series of rhodanine derivatives bearing chalcone (, I), 4-(2-oxo-2-phenylethoxy)benzene (, II) and 5-aryloxy pyrazole moieties (, III), respectivelyCitation16–23. Their anti-bacterial tests in vitro showed that these compounds all exhibited good inhibitory activity against the Gram-positive microorganisms especially for the multi-drug-resistant clinical isolates such as MRSA and quinolone-resistant S. aureus (QRSA). Especially in the series of I, compound 6q (A moiety was a naphthalene nucleus) showed the strongest activity with a minimum inhibitory concentration (MIC) value of 2 µg/mL against the multi-drug-resistant clinical isolatesCitation23. In the present work, as a part of our ongoing research, a new series of rhodanine derivatives (4a–i) were designed using 6q as the lead compound, in which the modification of 6q was focused on reserving the naphthalene nucleus, substituting the acetic acid group on the three-position of the rhodanine with different amino acid side chains (including D- or L-phenylalanine, D- or L-tyrosine, D- or L-valine, D- or L-leucine and L-isoleucine) as shown in . Moreover, another series of rhodanine derivatives (5a–i) were also been designed, in which the naphthalene nucleus was replaced by a biphenyl group in order to discuss the structure–activity relationship better. Both R- and S-configuration were involved in the two series of compounds so as to investigate the contribution of configuration on their anti-bacterial activity (). Thus, two series of rhodanine derivatives, including 17 new compounds were synthesized and screened for their anti-bacterial activities.
Figure 1. The structures of compounds I, II and III.
Figure 2. Lead compound and structure-based design of the target compounds.
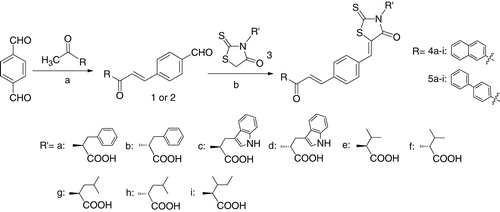
The target compounds (4a–i and 5a–i) were synthesized according to the route described in . The intermediates 1 and 2 were prepared by reacting acetophenones with terephthalaldehyde in the ethanol–NaOH/H2O conditionCitation24. Compounds 4a–i and 5a–i were obtained in good yields via a Knoevenagel condensation of compounds 1 (or 2) and N-substituted rhodanines using the reported procedureCitation25. The newly synthesized compounds were characterized by IR, 1H-NMR and mass spectra. Taking compound 4b as an example, the mass spectroscopy of 4b displayed an M + H signal at m/z 550, which was corresponding to its molecular weight of 549. In the 1H-NMR spectrum of compound 4b, in addition to the aromatic protons of benzene ring protons (δ = 7.15–8.97 ppm), a sharp singlet due to vinylic hydrogen was observed at 8.08 ppm, and a broad singlet due to methyne hydrogen linked to nitrogen-atoms was observed at 5.90 ppm. Meanwhile, it has been reported that the (Z)- or (E)-geometry was readily identified by 1H-NMR, as the vinylic proton is more de-shielded in the (Z)-isomer than the (E)-isomer. In (Z)-form, vinylic proton appeared at 7.21 ppm due to the magnetic anisotropy effects of the carbonyl group on the vinylic proton, while in (E)-form the resonance should be around 6.50 ppmCitation26,Citation27. In our 1H-NMR spectra, only a set of signals around 8.08 ppm was appeared, which confirmed our products were only in (Z)-configuration as thermodynamically favored structures.
Scheme 1. Synthetic scheme for the synthesis of compounds 4a–i and 5a–i.
The physicochemical properties of them are presented in the “Chemistry” section. Their anti-bacterial activities were all evaluated by a serial dilution method to obtain the MIC with different strains, including multi-drug-resistant clinical isolates.
Experimental protocols
Chemistry
Melting points were determined in open capillary tubes and were uncorrected. Reaction courses were monitored by TLC on silica gel-precoated F254 Merck plates. Developed plates were examined with ultra violet lamps (254 nm). IR spectra were recorded (in KBr) on a FTIR1730. 1H NMR spectra were measured on a Bruker AV-300 spectrometer using tetramethylsilane as the internal standard. Mass spectra were measured on an matrix assisted laser desorption ionization-time of flight/time of flight (MALDI-TOF/TOF) mass spectrometer (Bruker Daltonik, Bremen, Germany). Specific optical rotation was measured on a Digital automatic polariscope JASCO P-1020 (JASCO, Tokyo, Japan). The major chemicals were purchased from Sigma-Aldrich (St. Louis, MO) and Fluka Companies (Milwaukee, MI).
The general synthesis route of compound 1 and 2
To a stirred solution of terephthalaldehyde (12 mmol) in 15 mL ethanol and 15 mL 5% NaOH, the solution of 1-(naphthalen-2-yl)ethanone or 1-([1,1′-biphenyl]-4-yl)ethanone (10 mmol) in 15 mL ethanol was added drop-wise about 30 min in an ice-bath. The mixture was stirred at room temperature for 8 h and the pH value of the mixture was adjusted to 3 using dilute HCl. After adding 25 mL water into the mixture, the resulting precipitate was filtered, washed with water and recrystallized with ethanol to get the yellow solid.
Synthesis of rhodanine-3-acids (3)
To a solution of appropriate amino acid (30.3 mmol) and sodium hydroxide (30.3 mmol) in 25 mL of water, carbon disulfide (30.3 mmol) was added, and the resulting mixture was stirred vigorously overnight. An aqueous solution of sodium chloroacetate (30.3 mmol) was added to the mixture and stirring was continued at 23 °C for 3 h. Then, the reaction mixture was acidified with dilute HCl until pH 1.0 and refluxed overnight. The reaction mixture was neutralized with saturated NaHCO3 solution. The resultant solution was acidified again with dilute HCl. The cyclized product was extracted with ethyl acetate, dried over anhydrous sodium sulfate, evaporated under vacuum, and the residue was purified by column chromatography (dichloromethane/methanol = 95:05) to afford a brown liquid.
General procedure for the preparation of compounds 4a–i
To a suspension of 6 (2 mmol) in dry ethanol (10 mL), (E)-4-(3-(naphthalen-2-yl)-3-oxoprop-1-en-1-yl)benzaldehyde 2 (2.1 mmol), catalytic amounts of piperidine (0.1 mmol) and glacial acetic acid (0.1 mmol) were added. The resulting mixture was stirred and refluxed overnight. After cooling, the solvent was evaporated in vacuo, dried, and purified by silica gel column chromatography (dichloromethane/methanol = 100:1). The yield, melting point and spectral data of each compound are given below.
(S)-2-((Z)-5-(4-((E)-3-(naphthalen-2-yl)-3-oxoprop-1-en-1-yl)benzylidene)-4-oxo-2-thioxothiazolidin-3-yl)-3-phenylpropanoic acid (4a)
Yellow solid; yield 62%; m.p. 218–219 °C. : −202.3 (c = 0.54, acetone). IR (KBr) cm−1: 3449 (OH), 1687 (C=O). 1H NMR (DMSO-d6, 300 MHz, ppm): δ 3.55 (d, J = 5.7 Hz, 2H, CH2), 5.76 (br.s, 1H, NCH), 7.82 (d, 1H, J = 15.7 Hz, CH=CH), 8.09 (s, 1H, C=CH), 8.23 (d, 1H, J = 15.7 Hz, CH=CH), 7.16–8.29 (m, 16H, Ar–H), 13.44 (s, 1H, COOH). MS m/z 572 ([M + Na]+). Electrospray ionization high resolution mass spectrometry (ESI-HRMS) calcd for C32H23NNaO4S2+ ([M + Na]+): 572.0961; found: 572.0979.
(R)-2-((Z)-5-(4-((E)-3-(naphthalen-2-yl)-3-oxoprop-1-en-1-yl)benzylidene)-4-oxo-2-thioxothiazolidin-3-yl)-3-phenylpropanoic acid (4b)
Yellow solid; yield 15%; m.p. 204–206 °C. : 211.9 (c = 0.62, acetone). IR (KBr) cm−1: 3424 (OH), 1709 (C=O). 1H NMR (DMSO-d6, 300 MHz, ppm): δ 3.52 (d, J = 5.5 Hz, 2H, CH2), 5.90 (br.s, 1H, NCH), 7.82 (d, 1H, J = 15.7 Hz, CH=CH), 8.08 (s, 1H, C=CH), 8.25 (d, 1H, J = 15.7 Hz, CH=CH), 7.15–8.97 (m, 16H, Ar–H), 13.44 (s, 1H, COOH). MS m/z 550 (M + H). ESI-HRMS calcd for C32H23NNaO4S2+ ([M + Na]+): 572.0961; found: 572.0960.
(S)-3-(1H-indol-3-yl)-2-((Z)-5-(4-((E)-3-(naphthalen-2-yl)-3-oxoprop-1-en-1-yl)benzylidene)-4-oxo-2-thioxothiazolidin-3-yl)propanoic acid (4c)
Yellow solid; yield 23%; m.p. 130–132 °C. : −237.9 (c = 0.30, acetone). IR (KBr) cm−1: 3386 (OH), 2889 (NH), 1711(C=O). 1H NMR (DMSO-d6, 300 MHz, ppm): δ 3.57–3.75 (m, 2H, CH2), 5.90 (br.s, 1H, N–CH), 7.82 (d, 1H, J = 15.5 Hz, CH=CH), 8.07 (s, 1H, C=CH), 8.24 (d, 1H, J = 15.5 Hz, CH=CH), 7.07–8.96 (m, 16H, Ar–H), 10.81 (s, 1H, NH), 13.21 (s, 1H, COOH). MS m/z 589 (M + H). ESI-HRMS calcd for C34H24N2NaO4S2+ ([M + Na]+): 611.1070; found: 611.1073.
(R)-3-(1H-indol-3-yl)-2-((Z)-5-(4-((E)-3-(naphthalen-2-yl)-3-oxoprop-1-en-1-yl)benzylidene)-4-oxo-2-thioxothiazolidin-3-yl)propanoic acid (4d)
Yellow solid; yield 17%; m.p. 120–122 °C. : 227.6 (c = 0.63, CH3CO2C2H5). IR (KBr) cm−1: 3406 (OH), 2909 (NH), 1713 (C=O). 1H NMR (DMSO-d6, 300 MHz, ppm): δ 3.55–3.78 (m, 2H, CH2), 5.89 (br.s, 1H, N–CH), 7.84 (d, 1H, J = 15.6 Hz, CH=CH), 8.09 (s, 1H, C=CH), 8.27 (d, 1H, J = 15.6 Hz, CH=CH), 6.89–8.99 (m, 16H, Ar–H), 10.83 (s, 1H, NH), 13.37 (s, 1H, COOH). MS m/z 589 (M + H). ESI-HRMS calcd for C34H24N2NaO4S2+ ([M + H]+): 589.1250; found: 589.1244.
(S)-3-methyl-2-((Z)-5-(4-((E)-3-(naphthalen-2-yl)-3-oxoprop-1-en-1-yl)benzylidene)-4-oxo-2-thioxothiazolidin-3-yl)butanoic acid (4e)
Yellow solid; yield 28%; m.p. 206–208 °C. : 45.2 (c = 0.59, CHCl3). IR (KBr) cm−1: 3406 (OH), 1713 (C=O). 1H NMR (DMSO-d6, 300 MHz, ppm): δ 0.75 (d, 3H, J = 6.8 Hz, CH–CH3), 1.20 (d, 3H, J = 6.5 Hz, CH–CH3), 2.69–2.75 (m, 1H, CH(CH3)2), 5.19 (br.s, 1H, N–CH), 7.84 (d, 1H, J = 15.6 Hz, CH=CH), 7.92 (s, 1H, C=CH), 8.26 (d, 1H, J = 15.6 Hz, CH=CH), 7.63–8.97 (m, 11H, Ar–H), 13.22 (s, 1H, COOH). MS m/z 502 (M + H). ESI-HRMS calcd for C28H23NNaO4S2+ ([M + Na]+): 524.0961; found: 524.0947.
(R)-3-methyl-2-((Z)-5-(4-((E)-3-(naphthalen-2-yl)-3-oxoprop-1-en-1-yl)benzylidene)-4-oxo-2-thioxothiazolidin-3-yl)butanoic acid (4f)
Yellow solid; yield 15%; m.p. 136–139 °C. : 45.2 (c = 0.59, CHCl3). IR (KBr) cm−1: 3437 (OH), 1714 (C=O). 1H NMR (DMSO-d6, 300 MHz, ppm): δ 0.77 (d, 3H, J = 6.5 Hz, CHCH3), 1.21 (d, 3H, J = 6.1 Hz, CHCH3), 2.70–2.77 (m, 1H, CH(CH3)2), 5.18 (br.s, 1H, N–CH), 7.86 (d, 1H, J = 15.7 Hz, CH=CH), 7.94 (s, 1H, C=CH), 8.28 (d, 1H, J = 15.7 Hz, CH=CH), 7.35–8.99 (m, 11H, Ar–H), 13.18 (s, 1H, COOH). MS m/z 502 (M + H). ESI-HRMS calcd for C28H23NNaO4S2+ ([M + Na]+): 524.0961; found: 524.0966.
(S)-4-methyl-2-((Z)-5-(4-((E)-3-(naphthalen-2-yl)-3-oxoprop-1-en-1-yl)benzylidene)-4-oxo-2-thioxothiazolidin-3-yl)pentanoic acid (4g)
Yellow solid; yield 31%; m.p. 150–152 °C. : −5.1 (c = 0.58, CHCl3). IR (KBr) cm−1: 3260 (OH), 1721 (C=O). 1H NMR (DMSO-d6, 300 MHz, ppm): δ 0.87 (d, 3H, J = 6.8 Hz, CHCH3), 0.92 (d, 3H, J = 6.3 Hz, CHCH3), 1.47–1.53 (m, 1H, CH(CH3)2), 2.02–2.23 (m, 2H, CH2), 5.61 (br.s, 1H, NCH), 7.84 (d, 1H, J = 15.7 Hz, CH=CH), 8.13 (s, 1H, C=CH), 8.27 (d, 1H, J = 15.7 Hz, CH=CH), 7.63–8.98 (m 11H, Ar–H), 13.37 (s, 1H, COOH). MS m/z 516 (M + H). ESI-HRMS calcd for C29H25NNaO4S2+ ([M + Na]+): 538.1117; found: 538.1130.
(R)-4-methyl-2-((Z)-5-(4-((E)-3-(naphthalen-2-yl)-3-oxoprop-1-en-1-yl)benzylidene)-4-oxo-2-thioxothiazolidin-3-yl)pentanoic acid (4h)
Yellow solid; yield 16%; m.p. 198–200 °C. : 6.0 (c = 0.63, CHCl3). IR (KBr) cm−1: 3441 (OH), 1721 (C=O). 1H NMR (DMSO-d6, 300 MHz, ppm): δ 0.86 (d, 3H, J = 6.8 Hz, CHCH3), 0.91 (d, 3H, J = 6.3 Hz, CHCH3), 1.46–1.52 (m, 1H, CH(CH3)2), 2.01–2.20 (m, 2H, CH2), 5.59 (br.s, 1H, NCH), 7.83 (d, 1H, J = 15.6 Hz, CH=CH), 8.12 (s, 1H, C=CH), 8.25 (d, 1H, J = 15.6 Hz, CH=CH), 7.64–8.97 (m, 11H, Ar–H), 13.37 (s, 1H, COOH). MS m/z 516 (M + H). ESI-HRMS calcd for C29H25NNaO4S2+ ([M + Na]+): 538.1117; found: 538.1118.
(2S)-3-methyl-2-((Z)-5-(4-((E)-3-(naphthalen-2-yl)-3-oxoprop-1-en-1-yl)benzylidene)-4-oxo-2-thioxothiazolidin-3-yl)pentanoic acid (4i)
Yellow solid; yield 36%; m.p. 160–162 °C. : −45.8 (c = 0.63, CHCl3). IR (KBr) cm−1: 2967 (OH), 1717 (C=O). 1H NMR (DMSO-d6, 300 MHz, ppm): δ 0.79 (t, 3H, J = 7.2 Hz, CH2CH3), 0.92–0.97 (m, 2H, CH2CH3), 1.15 (d, 3H, J = 6.4 Hz, CHCH3), 1.20–1.24 (m, 1H, CHCH3), 5.23 (br.s, 1H, NCH), 7.83 (d, 1H, J = 15.8 Hz, CH=CH), 8.15 (s, 1H, C=CH), 8.26 (d, 1H, J = 15.8 Hz, CH=CH), 7.62–8.97 (m, 11H, Ar–H), 12.95 (s, 1H, COOH). MS m/z 538 (M + Na+). ESI-HRMS calcd for C29H25NNaO4S2+ ([M + H]+): 516.1298; found: 516.1298.
General procedure for the preparation of compounds 5a–i
To a suspension of 6 (2 mmol) in dry ethanol (10 mL), (E)-4-(3-([1,1′-biphenyl]-4-yl)-3-oxoprop-1-en-1-yl)benzaldehyde 3 (2.1 mmol), catalytic amounts of piperidine (0.1 mmol) and glacial acetic acid (0.1 mmol) were added. The resulting mixture was stirred and refluxed overnight. After cooling, the solvent was evaporated in vacuo, dried and purified by silica gel column chromatography (dichloromethane/methanol = 100:1). The yield, melting point and spectral data of each compound are given below.
(S)-2-((Z)-5-(4-((E)-3-([1,1′-biphenyl]-4-yl)-3-oxoprop-1-en-1-yl)benzylidene)-4-oxo-2-thioxothiazolidin-3-yl)-3-phenylpropanoic acid (5a)
Yellow solid; yield 38%; m.p. 123–125 °C. : −206.5 (c = 0.61, CH3CO2C2H5). IR (KBr) cm−1: 3005 (OH), 1711 (C=O). 1H NMR (DMSO-d6, 300 MHz, ppm): δ 3.54 (s, 2H, J = 5.6 Hz, CH2), 5.91 (br.s, 1H, NCH), 7.16–8.30 (m, 20H, Ar–H), 7.78 (s, 1H, C=CH), 13.53 (s, 1H, COOH). MS m/z 576 (M + H). ESI-HRMS calcd for C34H25NNaO4S2+ ([M + Na]+): 598.1117; found: 598.1126.
(R)-2-((Z)-5-(4-((E)-3-([1,1′-biphenyl]-4-yl)-3-oxoprop-1-en-1-yl)benzylidene)-4-oxo-2-thioxothiazolidin-3-yl)-3-phenylpropanoic acid (5b)
Yellow solid; yield 21%; m.p. 98–100 °C. : 187.2 (c = 0.63, CH3CO2C2H5). IR (KBr) cm−1: 3028 (OH), 1713 (C=O). 1H NMR (DMSO-d6, 300 MHz, ppm): δ 3.51 (s, 2H, J = 5.5 Hz, CH2), 5.83 (br.s, 1H, NCH), 7.10–8.21 (m, 20H, Ar–H), 7.77 (s, 1H, C=CH), 13.32 (s, 1H, COOH). MS m/z 576 (M + H). ESI-HRMS calcd for C34H25NNaO4S2+ ([M + Na]+): 598.1117; found: 598.1122.
(S)-2-((Z)-5-(4-((E)-3-([1,1′-biphenyl]-4-yl)-3-oxoprop-1-en-1-yl)benzylidene)-4-oxo-2-thioxothiazolidin-3-yl)-3-(1H-indol-3-yl)propanoic acid (5c)
Yellow solid; yield 35%; m.p. 108–112 °C. : −269.1 (c = 0.61, CH3CO2C2H5). IR (KBr) cm−1: 3398 (OH), 2890 (NH), 1706 (C=O). 1H NMR (DMSO-d6, 300 MHz, ppm): δ 3.55–3.80 (m, 2H, CH2), 5.89 (br.s, 1H, NCH), 7.08–8.29 (m, 20H, Ar–H), 7.78 (s, 1H, C=CH), 10.83 (s, 1H, NH), 13.53 (s, 1H, COOH). MS m/z 615 (M + H). ESI-HRMS calcd for C36H26N2NaO4S2+ ([M + Na]+): 637.1226; found: 637.1246.
(R)-2-((Z)-5-(4-((E)-3-([1,1′-biphenyl]-4-yl)-3-oxoprop-1-en-1-yl)benzylidene)-4-oxo-2-thioxothiazolidin-3-yl)-3-(1H-indol-3-yl)propanoic acid (5d)
Yellow solid; yield 19%; m.p. 126–128 °C. : 271.1 (c = 0.60, CHCl3). IR (KBr) cm−1: 3410 (OH), 2897 (NH), 1709 (C=O). 1H NMR (DMSO-d6, 300 MHz, ppm): δ 3.55–3.76 (m, 2H, CH2), 5.90 (br.s, 1H, NCH), 7.49–8.07 (m, 20H, Ar–H), 7.77 (s, 1H, C=CH), 10.83 (s, 1H, NH), 13.47 (s, 1H, COOH). MS m/z 615 (M + H). ESI-HRMS calcd for C36H26N2NaO4S2+ ([M + Na]+): 637.1226; found: 637.1236.
(S)-2-((Z)-5-(4-((E)-3-([1,1′-biphenyl]-4-yl)-3-oxoprop-1-en-1-yl)benzylidene)-4-oxo-2-thioxothiazolidin-3-yl)-3-methylbutanoic acid (5e)
Yellow solid; yield 26%; m.p. 120–123 °C. : −31.4 (c = 0.57, CHCl3). IR (KBr) cm−1: 2942 (OH), 1719 (C=O). 1H NMR (DMSO-d6, 300 MHz, ppm): δ 0.75 (d, 3H, J = 6.8 Hz, CHCH3), 1.19 (d, 3H, J = 6.4 Hz, CHCH3), 2.70–2.76 (m, 1H, CH(CH3)2), 5.17 (br.s, 1H, NCH), 7.49–8.29 (m, 15H, Ar–H), 7.77 (s, 1H, C=CH), 13.14 (s, 1H, COOH). MS m/z 528 (M + H). ESI-HRMS calcd for C30H25NNaO4S2+ ([M + H]+): 528.1298; found: 528.1310.
(R)-2-((Z)-5-(4-((E)-3-([1,1′-biphenyl]-4-yl)-3-oxoprop-1-en-1-yl)benzylidene)-4-oxo-2-thioxothiazolidin-3-yl)-3-methylbutanoic acid (5f)
Yellow solid; yield 20%; m.p. 89–92 °C. : 28.5 (c = 0.38, CHCl3). IR (KBr) cm−1: 2959 (OH), 1721 (C=O). 1H NMR (DMSO-d6, 300 MHz, ppm): δ 0.90 (d, 3H, J = 7.3 Hz, CHCH3), 1.19 (d, 3H, J = 6.5 Hz, CHCH3), 2.69–2.74 (m, 1H, CH(CH3)2), 5.17 (br.s, 1H, NCH), 7.43–8.29 (m, 15H, Ar–H), 7.77 (s, 1H, C=CH), 13.03 (s, 1H, COOH). MS m/z 528 (M + H). ESI-HRMS calcd for C30H25NNaO4S2+ ([M + H]+): 528.1298; found: 528.1294.
(S)-2-((Z)-5-(4-((E)-3-([1,1′-biphenyl]-4-yl)-3-oxoprop-1-en-1-yl)benzylidene)-4-oxo-2-thioxothiazolidin-3-yl)-4-methylpentanoic acid (5g)
Yellow solid; yield 32%; m.p. 110–112 °C. : −16.9 (c = 0.59, CH3CO2C2H5). IR (KBr) cm−1: 3028 (OH), 1713 (C=O). 1H NMR (DMSO-d6, 300 MHz, ppm): δ 0.87 (d, 3H, J = 6.5 Hz, CHCH3), 0.92 (d, 3H, J = 6.5 Hz, CHCH3), 1.47–1.53 (m, 1H, CH(CH3)2), 2.02–2.21 (m, 2H, CH2), 5.60 (br.s, 1H, N–CH), 7.42-8.29 (m, 15H, Ar–H), 7.78 (s, 1H, C=CH), 13.37 (s, 1H, COOH). MS m/z 542 (M + H). ESI-HRMS calcd for C31H27NNaO4S2+ ([M + Na]+): 564.1274; found: 564.1291.
(R)-2-((Z)-5-(4-((E)-3-([1,1′-biphenyl]-4-yl)-3-oxoprop-1-en-1-yl)benzylidene)-4-oxo-2-thioxothiazolidin-3-yl)-4-methylpentanoic acid (5h)
Yellow solid; yield 13%; m.p. 96–99 °C. : 19.3 (c = 0.45, CH3CO2C2H5). IR (KBr) cm−1: 3045 (OH), 1712 (C=O). 1H NMR (DMSO-d6, 300 MHz, ppm): δ 0.86 (d, 3H, J = 6.5 Hz, CHCH3), 0.89 (d, 3H, J = 6.4 Hz, CHCH3), 1.46–1.52 (m, 1H, CH(CH3)2), 2.01–2.23 (m, 2H, CH2), 5.60 (br.s, 1H, NCH), 7.45–8.59 (m, 15H, Ar–H), 7.76 (s, 1H, C=CH), 13.51(s, 1H, COOH). MS m/z 564 ([M + Na]+). ESI-HRMS calcd for C30H25NNaO4S2+ ([M + Na]+): 564.1274; found: 564.1279.
(2S)-2-((Z)-5-(4-((E)-3-([1,1′-biphenyl]-4-yl)-3-oxoprop-1-en-1-yl)benzylidene)-4-oxo-2-thioxothiazolidin-3-yl)-3-methylpentanoic acid (5i)
Yellow solid; yield 25%; m.p. 108–111 °C. : −27.2 (c = 0.61, CH3CO2C2H5). IR (KBr) cm−1: 3426 (OH), 1717 (C=O). 1H NMR (DMSO-d6, 300 MHz, ppm): δ 0.80 (t, 3H, J = 7.0 Hz, CH2CH3), 0.94–0.97 (m, 2H, CH2CH3), 1.16 (d, 3H, J = 6.3 Hz, CHCH3), 1.27–1.33 (m, 1H, CHCH3), 5.24 (br.s, 1H, NCH), 7.42–8.30 (m, 15H, Ar–H), 7.78 (s, 1H, C=CH), 13.29 (s, 1H, COOH). MS m/z 542 (M + H). ESI-HRMS calcd for C30H25NNaO4S2+ ([M + Na]+): 564.1274; found: 564.1284.
Pharmacology
Evaluation of anti-bacterial activity in vitro
The micro-organisms used in the present study were S. aureus (S. aureus RN 4220, S. aureus KCTC 209 and S. aureus KCTC 503) and Escherichia coli (E. coli 1356). The strains of multi-drug-resistant clinical isolates were MRSA (MRSA CCARM 3167 and MRSA CCARM 3506) and QRSA (QRSA CCARM 3505 and QRSA CCARM 3519). Clinical isolates were collected from various patients hospitalized in several clinics.
Test bacteria were grown to mid-log phase in Mueller-Hinton broth (MHB) and diluted 1000-fold in the same medium. The bacteria of 105 CFU/mL were inoculated into MHB and dispensed at 0.2 mL/well in a 96-well microtiter plate. As positive controls, oxacillin and norfloxacin were used. Test compounds were prepared in DMSO, the final concentration of which did not exceed 0.05%. A twofold serial dilution technique28 was used to obtain final concentrations. The MIC was defined as the concentration of a test compound that completely inhibited bacteria growth during 24 h incubation at 37 °C. Bacteria growth was determined by measuring the absorption at 650 nm using a microtiter enzyme-linked immunosorbent assay (ELISA) reader. All experiments were carried out three times.
Evaluation of cytotoxicity in vitro
Human cervical (Hela) cell monolayers were used as an in vitro model of cervicovaginal epithelium for testing the cytotoxicity of the new compounds. Hela cells were grown in Dulbecco-modified Eagle medium (DMEM) supplemented with fetal bovine serum (10%), and antibiotics (penicillin–streptomycin mixture [100 U/mL]). Cells at 80–90% confluence were split by trypsin (0.25% in phosphate buffered saline (PBS); pH 7.4), and the medium was changed at 24 h intervals. The cells were cultured at 37 °C in a 5% CO2 incubator. The cells were grown to three passages and approximately 1 × 104 cells were seeded into each well of a 96-well plate and allowed to incubate overnight to allow cells to attach to the substrate. After 24 h, the medium was replaced with DMEM supplemented with 10% fetal bovine serum (FBS) containing various concentrations of test compounds and incubated for 48 h. Then, 10 µl of methyl thiazolyl tetrazolium (MTT) solution (5 mg/mL in PBS) was added to each well. After incubation for 4 h, the medium was removed and the resulting formazan crystals were dissolved with 100 µl DMSO. After shaking 10 min, the optical density was measured at 570 nm using a microtiter ELISA reader. The assay was conducted four times. The IC50 values were defined as the concentrations inhibiting 50% of cell growth.
Results and discussion
Anti-bacterial activity
The anti-microbial assay was carried out using the following bacterial strains S. aureus RN 4220, S. aureus KCTC 209, S. aureus KCTC 503, MRSA CCARM 3167 and 3506, QRSA CCARM 3505 and 3519 and E. coli 1356. The in vitro anti-bacterial activity was evaluated using a 96-well microtiter plate and a serial dilution method to obtain the MIC. Oxacillin, norfloxacin, gatifloxacin and moxifloxacin were used as positive controls.
summarized the results obtained for the MICs of the 18 target compounds (4a–i and 5a–i) against three Gram-positive strains (S. aureus RN 4220, S. aureus KCTC 209 and S. aureus KCTC 503) and one Gram-negative strain (E. coli 1356). For S. aureus RN 4220, it could be found that most of the compounds (except 4b, 4c, 4d, 4i, 5c and 5d) showed good inhibition with MIC values in the range of 1–16 µg/mL. In particular, compounds 4g and 4h (MIC = 1 µg/mL) had a twofold more potent activity than the positive control norfloxacin (MIC = 2 µg/mL), and comparable activity to the positive control oxacillin (MIC = 1 µg/mL). For S. aureus KCTC 209, most of the compounds (except 4b, 4c, 4d, 4i, 5c and 5d) displayed poor inhibition with MICs ranging from 4 to 32 µg/mL, while these compounds showed better inhibitory activity against S. aureus KCTC 503, whose MICs were mostly in the range of 2–8 µg/mL. Unfortunately, all of the test compounds did not exhibit any inhibition against the Gram-negative strain E. coli 1356 (MICs > 64 µg/mL).
Table 1. Inhibitory activity (MIC*, µg/mL) of compounds 4a–i and 5a–i against bacteria.
The compounds 4a–i and 5a–i were also evaluated for their inhibitory activity against MRSA and QRSA. As shown in , compounds 4a–i and 5a–i (except 4b, 4c, 4d, 4i, 5c and 5d) presented the high levels of activity with MIC values of 2–8 µg/mL. For MRSA 3167 and 3506, most compounds presented comparable or much more potent activities than norfloxacin. Among them, compound 4g, with MIC value of 1 µg/mL, showed eightfold more potent than norfloxacin (MIC = 8 µg/mL) and 64-fold more potent than oxacillin (MIC > 64 µg/mL), twofold more potent than gatifloxacin (MIC = 2 µg/mL), and comparable with moxifloxacin (MIC = 1 µg/mL). For QRSA 3505 and 3519, compounds 4e, 4g, 4h and 5h also presented higher levels of activity (MIC = 2 µg/mL), which were slightly less active than oxacillin (MIC = 1 µg/mL) but much more potent than norfloxacin (MIC > 64 µg/mL), gatifloxacin (MIC = 8 or 4 µg/mL) and moxifloxacin (MIC = 4 µg/mL).
Table 2. Inhibitory activity (MIC*, µg/mL) of compounds 4a–i and 5a–i against clinical isolates of multidrug-resistant Gram-positive strains.
Most of synthesized compounds (4a–i and 5a–i) exhibit strong anti-bacterial activity, although the mechanism of action is not yet clearly understood. However, in this study, some preliminary remarks on the structure–activity relationship can be drawn from the results of bioactivities. (1) There is no remarkable difference of anti-bacterial activity between 4a–i and 5a–i. (2) The activity of S-configuration compound is a little better than the R-configuration compound. For example, the activity of compound 4g (S-configuration) is higher than that of compound 4h (R-configuration) generally. (3) The MIC values of the compounds derived from different amino acids differed greatly. For example, the compounds bearing tryptophan-derived rhodanines (4c, 4d, 5c and 5d) did not show any inhibition against all of the selected bacteria at 64 µg/mL. While the compounds bearing leucine-derived rodanines (4g, 4h, 5g and 5h) exhibited a strong inhibitory activity with MICs of 1–4 µg/mL against the four drug-resistance bacterial strains. The cytotoxicity of the selected compound (4g) was also evaluated. The IC50 value (HeLa cells) of 4g is 2.41, which is comparable with its MIC value.
Conclusion
Based on our previous work, we synthesized two new series of rhodanine derivatives (4a–i and 5a–i) and evaluated their anti-bacterial activities. Most of the compounds showed good anti-bacterial activities against Gram-positive bacteria as well as multi-drug-resistant strains of clinical isolates. Among them, compound 4g, with MIC value of 1 µg/mL, showed eightfold more potent than norfloxacin (MIC = 8 and 4 µg/mL) and 64-fold more potent than oxacillin (MIC > 64 µg/mL). Efforts to determine the reason for its anti-bacterial activity are ongoing and will be reported in due course.
Declaration of interest
This work was supported by the National Science Foundation of China (grant number 81260468). The authors report no conflicts of interest. The authors alone are responsible for the content and writing of this article.
References
- Patrick GL. An introduction to medicinal chemistry. 2nd ed. Oxford: Oxford University Press; 2001
- Mangalagiu II. Recent achievements in the chemistry of 1,2-diazines. Curr Org Chem 2011;15:730–52
- Moellering RC Jr. Current treatment options for community-acquired methicillin-resistant Staphylococcus aureus infection. Clin Infect Dis 2008;46:1032–7
- Gualtieri M, Bastide L, Villain-Guillot P, et al. In vitro activity of a new antibacterial rhodanine derivative against Staphylococcus epidermidis biofilms. J Antimicrob Chemother 2006;58:778–83
- Sim MM, Ng SB, Buss AD, et al. Benzylidene rhodanines as novel inhibitors of UDP-N-acetylmuramate/L-alanine ligase. Bioorg Med Chem Lett 2002;12:697–9
- Petrikaite V, Tarasevicius E, Pavilonis A. New ethacridine derivatives as the potential antifungal and antibacterial preparations. Medicina 2007;43:657–63
- Sortino M, Delgado P, Juárez S, et al. Synthesis and antifungal activity of (Z)-5-arylidenerhodanines. Bioorg Med Chem 2007;15:484–94
- Murugan R, Anbazhagan S, Sriman Narayanan S. Synthesis and in vivo antidiabetic activity of novel dispiropyrrolidines through [3 + 2] cycloaddition reactions with thiazolidinedione and rhodanine derivatives. Eur J Med Chem 2009;44:3272–9
- Chandrappa S, Kavitha CV, Shahabuddin MS, et al. Synthesis of 2-(5-((5-(4-chlorophenyl)furan-2-yl)methylene)-4-oxo-2-thioxothiazolidin-3-yl)acetic acid derivatives and evaluation of their cytotoxicity and induction of apoptosis in human leukemia cells. Bioorg Med Chem 2009;17:2576–84
- Rajamaki S, Innitzer A, Falciani C, et al. Exploration of novel thiobarbituric acid-, rhodanine- and thiohydantoin-based HIV-1 integrase inhibitors. Bioorg Med Chem Lett 2009;19:3615–8
- Ozkirimli S, Kazan F, Tunali Y. Synthesis, antibacterial and antifungal activities of 3-(1,2,4-triazol-3-yl)-4-thiazolidinones. J Enzym Inhib Med Chem 2009;24:447–52
- Ergenç N, Capan G, Günay NS, et al. Synthesis and hypnotic activity of new 4-thiazolidinone and 2-thioxo-4,5-imidazolidinedione derivatives. Arch Pharm (Weinheim) 1999;332:343–7
- Verma A, Saraf SK. 4-Thiazolidinone – a biologically active scaffold. Eur J Med Chem 2008;43:897–905
- Kumar G, Parasuraman P, Sharma SK, et al. Discovery of a rhodanine class of compounds as inhibitors of Plasmodium falciparum enoyl-acyl carrier protein reductase. J Med Chem 2007;50:2665–75
- Hardej D, Ashby CR, Jr Khadtare NS. The synthesis of phenylalanine-derived C5-substituted rhodanines and their activity against selected methicillin-resistant Staphylococcus aureus (MRSA) strains. Eur J Med Chem 2010;45:5827–32
- Song MX, Zheng CJ, Deng XQ, et al. Synthesis and bioactivity evaluation of rhodanine derivatives as potential anti-bacterial agents. Eur J Med Chem 2012;54:403–12
- Song MX, Zheng CJ, Sun LP, et al. Synthesis and biological evaluation of 5-aryloxypyrazole derivatives bearing a rhodanine-3-aromatic acid as potential antimicrobial agents. Bioorg Med Chem Lett 2012;22:7024–8
- Song MX, Zheng CJ, Deng XQ, et al. Synthesis and antibacterial evaluation of rhodanine-based 5-aryloxy pyrazoles against selected methicillin resistant and quinolone-resistant Staphylococcus aureus (MRSA and QRSA). Eur J Med Chem 2013;60:376–85
- Meng FL, Zheng CJ, Li YJ, et al. Synthesis and anti-bacterial activity of novel chalcone derivatives containing 2,4-thiazolidinedione-3-acetic acid moiety. Chin J Org Chem 2012;32:183–8 (in Chinese)
- Xu LL, Zheng CJ, Sun LP, et al. Synthesis of novel 1,3-diaryl pyrazole derivatives bearing rhodanine-3-fatty acid moieties as potential antibacterial agents. Eur J Med Chem 2012;48:174–8
- Liu XF, Zheng CJ, Liu XK, et al. Synthesis of new chalcone derivatives bearing 2,4-thiazo lidinedione and benzoic acid moieties as potential anti-bacterial agents. Eur J Med Chem 2011;46:3469–73
- Chen ZH, Zheng CJ, Sun LP, et al. Synthesis of new chalcone derivatives containing a rhodanine-3-acetic acid moiety with potential anti-bacterial activity. Eur J Med Chem 2010;45:5739–43
- Jin X, Zheng CJ, Song MX, et al. Synthesis and antimicrobial evaluation of L-phenylalanine-derived C5-substituted rhodanine and chalcone derivatives containing thiobarbituric acid or 2-thioxo-4-thiazolidinone. Eur J Med Chem 2012;56:203–9
- Jung SH, Park SY, Kim-Pak Y, et al. Synthesis and PPAR-γ ligand-binding activity of the new series of 2′-hydroxychalcone and thiazolidinedione derivatives. Chem Pharm Bull 2006;54:368–71
- Deep A, Jain S, Sharma PC. Synthesis and anti-inflammatory activity of some novel biphenyl-4-carboxylic acid 5-(arylidene)-2-(aryl)-4-oxothiazolidin-3-yl amides. Acta Pol Pharm 2010;67:63–7
- Penthala NR, Yerramreddy TR, Crooks PA. Microwave assisted synthesis and in vitro cytotoxicities of substituted (Z)-2-amino-5-(1-benzyl-1H-indol-3-yl)methylene-1-methyl-1H-imidazol-4(5H)-ones against human tumor cell lines. Bioorg Med Chem Lett 2010;20:591–3
- Barros FW, Silva TG, da Rocha Pitta MG, et al. Synthesis and cytotoxic activity of new acridine-thiazolidine derivatives. Bioorg Med Chem 2012;20:3533–9
- Clinical and Laboratory Standards Institute. Methods for dilution antimicrobial susceptibility tests for bacteria that grow aerobically. Approved standard M7-A6. Wayne (PA): Clinical and Laboratory Standards Institute; 2003