
Abstract
A series of lipophilic ester derivatives (2a–g) of (S)-1-(pent-4′-enoyl)-4-(hydroxymethyl)-azetidin-2-one has been synthesised in three steps from (S)-4-(benzyloxycarbonyl)-azetidin-2-one and evaluated as novel, reversible, β-lactamic inhibitors of endocannabinoid-degrading enzymes (human fatty acid amide hydrolase (hFAAH) and monoacylglycerol lipase (hMAGL)). The compounds showed IC50 values in the micromolar range and selectivity for hFAAH versus hMAGL. The unexpected 1000-fold decrease in activity of 2a comparatively to the known regioisomeric structure 1a (i.e. lipophilic chains placed on N1 and C3 positions of the β-lactam core) could be explained on the basis of docking studies into a revisited model of hFAAH active site, considering one or two water molecules in interaction with the catalytic triad.
Introduction
Nowadays, the endocannabinoid (EC) system is considered as one of the promising fields in pain and central nervous system disorder treatmentsCitation1,Citation2. The EC system is formed by the cannabinoid receptors (CB1, CB2), their endogenous ligands (anandamide (AEA), 2-arachidonoylglycerol (2-AG)) and enzymes/proteins involved in the EC biosynthesis, transport and regulationCitation3,Citation4. Amongst these enzymes, the EC-degrading enzymes, namely fatty acid amide hydrolase (FAAH) and monoacylglycerol lipase (MAGL), are of particular interest. Indeed, FAAH and MAGL hydrolyse, respectively, AEA and 2-AG, with the consequence of the termination of their action on CB1/CB2. Because the activation of the cannabinoid receptors results in multiple beneficial effects, a prolonged action of the cannabinoids is often desirable. The best way to achieve this goal consists in the inhibition of the EC-degrading enzymesCitation5–7. The search of selective inhibitors of FAAH and MAGL has already produced very active compounds featuring various chemical structures, and is still a growing research domain in medicinal chemistryCitation8–10.
FAAH is a serine amidase which possesses the non-traditional Ser-Ser-Lys catalytic triad. The processing of natural substrates involves the nucleophilic attack of Ser241 residue on the sessile amide carbonyl, leading to an unstable acyl-enzyme covalent intermediate which is rapidly hydrolysed for regenerating the active enzyme. The 3D-structure of the human FAAH (hFAAH) is not known. However, the rat isoforme (rFAAH) has been crystallised in the presence of MAFP (methylarachidonoyl fluorophosphonate) and the structure of the resulting enzyme-inhibitor complex has been resolvedCitation11–13. More recently, an engineered humanised rat FAAH (h/rFAAH) has been produced and successfully crystallised in the presence of three inhibitors, representative of the major classes of FAAH covalent inhibitorsCitation14–17. MAGL is also a serine hydrolase characterised by the classical Ser-His-Asp catalytic triad and two non-catalytic Cys residues located in the vicinity of the active site. The 3D-structure of the human MAGL (hMAGL) has been disclosed in 2010, independently by two research groupsCitation7,Citation18.
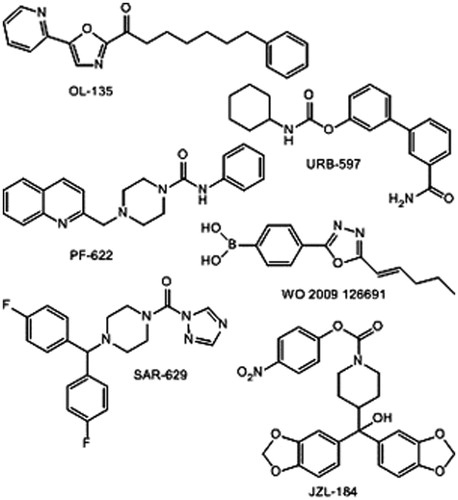
The mechanism-based design of hFAAH inhibitors focuses on electrophilic entities susceptible to form stable tetrahedal- or acyl-enzyme covalent intermediates, as usually considered for the long-term (or irreversible) inhibition of serine enzymes. Accordingly, the main classes of hFAAH inhibitors are as follows: α-keto-heterocycles (OL-135), carbamates (URB-597), ureas (PF-622) and boronic acids (WO 2009 126691)Citation8. Studies on the inhibition of hMAGL reveal the same trends with urea-(SAR-629) and carbamate-based compounds (JZL-184) ()Citation8–10. Surprisingly, the β-lactone and β-lactam templates are scarcely described in the field of EC system controlCitation19–21, although oxetan-2-onesCitation22 and mainly azetidin-2-onesCitation23–27 are well-known inhibitors of D,D-peptidases, β-lactamases, leukocyte elastase, thrombin, prostate specific antigen and human cytomegalovirus protease and among others.
Figure 1. Representative inhibitors of hFAAH and hMAGL.
A β-lactone (OMDM169)Citation19) inspired of a serine hydrolase inhibitor, tetrahydrolipstatin, has been disclosed by Di Marzo’s group in 2008 for the inhibition of hMAGL (). Since 2008, our group is interested in the development of β-lactams as selective inhibitors of hFAAHCitation28. The lead compound 1a (IC50 = 5.3 nM) displays two lipophilic chains anchored on the N1 and C3 positions of the azetidin-2-one ring (). The origin of the β-lactam template is the chiral precursor of carbapenem antibiotics which is commercially available, i.e. (3 R,4 R)-4-acetoxy-3-((1′R)-(t-butyldimethylsilyloxy)-ethyl)-azetidin-2-oneCitation29, named here chiron A ().
Figure 2. β-Lactone and β-lactam inhibitors of hMAGL and hFAAH, respectively.
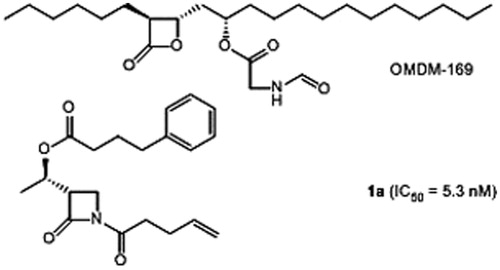
Figure 3. General structures of studied hFAAH inhibitors and their respective precursors.
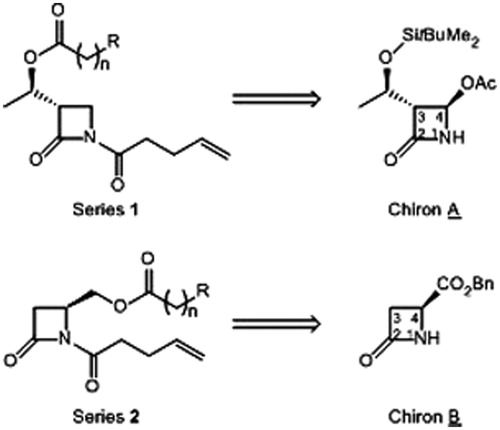
A random screening of our in-house library of β-lactamsCitation30, made from the same precursor A, showed that trisubstituted derivatives (at positions N1, C3 and C4) exhibit only moderate activity against hFAAH (see Supplementary Material). Therefore, we envisaged the synthesis of a novel family of disubstituted β-lactams, featuring the lipophilic chains on the positions N1 and C4 (, series of compounds 2). The (S)-configuration of the C4 carbon has been selected by comparison of molecules 1a and 2a (n = 3, R = Ph; ) positioned into the homology model of hFAAH active site previously publishedCitation28. The starting material of the series 2 of potential inhibitors is a known chiral β-lactam (named chiron B), used for the total synthesis of isocephem antibiotics, i.e. (S)-4-(benzyloxycarbonyl)-azetidin-2-one; B is readily accessible in large scale from (S)-aspartic acidCitation31,Citation32.
In this article, we describe the synthesis of representative members of the β-lactam family 2 and their evaluation for the inhibition of hFAAH and hMAGL. A comparison of inhibitors 1a and 2a has been performed by molecular modelling studies, guided by the recent crystallographic structures of h/rFAAH. Our previous model of active site has been completed, considering water molecules in interaction with the inhibitors and amino acid residues.
Experimental
Synthetic chemistry
Generalities
The solvents (technical and anhydrous) and reagents were purchased from Acros Organics (Thurnout, Belgium), Alfa Aesar (Bischeim, France), Cayman Chemical (Monluçon, France), Sigma-Aldrich-Fluka (Diegem, Belgium) or VWR (Leuven, Belgium), and used as received. The reactions in dry conditions were performed under argon atmosphere, in flame-dried vessel. Thin layer chromatography was realised on silica gel plate Merck 60F254, and revealed under UV light. Preparative chromatography was made with flash silica gel from Rocc.
The mass spectra (MS) were acquired on Thermo Finnigan LQC spectrometer, in electrospray ionisation (ESI), negative mode. 1H and 13C nuclear magnetic resonance (NMR) spectra were recorded on Bruker Avance 300 (Eke, Belgium) (300 MHz, for proton and 75 MHz for carbon) and Bruker Avance 500 equipments (500 MHz for proton and 125 MHz for carbon), using CDCl3 or CD3OD as solvent and tetramethylsilane as internal standard. Chemical shifts are reported in ppm (residual non-deuterated solvent: CDCl3 at 7.26 (H) and 77.16 (C) ppm; CD3OD at 3.31 (H) and 40.00 (C) ppm). Coupling constants (J) are given in Hertz (Hz). The infra-red spectra were acquired on Shimadzu FTIR-8400S apparatus (Deurne Antwerpen, Belgium). Samples were deposited on the Se–Zn crystal by evaporation from dichloromethane (DCM) solution to give a film. High resolution MS (HRMS) were measured with a QExactive spectrometer (Thermo Scientific, Bruxelles, Belgium). Rotations were obtained with a polarimeter Atago Ap-100 at 25 °C. Samples were dissolved in CHCl3 (concentrations given in percentages, i.e. g 100/mL). The purity of the tested compounds (≥95%) was controlled by HPLC.
Procedure for the reduction of (S)-4-(benzyloxycarbonyl)-2-azetidinone (B)
(S)-4-(Hydroxymethyl)-2-azetidinone (3)
To a solution of (S)-4-(benzyloxycarbonyl)-2-azetidinone (1.00 g, 4.87 mmol, 1 equiv.) in methanol (25 mL) was added NaBH4 (556 mg, 14.6 mmol, 3 equiv.), in small portions, at 0 °C and under argon atmosphere. The mixture was stirred for 15 min at 0 °C and then for 2 h at 20 °C. After neutralisation by the addition of acetic acid (10% aqueous solution, 15 mL), and concentration under vacuum, the crude alcohol was purified by filtration over a silica gel pad with DCM/methanol (9:1, v/v) as the eluent. 3 was recovered as a colourless oil (495 mg, 100% yield). Rf = 0.244 (DCM/MeOH, 9:1, v/v). δH (300 MHz; CD3OD) 2.67 (1 H, dd, J = 2.0, 14.9), 2.90 (1 H, dd, J = 4.6, 14.9), 3.54–3.61 (1 H, m) and 3.67–3.72 (2 H, m) ppm. δC (75 MHz; CD3OD) 40.0, 49.7, 64.7 and 171.1 ppm.
General procedure of esterification according to Method (ii)
To a solution of 3 (1 equiv.), dicyclohexylcarbodiimide (DCC, 1.1 equiv.) and 4-dimethylaminopyridine (DMAP, 0.05 equiv.) in dry DCM (10 mL/mmol), was added the appropriate carboxylic acid (1.1 equiv.) in DCM (5 mL/mmol), drop-wise under argon atmosphere. The mixture was stirred overnight at room temperature, then cooled at 0 °C (for precipitation of dicyclohexylurea) and filtered. The filtrate was concentrated under vacuum and the residue was purified by flash chromatography (elution with DCM/MeOH, 9:1, v/v) to furnish the ester 4 (for 4d, 4e and 4g, see Supplementary Material).
(S)-(4-Oxoazetidin-2-yl)methyl 2-(biphenyl-4-yl)acetate (4c)
The title compound (234 mg, 87% yield) was obtained as a white gummy solid from 3 (0.92 mmol). Rf = 0.35 (DCM/EtOAc, 1:1, v/v). νmax/cm 1732, 1714, 1487, 1413, 1242, 1222, 1149 and 1008. δH (500 MHz; CDCl3) 2.73 (1 H, ddd, J = 1.5, 2.4, 14.9), 3.05 (1 H, ddd, J = 2.0, 5.2, 14.9), 3.70 (2 H, s), 3.84–3.88 (1 H, m), 4.07 (1 H, dd, J = 6.4, 11.7), 4.43 (1 H, dd, J = 3.7, 11.7), 6.05 (1 H, br s), 7.32–7.38 (3 H, m), 7.41–7.46 (2 H, m) and 7.55–7.60 (4 H, m). δC (125 MHz; CDCl3) 40.85, 40.87, 46.1, 66.0, 127.2, 127.55, 127.49, 128.9, 129.7, 132.6, 140.4, 140.7, 167.0 and 171.5. m/z (ESI) 296.15 (M + H)+, 318.11(M + Na)+. HRMS m/z found 295.12041, calc. for C18H17NO3 295.12029.
(S)-(4-Oxoazetidin-2-yl)methyl 3-(1H-indol-3-yl)propanoate (4f)
The title compound (249 mg, 92% yield) was obtained as a yellow oil from 3 (0.99 mmol). Rf = 0.45 (DCM/MeOH, 9:1, v/v). [α]D +5.97 (c 1.8 in CHCl3). νmax/cm 2922, 1730 (br), 1456, 1340 and 1155. δH (300 MHz; CDCl3) 2.58 (1 H, ddd, J = 1.9, 5.2, 15), 2.77 (2 H, t, J = 7.3), 2.92 (1 H, ddd, J = 1.2, 2.1, 15), 3.12 (2 H, t, J = 7.3), 3.65 (1 H, m), 3.94 (1 H, dd, J = 6.8, 11.7), 4.28 (1 H, dd, J = 3.7, 11.7), 5.62 (1 H, br s), 6.96 (1 H, d, J = 2.1), 7.12 (1 H, td, J = 6.9, 0.9), 7.20 (1 H, td, J = 6.9, 1.1), 7.36 (1 H, d, J = 7.9), 7.59 (1 H, d, J = 7.8) and 8.06 (1 H, br s) ppm. δC (75 MHz; CDCl3) 20.9, 34.9, 40.7, 46.0, 65.7, 111.4, 114.6, 118.8, 119.5, 121.7, 122.3, 127.2, 136.3, 167.0 and 173.3 ppm. m/z (ESI) 273.12 (M + H)+, 295.10 (M + Na)+. HRMS m/z found 273.12338, calc. for C15H17N2O3 273.12337.
General procedure of esterification according to Method (iii)
To a solution of 3 (1 equiv.) in dry DCM solution (15 mL/mmol) at 20 °C, were added successively pyridine (1 equiv.) and the appropriate acid chloride (1 equiv.) under argon atmosphere. The mixture was stirred overnight, then diluted with DCM (15 mL/mmol) and washed with aqueous Na2CO3 (10%). The organic layer was washed with 1 N HCl and brine, dried over MgSO4, filtered and concentrated under vacuum. The residue was purified by flash chromatography (elution with DCM/EtOAc, 7:3, v/v) to furnish the ester 4 (for 4h, see Supplementary Material).
(S)-(4-Oxoazetidin-2-yl)methyl 4-phenylbutanoate (4a)
The title compound (88 mg, 72% yield) was obtained as a yellow oil from 3 (0.49 mmol). Rf = 0.38 (DCM/MeOH, 95:5, v/v). [α]D = +10.07 (c = 1.3). νmax/cm 3329, 2943, 1730, 1688, 1616, 1496 and 1142. δH (300 MHz; CDCl3) 1.96 (2 H, quint, J = 7.5), 2.36 (2 H, t, J = 7.5), 2.65 (2 H, t, J = 7.5), 2.75 (1 H, ddd, J = 1.4, 2.4, 14.9), 3.09 (1 H, ddd, J = 2.0, 5.2, 14.9), 3.85 (1 H, m), 4.01 (1 H, dd, J = 6.5, 11.7), 4.37 (1 H, dd, J = 3.8, 11.7), 6.06 (1 H, br s), and 7.13–7.33 (5 H, m) ppm. δC (75 MHz; CDCl3) 26.4, 33.4, 35.1, 40.9, 46.2, 65.5, 126.2, 128.5, 128.6, 141.2, 167.0 and 173.3 ppm. m/z (ESI) 248.13 (M + H)+, 270.11 (M + Na)+. HRMS m/z found 248.12823, calc. for C14H18NO3 248.12812.
(S)-(4-Oxoazetidin-2-yl)methyl biphenyl-4-carboxylate (4b)
The title compound (122 mg, 78% yield) was obtained as a white amorphous powder from 3 (0.55 mmol). Rf = 0.27 (Cyclohexane/EtOAc, 2:8, v/v). νmax/cm 2910, 2850, 1738, 1714, 1606, 1406, 1286, 1269 and 1123. δH (300 MHz; CDCl3) 2.90 (1 H, ddd, J = 1.6, 2.2, 14.9), 3.17 (1 H, ddd, J = 1.9, 5.2, 14.9), 4.03 (1 H, m), 4.34 (1 H, dd, J = 6.3, 11.7), 4.64 (1 H, dd, J = 3.7, 11.7), 6.16 (1 H, br s), 7.36–7.53 (3 H, m), 7.57–7.74 (4 H, m) and 8.10 (2 H, d, J = 8.3) ppm. δC (75 MHz; CDCl3) 41.0, 46.4, 66.1, 127.3 (2 C), 127.4, 128.4, 129.1, 130.3, 139.9, 146.3, 166.3 and 167.0 ppm. m/z (ESI) 304.09 (M + Na)+, 585.19 (2 M+ Na)+. HRMS m/z found 304.09475, calc. for C17H15NO3Na 304.09441.
General procedure for N-acylation according to Method (iv)
To a solution of NH-azetidinone 4 (1 equiv.) in DCM (8 mL/mmol) were added successively pyridine (2 equiv.) and 4-pentenoyl chloride (2 equiv.) under argon atmosphere. The mixture was refluxed during 24 h, then diluted with DCM (20 mL/mmol). After washing with aqueous Na2CO3 (10%) (three times), the organic layer was washed with 3 N HCl and brine, dried over MgSO4, filtered and concentrated under vacuum. The residue was purified by flash chromatography (elution with DCM/EtOAc, 98:2, v/v) to furnish the N-acyl azetidinone 2 (for 2d and 2e, see Supplementary Material).
(S)-(4-Oxo-1-pent-4-enoylazetidin-2-yl)methyl 4-phenylbutanoate (2a)
The title compound (26 mg, 43% yield) was obtained as a colourless oil from 4a (0.18 mmol). Rf = 0.5 (DCM/MeOH, 9:1, v/v). [α]D = −10.63 (c = 3.1). νmax/cm 2956, 2924, 1791, 1738, 1699, 1454, 1383, 1313 and 1144. δH (300 MHz; CDCl3) 2.01 (2 H, quint, J = 7.4), 2.34–2.50 (4 H, m), 2.69 (2 H, t, J = 7.4), 2.84 (2 H, t, J = 7.4), 2.95 (1 H, dd, J = 3.4, 16.3), 3.17 (1 H, dd, J = 6.3, 16.3), 4.31 (1 H, m), 4.43 (1 H, dd, J = 3.1, 12.0), 4.57 (1 H, dd, J = 4.1, 12.0), 5.01–5.15 (2 H, m), 5.85 (1 H, m), 7.18–7.26 (3 H, m) and 7.29–7.36 (2 H, m) ppm. δC (75 MHz; CDCl3) 26.4, 28.0, 33.4, 35.1, 35.8, 39.2, 48.5, 61.6, 115.9, 126.2, 128.5 (2 C), 141.2, 136.4, 164.2, 170.7 and 173.1 ppm. m/z (ESI) 330.17 (M + H)+, 352.15 (M + Na)+. HRMS m/z found 352.1519, calc. for C19H23NO4Na 352.15193.
(S)-(4-Oxo-1-pent-4-enoylazetidin-2-yl)methyl biphenyl-4-carboxylate (2b)
The title compound (25 mg, 55% yield) was obtained as a colourless oil from 4b (0.12 mmol). Rf = 0.38 (DCM/EtOAc, 95:5, v/v). [α]D = −27.31 (c = 0.8). νmax/cm 2921, 1790, 1716, 1703, 1608, 1383, 1269 and 1113. δH (300 MHz; CDCl3) 2.37–2.48 (2 H, m), 2.79–2.91 (2 H, m), 3.07 (1 H, dd, J = 3.4, 16.3), 3.23 (1 H, dd, J = 6.3, 16.3), 4.42 (1 H, m), 4.64 (1 H, dd, J = 3.1, 12.0), 4.82 (1 H, dd, J = 4.0, 12.0), 4.94–5.11 (2 H, m), 5.85 (1 H, m), 7.36–7.51 (3 H, m), 7.58–7.71 (4 H, m) and 8.06 (2 H, d, J = 8.5) ppm. δC (75 MHz; CDCl3) 28.1, 35.8, 39.3, 48.7, 62.2, 116.0, 127.37, 127.42, 128.4, 128.2, 129.1, 130.3, 136.4, 139.9, 146.3, 164.3, 166.0 and 170.7 ppm. m/z (ESI) 364.15 (M + H)+, 386.14 (M + Na)+. HRMS m/z found 364.15462, calc. for C22H22NO4 364.15433.
(S)-(4-Oxo-1-pent-4-enoylazetidin-2-yl)methyl 3-(1H-indol-3-yl)propanoate (2f)
The title compound (82 mg, 63% yield) was obtained as a yellow oil from 4f (0.37 mmol). Rf = 0.60 (DCM/MeOH, 95:5, v/v). [α]D = −12.42 (c = 0.9). νmax/cm 2934, 1790, 1736, 1698, 1641, 1458, 1315 and 1155. δH (300 MHz; CDCl3) 2.38 (2 H, m), 2.66–2.79 (5 H, m), 2.97 (1 H, dd, J = 6.4, 16.3), 3.11 (2 H, t, J = 7.4), 4.18 (1 H, m), 4.38 (1 H, dd, J = 3.1, 12, 4.50 (1 H, ddd, J = 2.1, 4.3, 12), 4.97–5.11 (2 H, m), 5.81 (1 H, m), 6.99 (1 H, d, J = 2.1), 7.09–7.26 (2 H, m), 7.35 (1 H, d, J = 8.0), 7.58 (1 H, d, J = 7.7) and 8.08 (1 H, br s) ppm. δC (75 MHz; CDCl3) 20.7, 27.9, 34.7, 35.7, 38.9, 48.4, 61.5, 111.3, 114.5, 115.9, 118.7, 119.4, 121.6, 122.2, 127.1, 136.3, 136.4, 164.3, 170.7 and 173.0 ppm. m/z (ESI) 355.16 (M + H)+, 377.15 (M + Na)+. HRMS m/z found 355.16541, calc. for C20H23N2O4 355.16523.
General procedure for N-acylation according to Method (v)
To a solution of NH-azetidinone 4 (1 equiv.) in dry tetrahydrofuran (THF) solution (10 mL/mmol) cooled at −78 °C, was added drop-wise, under argon atmosphere, a solution of LiHMDS (lithium hexamethyldisilazide) (1 M in THF, 1.1 equiv.) and then DMF (dimethylformamide) (0.1 mL/mmol). The mixture was stirred for 30 min. at −78 °C, then 4-pentenoyl chloride was added drop-wise (1.1 equiv.). After 1 h stirring at −78 °C, the solution was allowed to warm at room temperature and left 2 h at 20 °C. After concentration under vacuum, the residue was dissolved in DCM (20 mL/mmol) and washed three times with brine. The organic layer was dried over MgSO4, concentrated and purified by flash chromatography (elution with DCM/EtOAc, 95:5, v/v) to furnish 2 (for 2g, see Supplementary Material).
(S)-(4-Oxo-1-pent-4-enoylazetidin-2-yl)methyl 2-(biphenyl-4-yl)acetate (2c)
The title compound (198 mg, 70% yield) was obtained as a yellow oil from 4c (0.75 mmol). Rf = 0.22 (DCM/EtOAc, 98:2, v/v). [α]D = −23.1 (c = 1.7). νmax/cm 3164, 1789, 1739, 1701, 1317 and 1153. δH (500 MHz; CDCl3) 2.33–2.39 (2 H, m), 2.66–2.80 (2 H, m), 2.83 (1 H, dd, J = 3.4, 16.2), 3.06 (1 H, dd, J = 6.4, 16.2), 3.69 (2 H, s), 4.22–4.27 (1 H, m), 4.39 (1 H, dd, J = 2.9, 12.1), 4.61 (1 H, dd, J = 3.8, 12.1), 4.96–5.07 (2 H, m), 5.74–5.84 (1 H, m), 7.27–7.40 (3 H, m), 7.40–7.51 (2 H, m) and 7.51–7.67 (4 H, m) ppm. δC (125 MHz; CDCl3) 27.9, 35.6, 38.9, 40.9, 48.4, 61.8,115.8, 127.1, 127.4, 127.5, 128.9, 129.7, 132.6, 136.4, 140.3, 140.7, 164.0, 170.6 and 171.0 ppm. m/z (ESI) 378.02 (M + H)+, 400.15 (M + Na)+. HRMS m/z found 400.1511, calc. for C23H23NO4Na 400.1525.
Enzyme inhibition
In vitro assays for hFAAH
Tubes containing the enzyme (10 mM Tris-HCl, 1 mM ethylenediamine tetraacetic acid (EDTA), 0.1% (w/v) bovine serum albumin (BSA), pH 7.4, 165 µL), test compounds in DMSO (dimethylsulfoxide) or DMSO alone for controls (10 µL) and [3H]-AEA (50 000 dpm, 2 µM final concentration, 25 µL) were incubated at 37 °C for 10 min. Reactions were stopped by rapidly placing the tubes in ice and adding 400 µL of ice-cold chloroform/methanol (1:1 v/v) followed by vigorous mixing. Phases were separated by centrifugation at 850 g, and aliquots (200 µL) of the upper methanol/buffer phase were counted for radioactivity by liquid scintillation counting (LSC). In all experiments, tubes containing buffer only were used as control for chemical hydrolysis (blank) and this value was systematically subtracted. Using these conditions, URB-597 inhibits hFAAH with an IC50 value of 40 nM.
In vitro assays for hMAGL
Tubes containing purified enzyme (10 mM Tris-HCl, 1 mM EDTA, 0.1% (w/v) BSA, pH 8.0, 165 µL), test compounds in DMSO or DMSO alone for controls (10 µL) and [3H]-2-OG (50 000 dpm, 2 µM final concentration, 25 µL) were preincubated at 20 °C for 30 min and incubated at 37 °C for 10 min. Reactions were stopped by rapidly placing the tubes in ice and adding 400 µL of ice-cold chloroform/methanol (1:1 v/v) followed by vigorous mixing. Phases were separated by centrifugation at 850 g, and aliquots (200 µL) of the lower chloroform phase were counted for radioactivity by LSC. In all experiments, tubes containing buffer only were used as control for chemical hydrolysis (blank) and this value was systematically subtracted.
Reversibility studies
Human recombinant FAAH (27.5 µg) was incubated in the presence of the inhibitors or DMSO during 1 h at room temperature, in a total of 15 µL assay buffer (Tris 10 mM, EDTA 1 mM, pH 7.4). Following this step, assay buffer was added to obtain a 100-fold dilution of the samples. A 175 µL aliquot was then taken after 0, 30 or 90 minutes and AEA, diluted in the same buffer supplemented with 0.1% BSA, was immediately added ([3H]-AEA; 25 µL; 50 000 dpm, 2 μM final AEA concentration). The hydrolysis reaction was allowed to proceed during 30 min at 37 °C and 400 μL of ice-cold chloroform/methanol (1:1 v/v) were added to stop the reaction, followed by vigorous mixing. Tubes were centrifuged at 850 g during 5 min and the radioactivity present in the upper aqueous phase was measured by LSC. Tubes lacking the enzyme were used as blanks, the value of which was systematically subtracted from other values. Results were expressed as per cent of control activity (three experiments performed in triplicate). Initial compound concentrations ([2a] = 100, 150 and 250 µM; [URB597] = 5 µM; [CAY10402] = 10 nM) were carefully selected in preliminary experiments omitting the dilution step, so that maximal and minimal inhibition should be observed at the concentrations obtained before and after the dilution, respectively. The concentrations of URB597 and CAY10402 correspond to about 100 times their respective IC50 determined in our laboratory.
The percentage of recovery of hFAAH activity after dilution was calculated according to the following formula:
where %act high []set up = percentage of activity in high inhibitor concentration (before dilution); %act low []set up = percentage of activity in low inhibitor concentration (after dilution).
Computational chemistry
The previously determined modelled structure of hFAAH was usedCitation28. Docking of the inhibitors into the active site of FAAH was performed using the GOLD programCitation33. It is based on a genetic algorithm (GA), performing docking of flexible ligands into proteins with partial flexibility in the neighbourhood of the active site. The binding region was defined as a 17 Å radius sphere centred on Phe192. Two explicit water molecules were allowed to switch on and off and to rotate around their three principal axes. For the 20 GA runs performed, a total of 100 000 genetic operations were carried out on 5 islands, each containing 100 individuals. The niche size was set to 2, and the value for the selection pressure was set to 1.1. Genetic operator weights for crossover, mutation and migration were set to 95, 95 and 10, respectively. The solutions were then ranked by GOLD score. The GOLD fitness function is made up of four components: protein-ligand hydrogen bond energy, protein-ligand van der Waals energy, ligand internal van der Waals energy and ligand torsional strain energy.
The compounds surrounded by the closer amino acids were optimised by a combined hybrid quantum mechanics/molecular mechanics method at the B3LYP/dzp/PM6 level using the GAUSSIAN 09 program packageCitation34. The figures were produced using PyMOLCitation35.
Results and discussion
Synthesis
The chiron B is prepared in four steps from (S)-aspartic acid according to the literatureCitation31,Citation32 (). Selective reduction of the benzyl ester function is obtained with sodium borohydride in quantitative yieldCitation36. The esterification reaction of the C4 hydroxymethyl substituent is performed via two methods, namely the condensation of a carboxylic acid in the presence of DCC and 4-DMAP, or the acylation by an acid chloride in the presence of pyridine. The choice of the end-chain substituent R and the chain length n is inspired from the series 1 of inhibitors previously publishedCitation37,Citation38. Except for 4e, the yields of esters 4a–h () are excellent after chromatographic purification. Most of the reagents are commercially available; 4-(pyridin-4-yl)-butanoic acid and 2-(pyridin-4-yl)-butanoic acid have been prepared by Michael addition of dimethyl malonate on vinylpyridines, followed by saponification and decarboxylation (see Supplementary Material)Citation39,Citation40.
Scheme 1. Synthesis of 1,4-disubstituted β-lactams: (i) NaBH4, MeOH, 0–20 °C, 3 h; (ii) carboxilic acid, DCC, DMAP (catal.), DCM, 20 °C, 24 h; (iii) acid chloride, pyridine or DMAP, DCM, 20 °C, 24 h; (iv) ClO-(CH2)2-CH=CH2, pyridine, DCM, reflux, 24 h; (v) ClCO-(CH2)2-CH=CH2, LiHMDS, THF-DMF, −78–20 °C, 1 h. See for yields of 4 and 2.
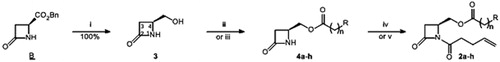
Table 1. Yields of compounds (%) of the .
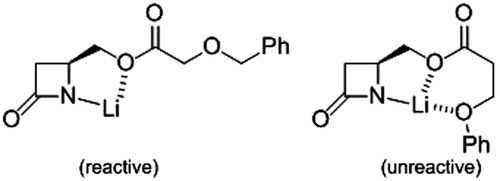
The next step is the N-acylation with 4-pentenoyl chloride to furnish the series of β-lactams 2a–h equipped with the N1 chain of the best inhibitors of the series 1 (). Deprotonation of 4 with lithium hexamethyldisilazide at low temperature and quenching of the anion by the acid chlorideCitation41 give the expected products 2, except 2h (). We postulate that the intermediate anion of 4h is inert, because of the formation of a stable O,O,N-complex with the lithium cation (). The regioisomeric structure, i.e. anion from 4g, should not form similar complex due to cyclic strain. Prolonged treatment of the precursors 4 with 4-pentenoyl chloride and pyridine in refluxing DCM leads also to the desired compounds 2 in few cases, but in low yields (). The β-lactams 2 have been purified by column chromatography and characterised by the usual spectroscopic methods. For instance, the 13C NMR spectra show three characteristic carbonyl signals, at 164 ppm (β-lactam), 170 ppm (imide) and 171–173 ppm (ester). In the 1H NMR spectra, the β-lactamic protons give a typical ABX pattern, with H3 at 2.9 ppm (dd, J3,4trans = 3 Hz and J3,3′ = 16 Hz), H3′ at 3.2 ppm (dd, J3′,4cis = 6 Hz and J3,3′ = 16 Hz) and H4 at 4.0 ppm (m). Another ABX pattern is visible, attributed to the CH2O motif fixed at C4: one doublet of doublet at 4.3 ppm (J = 6 and 12 Hz) and another at 4.6 ppm (J = 4 and 12 Hz).
Figure 4. Intermediates resulting from the deprotonation of 4g and 4h, respectively.
Enzyme inhibition
Human recombinant enzymes, namely, hFAAHCitation42 and hMAGLCitation43, are prepared according to the literature. The β-lactams 2 are tested in a competitive hydrolytic assay using [3H]-labelled substrates, i.e. [3H]-AEA in the case of hFAAH and [3H]-2-OG in the case of hMAGL. Briefly, tested compound, enzyme and labelled substrate are incubated at 37 °C during a few minutes and the extent of enzyme inhibition is then evaluated by LSC of [3H]-ethanolamine and [3H]-glycerol resulting from the hydrolysis of [3H]-AEA and [3H]-2-OG, respectively ().
Table 2. hFAAH and hMAGL inhibition.
All compounds 2 present a micromolar activity against hFAAH, with IC50 values ranging from 0.9 to 173 µM. The less active inhibitors feature a pyridine motif in the C4 side-chain. Comparatively to the lead compound 1a of the previous series, 2a appears 1000 times less potent for inhibiting hFAAH. Similarly, compounds 2c, 2e and 2g are, respectively, 650, 560 and 1370 times less active than the corresponding inhibitors 1c, 1e and 1gCitation28,Citation37,Citation38. Compounds 2 behave as quite selective inhibitors of hFAAH versus hMAGL: the selectivity factor of 2a and 2f is about 20. However, the selectivity is more pronounced for the lead compound 1a (S = 800)Citation28,Citation37,Citation38.
We have previously demonstrated that 1a is a reversible inhibitor of hFAAH: it is not processed by the enzyme according to HPLC/HRMS studiesCitation37, and the enzyme activity is recovered after a rapid and large dilution of enzyme/inhibitor mixturesCitation28. The mode of action of 2a has been similarly controlled by wash-out experiments, using URB-597 (belonging to the carbamate family of FAAH inhibitors, ) and CAY-10402 (belonging to the α-keto-heterocycle family of FAAH inhibitors) as standards for the irreversible and reversible inhibitions, respectivelyCitation38. Briefly, hFAAH and 2a (at 100, 150 and 200 µM) are incubated (1 h, 25 °C), then 100-fold diluted with buffer. Aliquots are taken (after 0, 30 and 90 min) for enzyme residual activity measurement. As shown in , the enzyme activity is totally recovered after rapid dilution of the 2a/hFAAH mixtures, indicating a reversible mode of action, like CAY-10402. On the contrary, the hFAAH activity remains largely inhibited in the case of URB-597.
Figure 5. Percentage of recovery of hFAAH activity after dilution with 2a versus controls.
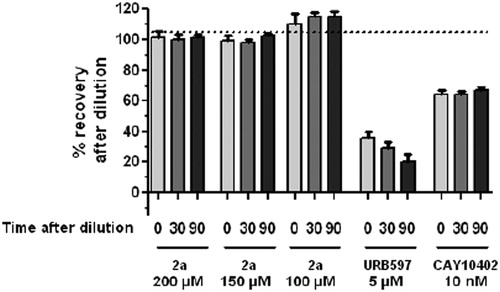
Thus, 2a and 1a exhibit very similar reversible inhibition profiles but the former β-lactam is 1000 times less potent, and 40 times less selective, than the latter. This unexpected and disappointing result could not be predicted from the docking of the novel compounds 2a in the simple model of hFAAH active site and the comparison with the previous inhibitor 1a.
Docking studies
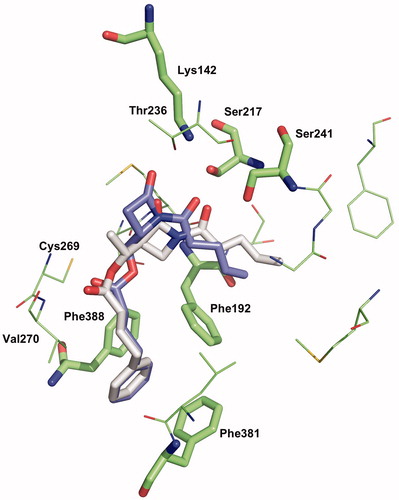
The position and confirmation of the amino acids of the modelled hFAAH active site we used in 2009Citation2Citation8 is close to the crystallographic structure of h/rFAAH published in 2011Citation44, where the authors have been able to identify both non-covalent and covalent states of a α-keto-heterocycle reversible inhibitor trapped into the catalytic pocket. The hFAAH active site is formed by a hydrophobic tunnel (named ACB for acyl chain binding), the hydrophilic catalytic triad (Ser241, Ser217 and Lys142), a second tunnel exposed to the solvent (named CA for cytoplasmic access) and a third channel, close to the ACB channel, composed of three phenylalanine residues (Phe338, Phe381 and Phe192) and named as “phenyl pocket”. In this model, the same binding mode (named mode 1)Citation28 is found for 1a and 2a docked as non-covalent complexes in the active site as they are reversible, competitive inhibitors (). The molecules feature the syn-conformation of the imide motif (formed by the β-lactam carbonyl C2=O and the exocyclic carbonyl N1-C=O). Hydrogen bonds are especially observed between Ser241 and Ser217 and the exocyclic CO, and between Thr236 and the β-lactam CO. The phenyl chain interacts with the “phenyl pocket” and the alkene chain lies at the beginning of ACB channel. The complexes of 1a and 2a have been optimised and their interaction energies have been calculated at the B3LYP-PM6 level. In this binding mode 1, the 2a complex is found more stable than the 1a complex (ΔE = 6.7 kcal/mol). This result is in disagreement with our experimental values of inhibition and thereby prompts us to consider water molecules into the active site model, which will induce different binding modes, allowing possibly the discrimination between the series of inhibitors.
Figure 6. Binding mode 1 of 1a and 2a in the active site of hFAAH. Hydrogens were removed for clarity.
The modelling and analysis of crystallographic data featuring waters in recently solved h/rFAAH structures have confirmed their presence as well as the importance of two water molecules being close to the catalytic triadCitation16. We, therefore, considered docking studies, including them in the active site by using the option of GOLD allowing water molecules to switch on and off and to rotateCitation45. Such strategy was repeatedly shown to better model the interactions of inhibitors into enzyme cavityCitation46,Citation47. Two models can be built: the first with only the water molecule closest to the triad (1 W model), and the second with both water molecules (2 W model).
The binding mode 1 appears still compatible with the presence of one water molecule (mode 1-1 W) in interaction with the imide function of the inhibitor (1a or 2a) and the active serine residue; in that position both complexes have the same stabilisation energy (ΔE = 0.7 kcal/mol) (see Figure S1 in Supplementary Material).
Another binding mode, named mode 2, can be found where the water molecule makes hydrogen bonds with the Ser241 residue and the ester function of the inhibitor (1a or 2a). In this pose, the imide conformation is anti and the phenyl chain lies in the ACB channel (), as observed in the crystallographic complex formed by h/rFAAH and OL-135Citation44. Here, computations suggest the 1a complex as the most favourable one (ΔE = 8.8 kcal/mol). With two water molecules in the active site (in interaction with Ser241), another binding mode (named mode 3) is revealed for both inhibitors 1a and 2a (). The β-lactam carbonyl is turned to Ser241, the phenyl chain is located in the “phenyl pocket” and the alkene chain occupies the CA channel. The binding mode 3 gives a more stable complex for 1a, compared with 2a (ΔE = 16.6 kcal/mol). This binding mode is not compatible with the anti-conformation of the imide motif. Binding modes 2 and 3 are therefore in agreement with the experimentally determined potency of the compounds.
Figure 7. Binding mode 2-1 W of 1a and 2a in the active site of hFAAH. Hydrogens were removed for clarity.
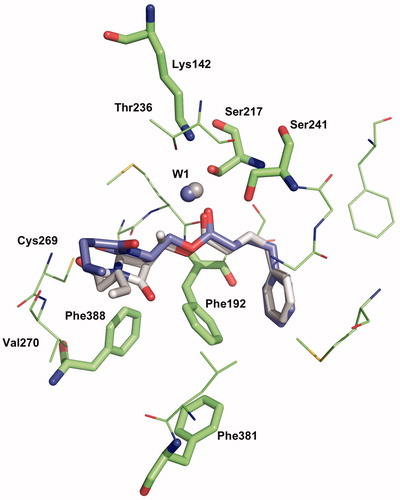
Figure 8. Binding mode 3-2 W of 1a and 2a in the active site of hFAAH. Hydrogens were removed for clarity.
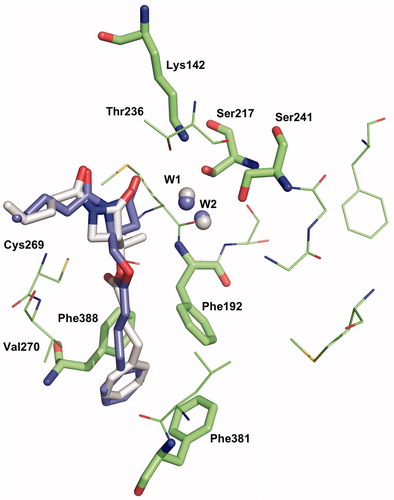
In the novel modes 2 and 3, like in mode 1, the C4 chain of β-lactams lies in a hydrophobic environment slightly compatible with a pyridinyl substituent. This may explain the drop of activity observed for compounds 2d and 2e. The reduced activity of 2g featuring an ether function in the phenyl chain (2g is an isostere of 2a) can be similarly justified.
Conclusion
The search for non-covalent, reversible, competitive inhibitors of hFAAH is an important topic in current drug discovery, already illustrated by various heterocyclic structuresCitation48, such as benzoxazolesCitation49, benzothiazolesCitation50, benzimidazolesCitation51, tetrahydropyridopyridinesCitation52 and piperidinesCitation53.
We previously demonstrated that β-lactams, which are usually covalent inhibitors of serine-enzymes by acylation reaction, behave as non-covalent inhibitors of hFAAH as exemplified with the lead compound 1aCitation28,Citation37,Citation38. Interestingly, OMDM169 (β-lactone) was also reported to be a reversible inhibitor of MAGLCitation20.
In this paper, our idea was to develop further efficient and selective hFAAH β-lactamic inhibitors, acting similarly to our previous lead compound 1a, and rapidly produced (three steps) from a cheap chiral starting material, available on large scale (precursor B). We could indeed easily obtain the novel series of β-lactams 2, but their inhibition activities against hFAAH were disappointing, although the molecules fitted perfectly into our first enzyme active site model, with an interaction energy even more favourable than 1a. Therefore, we questioned about the hFAAH model relevance as a predictive tool for the design of novel active β-lactams.
Since hFAAH is not yet crystallised, we considered the recent crystallographic data of h/rFAAH suggesting to take into account the presence of linked water molecules into the active site. Two water molecules identified in the free enzyme are still present in the enzyme/inhibitor complex (acyl-enzyme intermediate)Citation16, or replaced by the inhibitor (tetrahedral intermediate)Citation14,Citation15,Citation44,Citation54 covalently fixed on Ser241. New docking experiments of β-lactams 1a and 2a have been performed by introducing two water molecules near the catalytic triad. Then, we have left the choice to remove one or two water molecules. The results show that at least one water molecule is conserved and new binding modes of the inhibitors have been identified. In particular, the positioning of the C4 phenyl chain in the “phenyl pocket” and the alkene chain in the CA channel, instead of the ACB tunnel, has revealed a significantly higher stabilisation of the 1a non-covalent complex compared with 2a. This binding mode 3 could possibly explain the micromolar activity of 2a regarding the nanomolar activity of 1a.
In the absence of available experimental data on co-crystals formed from hFAAH and β-lactamic inhibitors, h/rFAAH active site models and related docking experiments can be used for the design of new inhibitors, though their outputs are mainly qualitative. At the present time, the biochemical screening of oriented libraries of compounds remains the most suitable strategy of lead discoveryCitation55,Citation56. This is particularly true in the field of β-lactam derivatives which involve a privileged structure (azetidin-2-one) commonly found in natural products and (semi)-synthetic drugs. In this context, our series of β-lactams could be advantageously tested against serine hydrolases, including diacylglycerol lipase also involved in the EC systemCitation19.
Declaration of interest
This work was supported by the UCL, F.R.S.-FNRS (Belgium) and partially by the PAI program PROFUSA P6/19. C. M., J. M.-B., R. R. and E. A. P. are senior research associates of the F.R.S.-FNRS (Belgium).
The authors have no conflicts of interest.
Supplementary material available online Preliminary screening of tri-substituted β-lactams, synthesis of 4- and 2-(pyridin-4-yl)butanoic acids and docking of 1a and 2a into hFAAH with binding mode 1-1w are given as Supplementary material.
Supplementary Material
Download PDF (783 KB)References
- Pillarisetti S, Alexander CW, Khanna I. Pain and beyond: fatty acid amides and fatty acid amide hydrolase inhibitors in cardiovascular and metabolic diseases. Drug Discov Today 2009;14:1098–111
- Gerra G, Zaimovic A, Gerra ML, et al. Pharmacology and toxicology of Cannabis derivatives and endocannabinoid agonists. Recent Pat CNS Drug Discov 2010;5:46–52
- Muccioli GG. Endocannabinoid biosynthesis and inactivation, from simple to complex. Drug Discov Today 2010;15:474–83
- Vandevoorde S, Lambert DM. The multiple pathways of endocannabinoid metabolism: a zoom out. Chem Biodivers 2007;4:1858–81
- Vandevoorde S. Overview of the chemical families of fatty acid amide hydrolase and monoacylglycerol lipase inhibitors. Curr Top Med Chem 2008;8:247–67
- Seierstad M, Breitenbucher JG. Discovery and development of fatty acid amide hydrolase (FAAH) inhibitors. J Med Chem 2008;51:7327–43
- Bertrand T, Augé F, Houtmann J, et al. Structural basis for human monoglyceride lipase inhibition. J Mol Biol 2010;396:663–73
- Feledziak M, Lambert DM, Marchand-Brynaert J, et al. Inhibitors of the endocannabinoid-degrading enzymes, or how to increase endocannabinoid’s activity by preventing their hydrolysis. Rec Patents CNS Drug Disc 2012;7:49–70
- Bisogno T, Maccarone M. Latest advances in the discovery of fatty acid amide hydrolase inhibitors. Expert Opin Drug Discov 2013;8:509–22
- Khanna IK, Alexander CW. Fatty acid amide hydrolase inhibitors – progress and potential. CNS Neurol Disord Drug Targets 2011;10:545–58
- Bracey MH, Hanson MA, Masuda KR, et al. Structural adaptations in a membrane enzyme that terminates endocannabinoid signaling. Science 2002;298:1793–6
- Mc Kinney MK, Cravatt BF. Structure and function of fatty acid amide hydrolase. Annu Rev Biochem 2005;74:411–32
- Michaux C, Labar G. Fatty acid amide hydrolase: from characterization to therapeutics. Chem Biodivers 2007;4:1882–902
- Mileni M, Johnson DS, Wang Z, et al. Structure-guided inhibitor design for human FAAH by interspecies active site conversion. Proc Natl Acad Sci USA 2008;105:12820–4
- Mileni M, Garfunkle J, DeMartino JK, et al. Binding and inactivation mechanism of a humanized fatty acid amide hydrolase by α-ketoheterocycle inhibitors revealed from cocrystal structures. J Am Chem Soc 2009;131:10497–506
- Mileni M, Garfunkle J, Ezzili C, et al. X-ray crystallographic analysis of α-ketoheterocycle inhibitors bound to a humanized variant of fatty acid amide hydrolase. J Med Chem 2010;53:230–40
- Mileni M, Kamtekar S, Wood DC, et al. Crystal structure of fatty acid amide hydrolase bound to the carbamate inhibitor URB597: discovery of a deacylating water molecule and insight into enzyme inactivation. J Mol Biol 2010;400:743–54
- Labar G, Bauvois C, Borel F, et al. Crystal structure of the human monoacylglycerol lipase, a key actor in endocannabinoid signaling. ChemBioChem 2010;11:218–27
- Ortar G, Bisogno, T, Ligresti A, et al. Tetrahydrolipstatin analogues as modulators of endocannabinoid 2-arachidonoylglycerol metabolism. J Med Chem 2008;51:6970--9
- Bisogno T, Ortar G, Petrosino S, et al. Development of a potent inhibitor of 2-arachidonoylglycerol hydrolysis with antinociceptive activity in vivo. Biochim Biophys Acta 2009;1791:53–60
- Urbach A, Muccioli GG, Stern E, et al. 3-Alkenyl-2-azetidinones as fatty acid amide hydrolase inhibitors. Bioorg Med Chem Lett 2008;18:4163–7
- Böttcher T, Sieber SA. β-Lactones as privileged structures for the active-site labeling of versatile bacterial enzyme classes. Angew Chem Int Ed 2008;47:4600–3
- Gérard S, Nollet G, Vande Put J, et al. 1-Alkoxycarbonyl-3-halogenoazetidin-2-ones as elastase (PPE) inhibitors. Bioorg Med Chem 2002;10:3955–64
- Gérard S, Galleni M, Dive G, et al. Synthesis and evaluation of N1/C4-substituted β-lactams as PPE and HLE inhibitors. Bioorg Med Chem 2004;12:129–38
- Gerona-Navarro G, Perez de Vega MJ, Garcia-Lopez MT, et al. Synthesis and anti-HCMV activity of 1-acyl-β-lactams and 1-acylazetidines derived from phenylalanine. Bioorg Med Chem Lett 2004;14:2253–6
- Gerona-Navarro G, Perez de Vega MJ, Garcia-Lopez MT, et al. From 1-acyl-β-lactam human cytomegalovirus protease inhibitors to 1-benzyloxycarbonylazetidines with improved antiviral activity. A straightforward approach to convert covalent to noncovalent inhibitors. J Med Chem 2005;48:2612–21
- Marchand-Brynaert J, Brulé C. Penicillins. In: Katritzky AR, Ramsden CA, Scriven EFV, Taylor RJK, eds. Comprehensive heterocyclic chemistry III. Vol. 2. Oxford: Elesvier Ltd; 2008:173–237
- Feledziak M, Michaux C, Urbach A, et al. β-Lactams derived from a carbapenem chiron are selective inhibitors of human fatty acid amide hydrolase versus human monoacylglycerol lipase. J Med Chem 2009;52:7054–68
- Berks AH. Preparations of two pivotal intermediates for the synthesis of 1-β-methyl carbapenem antibiotics. Tetrahedron 1996;52:331–75
- Urbach A, Dive G, Marchand-Brynaert J. Novel large-ring 1,3-bridged 2-azetidinones as potential inhibitors of penicillin-binding proteins. Eur J Org Chem 2009;11:1757–70
- Salzman TN, Ratcliffe RW, Christensen BG, Bouffard FA. A stereocontrolled synthesis of (+)-thienamycin. J Am Chem Soc 1980;102:6161–3
- Baldwin JE, Adlington RM, Callins DW, et al. Stereospecific synthesis of dealanylalahopcin. Tetrahedron 1990;46:4733–48
- Jones G, Willett P, Glen RC, et al. Development and validation of a genetic algorithm for flexible docking. J Mol Biol 1997;267:727–48
- Frisch MJ, Trucks GW, Schlegel HB, et al. Gaussian 09, Revision A.02. Wallingford (CT): Gaussian, Inc.; 2009
- De Lano WL. The PyMOL molecular graphics system, 0.99. San Carlos: DeLano Scientific; 2002
- Gérard S, Marchand-Brynaert J. Protecting group migration in the chemistry of 1-t-butyldimethylsilyl-4-hydroxymethyl-2-azetidinone. Tetrahedron Lett 2003;44:6339–42
- Feledziak M, Muccioli GG, Lambert DM, et al. SAR and LC/MS studies of β-lactamic inhibitors of human fatty acid amide hydrolase (hFAAH): evidence of a nonhydrolytic process. J Med Chem 2011;54:6812–23
- Feledziak M, Michaux C, Lambert DM, et al. An unprecedented reversible mode of action of β-lactams for the inhibition of human fatty acid amide hydrolase (hFAAH). Eur J Med Chem 2013;60:101–11
- Klein SI, Molino BF, Czekaj M, et al. Design of a new class of orally active fibrinogen receptor antagonists. J Med Chem 1998;41:2492–502
- Menghin S, Pertz HH, Kramer K, et al. Nα-imidazolylalkyl and pyridylalkyl derivatives of histaprodifen: synthesis and in vitro evaluation of highly potent histamine H1-receptor agonists. J Med Chem 2003;46:5458–70
- Gérard S, Dive G, Clamot B, et al. Synthesis, hydrolysis, biochemical and theoretical evaluation of 1,4-bis(alkoxycarbonyl)azetidin-2-ones as potential elastase inhibitors. Tetrahedron 2002;58:2423–33
- Labar G, Vliet FV, Wouters J, et al. A MBP-FAAH fusion protein as a tool to produce human and rat fatty acid amide hydrolase: expression and pharmacological comparison. Amino Acids 2008;34:127–33
- Labar G, Bauvois C, Muccioli GG, et al. Disulfiram is an inhibitor of human purified monoacylglycerol lipase, the enzyme regulating 2-arachidonoylglycerol signaling. ChemBioChem 2007;8:1293–7
- Mileni M, Garfunkle J, Ezzili C, et al. Fluoride-mediated capture of a noncovalent bound state of a reversible covalent enzyme inhibitor: X-ray crystallographic analysis of an exceptionally potent α-ketoheterocycle inhibitor of fatty acid amide hydrolase. J Am Chem Soc 2011;133:4092–100
- Verdonk ML, Chessari G, Cole JC, et al. Modeling water molecules in protein-ligand docking using GOLD. J Med Chem 2005;48:6504–15
- Huang W, Gherib R, Gauld JW. An active site water broadens substrate specificity in S-ribosylhomocysteinase (LuxS): a docking, MD, and QM/MM study. J Phys Chem B 2012;116:8916–29
- Yoshikawa Y, Kobayashi K, Oishi S, et al. Molecular modeling study of cyclic pentapeptide CXCR4 antagonists: new insight into CXCR4-FC131 interactions. Bioorg Med Chem Lett 2012;22:2146–50
- Otrubova K, Boger DL. α-Ketoheterocycle-based inhibitors of fatty acid amide hydrolase (FAAH). ACS Chem. Neuroscience 2012;3:340–8
- Estiarte MA, Johnson RJ, Kaub CJ, et al. 2-Amino-5-arylbenzoxazole derivatives as potent inhibitors of fatty acid amide hydrolase (FAAH). MedChemComm 2012;3:611–19
- Tian G, Paschetto KA, Gharahdaghi F, et al. Mechanism of inhibition of fatty acid amide hydrolase by sulfonamide-containing benzothiazoles: long residence time derived from increased kinetic barrier and not exclusively from thermodynamic potency. Biochemistry 2011;50:6867–78
- Min X, Thibault ST, Porter AC, et al. Discovery and molecular basis of potent noncovalent inhibitors of fatty acid amide hydrolase (FAAH). Proc Natl Acad Sci USA 2011;108:7379–84
- Gowlugari S, DeFalco J, Nguyen MT, et al. Discovery of potent, non-carbonyl inhibitors of fatty acid amide hydrolase (FAAH). MedChemComm 2012;3:1258–63
- Caprioli A, Coccurello R, Rapino C, et al. The novel reversible fatty acid amide hydrolase inhibitor ST4070 increases endocannabinoid brain levels and counteracts neuropathic pain in different animal models. J Pharmacol Exper Therapeutics 2012;342:188–95
- Ezzili C, Mileni M, McGlinchey N, et al. Reversible competitive α-ketoheterocycle inhibitors of fatty acid amide hydrolase containing additional conformational constraints in the acyl side chain: orally active, long-acting analgesics. J Med Chem 2011;54:2805–22
- Swinney DC, Anthony J. How were new medicines discovered? Nature Rev Drug Discov 2011;10:507--19
- Kumar K, Waldmann H. Synthesis of natural product inspired compound collections. Angew Chem Int Ed 2009;48:3224–42