
Abstract
Curcumin (diferuloylmethane), the active ingredient in the eastern spice turmeric (Curcuma longa), has been shown to inhibit the activities of numerous enzymes and signaling molecules involved in cancer, bacterial and viral infections and inflammatory diseases. We have investigated the inhibitory activities of curcumin and chemically modified curcumin (CMC) derivatives toward lethal factor (LF), the proteolytic component of anthrax toxin produced by the bacterium Bacillus anthracis. Curcumin (Compound 1) appears to inhibit the catalytic activity of LF through a mixture of inhibitory mechanisms, without significant compromise to the binding of oligopeptide substrates, and one CMC derivative in particular, Compound 3 (4-phenylaminocarbonylbis-demethoxycurcumin), is capable of inhibiting LF with potency comparable with the parent compound, while also showing improved solubility and stability. The quantitative reduction in catalytic activity achieved by the different CMC derivatives appears to be a function of the proportion of the multiple mechanisms through which they inhibit the enzyme.
Introduction
Bacillus anthracis and anthrax
Bacillus anthracis is the bacterium responsible for the disease anthrax in humans and other animals, and is most often encountered in nature in the form of spores that are highly resistant to environmental damage. Though cutaneous or gastrointestinal anthrax infection can be fatal if not treated promptly, infection is most deadly when these spores are inhaled. Macrophages and dendritic cells take up the spores at the site of infection and are then mobilized to the nearby lymph nodes, where the spores begin to germinate within the phagolysosomesCitation1,Citation2. Once the lymph node has been incapacitated, the bacteria continue to infect additional lymph nodes and eventually enter the blood streamCitation1. This process of quickly taking up residence in the lymph nodes and destroying or inactivating the local population of macrophages and dendritic cells is thought to depress the initial immune response and increase the virulence of B. anthracis infectionCitation1–3. A major contributing factor to the morbidity of anthrax infection is that it is often associated with no or only mild symptoms until the bacteria have left the lymph nodes and spread to the blood, at which point they begin to multiply at a rate that is very difficult to overcome by antibiotics aloneCitation4,Citation5. Especially after the terrorist events of 2001, the threat of anthrax as a biological weapon has made development of more effective treatments for the infection a top priority, though only limited progress has been made over this time.
The primary toxic effects of B. anthracis are caused by secretion of three proteins acting synergistically: lethal factor (LF), the metalloproteinase component; edema factor (EF), a Ca2+/calmodulin-dependent adenylate cyclase and protective antigen (PA), a carrier protein for the former two proteins that binds cell surface receptors and polymerizes to form a poreCitation3,Citation6. In isolation, LF and EF are benign to cells: they require binding to PA before they can enter cells and exert their cytotoxicity. The complexes with PA are referred to as lethal toxin (LeTx) and edema toxin, respectivelyCitation6,Citation7. Ultimately, evidence shows that the biological activity of LF is the main and direct cause of anthrax toxicity.
Once secreted, PA molecules bind to a class of cell surface receptors known as anthrax toxin receptors, which allows a furin-like protease on the cell surface to release a 20 kDa portion of the PA amino terminus, leaving the activated form of PA behind in the plasma membrane. Once activated, PA molecules come together to form a heptamer, and are then capable of binding both LF and EF. Evidence has shown that PA also exists in its cleaved, active form in the serum, in complex with LF or EFCitation1,Citation8,Citation9. These complexes localize in lipid rafts within the cell membrane, and are subsequently endocytosed into acidic compartments, where the endosomal change in pH causes the PA components of the toxin to undergo a conformational change, forming a pore and releasing bound LF or EF into the cytosol, where they exert their effects directly on target proteinsCitation10.
Until recently, LF had only one known group of natural substrates: the mitogen-activated protein kinase kinase (MAPKK), or MEK, family of protein kinases, a central component of the mitogen-activated protein kinase (MAPK) cascade that is involved in transcription of genes controlling the cell cycle, cell growth, differentiation, development and multiple arms of the inflammatory response. LF specifically cleaves the MEKs at a consensus sequence near their amino termini, at which they normally bind to their own downstream targets, the MAPK, or ERK, family of protein kinases. Disruption of the MAPK signaling pathway by LF results in cell death via apoptosis in some cases, and down regulation of inflammatory biomarkers, leading to a general suppression of the innate immune responseCitation3.
Recently, a second target of LF from the family of pathogen-associated molecular pattern recognition proteins known as the NOD-like receptors (NLRs) was discovered in rat and mouse macrophagesCitation11,Citation12. Activation of these NLRs, specifically Nlrp1 in ratsCitation12 and Nlrp1b in miceCitation11, occurs when LF cleaves a sequence of residues near their amino termini, as with the MAPKKs. Cleavage of Nlrp1/Nlrp1b causes it to oligomerize with other activated Nlrp1/Nlrp1b proteins, and to subsequently aid in the recruitment of caspase-1 to a multi-protein complex known as the inflammasomeCitation12. Activation of caspase-1 by the inflammasome causes rapid programmed cell death, or pyroptosis, along with the production of inflammatory cytokinesCitation11,Citation12 IL-1β and IL-18. Though this signaling pathway has not been characterized as extensively as the MAPK pathway, and the relationship between LF and Nlrp1 has thus far only been studied in rats and mice, humans express an ortholog of Nlrp1, and this role for LF may contribute to the rapid, systemic toxicity of anthrax infection that could not previously be explained by the cleavage of the MAPKKs alone.
The LF protein itself has four domains, which appear to have arisen through a series of duplications, mutations and fusions over timeCitation6,Citation13. Domain I is the protein’s PA-binding domain, which allows LF to complex with the carrier protein. Domains II, III, and IV together form a groove in which substrate binding and catalysis occur. Domain II resembles the ADP-ribosylating toxin of the closely related bacterium Bacillus cereus. Domain III folds into Domain II, and may be a product of a duplication of regions of Domain II. Domain IV is the catalytic domain of LF, a metalloproteinase domain classified as a member of the zincin family of zinc metalloproteinases, in the subfamily known as gluzincinsCitation14. This domain also bears structural similarities to Domain I.
It is important to point out that the results of intoxication with LF have been shown to differ considerably from the systemic effects of clinical anthrax infection. Work done by Popov et al.Citation15 has shown that B. anthracis secretes a number of metalloproteinases in addition to LeTx, and these, as well as cell wall components released by dying bacteria, may play a role in the systemic effects of anthrax not seen when experimental animals are challenged with LeTx aloneCitation15,Citation16. Nevertheless, LF may be responsible for the initial, primary physiological insult, incapacitating macrophages and dendritic cells within the lymph nodes, and allowing the bacteria to reach a threshold where they may then spread and cause these systemic effects, such as diffuse vasculitis, hemorrhagic lesions and necrosis in multiple tissuesCitation2,Citation15. By limiting the initial insult through inhibition of LF, it may be possible to overcome the acute danger of anthrax, make treatment more effective, and decrease fatalities from infection.
Curcumin: history, chemistry and evidence for inhibition of metalloproteinases
Curcumin is a naturally occurring compound of the curcuminoid family, isolated originally from the plant Curcuma longa. The rhizome of this plant, specifically, is used to create the spice known as turmeric, and is a major component of the daily diet in many Asian countries. Even before the modern characterization of curcumin’s molecular structure and functionality, it has long been used in traditional eastern medicines.
With its natural medicinal history in mind, curcumin has been studied extensively over the past few decades in a wide variety of systems, and has been found to exhibit significant pleiotropic effects. These effects may be attributed to the chemistry of curcumin, consisting of two hydroxylated phenyl rings joined by a conjugated, flexible linker region with a β-diketone moiety at its center (). The β-diketone moiety is capable of undergoing keto–enol tautomerization, though the enol form is more stable in both the solid phase and in solution, and is the dominant species at physiological pHCitation17,Citation18. The biological activities of curcumin are wide ranging: beyond having intrinsic antioxidant properties, it has been found to bind a wide spectrum of cellular constituents in vitro and in vivo, including inflammatory molecules, protein kinases, carrier proteins, cell survival proteins, structural proteins, prion protein, antioxidant response elements, metal ions and moreCitation17. In addition, curcumin has been shown to localize in the plasma membrane, cytoplasm, nucleus and mitochondria, and shows virtually no toxicity in humansCitation17,Citation19,Citation20.
Figure 1. Molecular structures of Compounds 1–5.
The structural homology among the bacterial metalloproteinases, including LF, botulinum toxin and thermolysin, as well as the eukaryotic matrix metalloproteinases, prompted us to consider whether inhibitors of the mammalian zinc-dependent proteolytic enzymes could also be used to inhibit the bacterial proteinases. Initial studies on compounds that inhibit both families of metalloproteinases were carried out by Kocer et al.Citation21 using non-antimicrobial tetracyclines, and showed that these chemically modified tetracyclines were capable of inhibiting LF in human macrophages and monocytoid cell lines when administered before or even hours after the addition of the toxin to the cellsCitation21. Given that the β-diketone moiety seems to be responsible for the tetracyclines’ inhibition of other metalloproteinases via zinc binding, the metal-chelating capacity of curcumin provided a basis for consideration that the compound could also inhibit these metalloenzymes by binding to the zinc atom in their active site through its own β-diketone moietyCitation22. In addition to the tetracyclines, other polyphenolic compounds, such as the catechin family of compounds, have also been shown to act as LF inhibitorsCitation23. These observations, when combined with the extensively documented overall anti-inflammatory properties of curcumin, provide support for efforts to explore the considerable potential of curcumin and its derivatives as candidates for drug development.
Chemical modification of curcumin
The presence of hyroxylated phenyl groups in many of the known inhibitors of LF suggests that conjugated hydroxyl or oxo groups may play an important role in the binding of these compounds to the enzymeCitation23–25. By chemically modifying these and other substituents of curcumin, and screening them for their individual effect on the level of inhibition, we can make better predictions about their importance to binding, and begin to formulate hypotheses of their mode of action as we move toward more rationally designed compounds with increased potencies.
Chemically modified curcumin (CMC) derivatives, all of which vary with respect to additions, subtractions, and modifications of the naturally occurring functional groups of the parent compound, have been synthesized by numerous groups, in an attempt to exploit their specific interactions in particular systems. In this study, four such CMCs (Compounds 2–5), synthesized as reported previouslyCitation22, were examined for inhibition of LF in vitro, in addition to curcumin itself ().
Some of these CMCs have been found to have inhibitory potencies greater than or equal to curcumin itself against several of the matrix metalloproteinasesCitation22, and this provided sufficient evidence that they may also exhibit comparable inhibition against LF. One of these CMCs in particular, Compound 3 (4-phenylaminocarbonyl bis-demethoxycurcumin, CMC2.24Citation22), has shown exceptional promise in other systems, and is thus given prominence in this and other papers.
Compound 3 shows improved solubility and even less toxicity in cell and tissue culture, as well as in in vivo studies, when compared with the parent compoundCitation22. The modifications to curcumin in synthesizing Compound 3 include subtraction of the methoxy groups from the 3′ positions of curcumin’s flanking aromatic rings, as well as the addition of a phenyl group, which is connected to the center of the molecule via a peptide bond. This modification provides Compound 3 with an additional carbonyl capable of participating in keto–enol tautomerization, as well as several additional resonance structures, and a third hydrophobic region at its periphery. Studies by Zhang et al. show that Compound 3 is nearly 10-fold more acidic than curcumin itselfCitation18, and exists largely as an enolate rather than an enol at physiological pH, which is likely a consequence of the additional electron-withdrawing group. This difference also seems to be responsible for Compound 3’s greater solubility and superior zinc-binding abilityCitation18.
Methods
Recombinant LF (native sequence) was purchased from List Biological Laboratories, Inc. (Campbell, CA), and used without further purification. The lyophilized enzyme was solubilized to 11.1 μM in 5 mM HEPES, 50 mM NaCl, and 1 mg/mL bovine serum albumin, at pH 7.5, and stored at −80 °C, as per the manufacturer’s protocol. MAPKKide, a FRET-quenched synthetic peptide analogous to the MAPKK N-terminal domain was also purchased in lyophilized form from List Biological Laboratories, Inc., solubilized to 5 mM in ≥99.9% A.C.S. spectrophotometric grade DMSO from Sigma-Aldrich (St. Louis, MO), and stored at −20 °C without further purification, as per the manufacturer’s protocol. Curcumin (99% purity) was purchased from Selleck Chemicals (Houston, TX), diluted to 50 mM in ≥99.9% DMSO (Sigma-Aldrich, St. Louis, MO), and stored at −20 °C. All CMC derivatives were synthesized as reported and characterized in a previously published paperCitation22, solubilized to 50 mM in ≥99.9% DMSO (Sigma-Aldrich, St. Louis, MO), and stored at −20 °C. No alterations or modifications to the previously published synthesis protocol were made. Purity of ≥99.5% was ensured for all synthesized compounds through 1H NMR, mass spectrometry, TLC and HPLC. The reaction buffer prepared for use in all assays was 20 mM HEPES with 0.125 mg/mL bovine serum albumin, adjusted to pH 7.4, and stored at 4 °C.
LF peptidolytic activity assay
Cleavage of the synthetic MAPKK substrate by LF results in separation of the donor and acceptor moieties and restoration of fluorescence. Development of fluorescence as read by a SpectraMax M2 multiwell microplate fluorometer (Ex/Em = 490 nm/523 nm) was used to measure the rate of LF-catalyzed substrate cleavageCitation21. All reagents were diluted as necessary for each assay just prior to use. Assays were repeated until conditions were optimized and consistent results were achieved. Reactions for data collection were then carried out in triplicate, in 96-well microplates, at 37 °C, with a final volume of 150 μL in each well: 50 μL of LF, 50 μL of substrate and 50 μL of inhibitor (or reaction buffer for control wells). LF was diluted with reaction buffer to a 100 nM final concentration in the well under all reaction conditions, while inhibitor and substrate were diluted to various concentrations, in reaction buffer, as described in the results. All LF and inhibitor/control pairs were incubated at 37 °C in the reaction wells for 20 min prior to the addition of substrate, and read by the fluorometer immediately after the addition of substrate. IC50 values for curcumin (Compound 1) and Compound 3 were determined empirically by plotting the percentage of inhibition versus inhibitor concentration. Additional graphical analysis was performed using nonlinear curve fitting software (EnzFitter, Elsevier-Biosoft).
Results
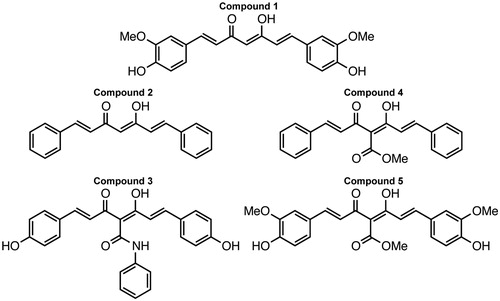
Curcumin (Compound 1) and the CMCs we tested, with the exception of Compound 2, were all found to inhibit LF peptidolytic activity in a dose-dependent fashion (). The linear time course of the reactions indicates that neither the assay conditions nor the extended presence of the inhibitor had any denaturing effect on the enzyme, and thus that the data acquired do not reflect progressive irreversible inactivation. R2 values for all data exceeded 0.99, as determined by Levenberg–Marquardt linear regression analysis, indicating a high level of goodness-of-fit.
Figure 2. (A) Lethal factor activity as measured by fluorescence generated over time. 100 nM lethal factor, 3 μM substrate and 20 μM inhibitors were used uniformly. Assay buffer was used in place of inhibitor as a control. (B) Percent residual activity of 100 nM lethal factor and 3 μM substrate after incubation with 20 μM inhibitors. All experiments were performed in triplicate. R2 values for all data exceeded 0.99, as determined by Levenberg–Marquardt linear regression analysis, indicating a high level of goodness-of-fit.
On average, in the presence of 20 μM curcumin, cleavage of 3 µM substrate with 100 nM LF was inhibited by 33–41% (Supplemental ). Under the same conditions, Compound 3 was capable of inhibiting LF to a comparable extent, resulting in 26–28% inhibition. The other CMCs exhibited somewhat weaker inhibitory potency, likely indicative of their specific chemical modifications (i.e. the removal of 3′ methoxy and 4′ hydroxyl groups for Compounds 2 and 4, believed to be important to binding). In the presence of Compound 2, a derivative of curcumin stripped of nearly all functional groups, the level of LF activity was equal to or greater than that observed in the absence of any curcuminoids. IC50 values for curcumin and Compound 3 with 100 nM LF and 3 μM substrate were determined to be approximately 23.8 and 33.1 μM, respectively, though it is worth noting that these values are significantly dependent on substrate concentration.
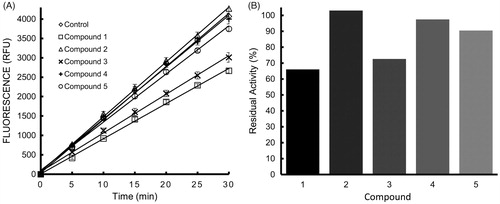
As the strongest inhibitor of LF among the CMCs tested, Compound 3 was assayed further, along with curcumin itself, in an attempt to begin to elucidate their mechanisms of action and potency. The same trend of dose-dependent inhibition was observed across four substrate concentrations tested for both curcumin and Compound 3 (Supplemental ), and unexpectedly, the extent of inhibition of LF after incubation with either curcumin or Compound 3 was unaffected or increased, with some variability across the spectrum of inhibitor concentrations, at increasing substrate concentrations (Supplemental ), suggesting that these compounds do not act through a simple competitive mechanism of inhibition, but rather that the curcuminoids act to inhibit LF through a mixture of inhibitory mechanisms. An increase in inhibition as a result of increasing substrate concentration, considered alone, would be evidence for uncompetitive inhibition; however, the fact that this trend is not entirely consistent indicates that multiple modes of inhibition are likely to account for the reduced enzyme activity, rather than a single classical inhibitory mechanism. The linearity of the progress curves for extent of substrate cleavage over time () suggests that the inhibitors are in rapidly reversible equilibrium with the enzyme.
The analysis of Lineweaver–Burk and Dixon plots () also supports the hypothesis that these compounds do not inhibit LF through a competitive mechanism, and as would be expected for a mixture of inhibitory mechanisms, the data fail to fit any one of the simple inhibitory patterns alone. Intersection of the lines in the second and third quadrant of a Lineweaver–Burk plot is an indication that curcumin and Compound 3 may actually act to increase the affinity of LF for substrate while they decrease catalytic activity. This again is strong evidence against competitive inhibition, as it would be unlikely for a compound to increase an enzyme’s affinity for substrate and to occupy its substrate-binding site simultaneously without postulating multiple inhibitor-binding sites on the enzyme. Instead, it appears that curcumin and Compound 3 act through mechanisms of both uncompetitive and non-competitive inhibition, with the relative contributions of these two modes of inhibition being functions of both substrate and inhibitor concentrations.
Figure 3. Lineweaver–Burk and Dixon plots of 100 nM lethal factor activity over four substrate concentrations and four inhibitor concentrations. (A) Lineweaver–Burk plot for Compound 1, (B) Lineweaver–Burk plot for Compound 3, (C) Dixon plot for Compound 1 and (D) Dixon plot for Compound 3.
If the mechanism of inhibition by these compounds was that of pure non-competitive inhibition, a Lineweaver–Burk plot should produce lines that intersect on the X-axis, while pure uncompetitive inhibition should produce parallel lines. Neither of these patterns were observed exclusively for curcumin or Compound 3, though it does appear that the lines are more parallel within the range of lower inhibitor concentrations for curcumin, and have a more parallel appearance overall for Compound 3, indicative of a predominantly uncompetitive inhibitory mode for the latter. Families of Dixon Plots for both curcumin and Compound 3 appeared as lines that intersect at a common point above the X-axis, as is seen for uncompetitive inhibition, with the exception of the lowest substrate concentration of both inhibitors assayed (3 μM), which deviates substantially from the trend. This provides additional support for the hypothesis that there are different inhibitory mechanisms at work over lower or higher relative substrate and inhibitor concentrations. Together, these patterns provide evidence that curcumin and Compound 3 may have more than one mode of binding to LF: one with a higher binding affinity and a second with a lower binding affinity that does not play a substantial role until a threshold concentration of inhibitor is reached.
The hypothesis that curcumin and Compound 3 act through a mixture of inhibitory mechanisms is not easily submitted to rigorous quantitation, though the preliminary data were analyzed according to models of pure non-competitive and pure uncompetitive inhibition, using nonlinear curve fitting algorithms that weigh the individual data points equally, unlike the linear transforms used in the Lineweaver–Burk and Dixon plots (Supplemental ). The analysis using such algorithms showed that the data for curcumin could be fit to a model of pure uncompetitive inhibition with an apparent Ki of 11.5 μM, while fitting the data to a model of purely non-competitive inhibition produced an apparent Ki of 20.3 μM – nearly double that of the uncompetitive fit. Fitting the data for Compound 3 to a model of purely uncompetitive inhibition produced an apparent Ki of 19.7 μM, while a fit to a model of pure non-competitive inhibition produced an apparent Ki of 34.5 μM. The R2 values for the fits of the data points to these two models were all greater than 0.92. These values suggest that aspects of both non-competitive and uncompetitive inhibition may play a part in the inhibition of LF activity by curcumin and Compound 3, and more specifically, that the compounds may be capable of binding both the enzyme–substrate complex (ES complex) and the free enzyme, but they appear to bind more tightly (nearly two-fold in the case of curcumin) to the ES complex, providing an explanation for the ratio of different inhibitory characteristics seen at varying substrate and inhibitor concentrations. Based on the preliminary data alone, however, it is still unclear whether these compounds actually bind to multiple sites on the enzyme, affecting substrate affinity and catalytic activity differentially at each of these putative sites.
Discussion
LF is just one component of the complex with PA known as LeTx, and further assays of the curcuminoids’ ability to inhibit this holotoxin must next be investigated to determine whether the compounds retain their inhibitory potency in cell-based assays that more closely model events in vivo. LF is known to bind to PA molecules after they have docked on the cell surface, but it has also been shown that LF and activated PA can bind in the extracellular spaceCitation1,Citation8,Citation9. LF and PA are certainly present in the extracellular space individually as well; thus, it is not entirely necessary for a LF inhibitor to be able to enter the cell on its own. Rather, it can suffice for an inhibitor to bind LF in the extracellular space and travel with it into the cell, or to prevent it from binding PA at all. In just one example, such mechanisms may apply to some hydroxamate inhibitors of LFCitation19.
Nevertheless, cellular uptake of curcumin has been quantitated and its localization throughout the cell demonstrated, particularly in the plasma membraneCitation20. Given the improved solubility and bioavailability of Compound 3Citation22, as well as the overall lipophilic nature of the parent compoundCitation20, evidence would suggest that cellular uptake of the CMCs should be comparable with the parent compounds. Moreover, considering the mixture of inhibitory mechanisms we believe to be at work in this system, it is entirely possible that the curcuminoids’ capacity to inhibit LF by binding and inducing a conformational change in the enzyme could also have an effect on its PA-binding domain, thus inhibiting not just the proteinase component of the toxin, but also the fundamental sequence of events that result in entry of LF into cells.
Curcumin has shown promise as a platform for the development of drugs to target many diseases and syndromes, including cancer and inflammatory diseases, as well as anthrax; however, one of the major obstacles to overcome in considering curcumin for further drug development has been its relatively low solubilityCitation15. Despite this, studies by Zhang et al. show that curcumin and Compound 3 bind fairly strongly to bovine serum albuminCitation18, and when considering normal plasma concentrations of serum albumin, this should provide sufficient capacity to carry high enough concentrations of curcumin or Compound 3 through the blood, increasing the half-time of their decomposition from mere minutes to tens of hours or days. In this same study, curcumin and Compound 3 administered by oral gavage to rats expressing pathologically excessive levels of MMPs showed no evidence of toxicity, even in doses as high as 500 mg/kg of body weightCitation22. Through chemical modification, it has now proven possible to synthesize derivatives of curcumin that have improved solubility, stability and potential bioavailability, while still retaining or improving upon the inhibitory potency and negligible toxicity of the parent compound.
The data obtained thus far are encouraging, though in general, in vivo experiments need to be devised to assess a specific curcuminoid’s value as an agent to be considered for post-exposure prophylactic management of anthrax. Though curcumin appears ostensibly to be a more potent inhibitor of LF in these in vitro studies, Compound 3’s superior stability, solubility and ability to bind serum albumin provide enough evidence that it should not be ruled out as a potentially valuable therapeutic agent for treating anthraxCitation18. If it can be shown that the curcuminoids are capable of inhibition of not just LF but LeTx as well, then experiments with cell lines should next be conducted to determine cell permeability of each individual candidate CMC. On the basis of such cell-based studies, tests of the compound in animal models can then be contemplated and improved methods for delivering the compound can be devised.
The hypothesis that the curcuminoids may actually function via a combination of inhibitory mechanisms, or may even bind to two physically distinct sites on LF, resulting in the apparent mixed mode by which the compounds inhibit the enzyme, should also be investigated further. Two scenarios are possible to explain these observations. One possibility is that a single curcumin or Compound 3 molecule may be capable of binding to an allosteric site on LF, and may affect both affinity for substrate and catalytic activity by inducing a conformational change in the enzyme. A second possibility is that there is more than one binding site for curcumin or Compound 3 molecules on LF, one of which may affect catalysis and the other of which may affect substrate binding; when both sites are occupied, they may both contribute to the conformational changes necessary to affect substrate binding and catalysis. The presence of zinc atoms at these allosteric sites could provide a chemical basis for inhibitor binding. Regardless, the data thus far point to an inhibitory mechanism other than competitive inhibition, a conclusion consistent with other reports of inhibition of LF by other inhibitors of LF with aromatic rings bearing hydroxyl or oxo groups, such as the gallocatechinsCitation5,Citation23–25.
The zinc-binding capacity of the curcuminoids was part of the basis for our initial motivation to explore their efficacy against LF, and though our data do not indicate that they inhibit the enzyme by binding the zinc atom in its active site, studies by Kochi et al. have shown evidence of up to three zinc atoms present in LFCitation26, any or all of which may be involved in the binding of the curcuminoids to LF at one or more allosteric sites. It also appears that the presence of hydroxyl groups at the 4′ positions of these molecules may play an important role in the binding of these compounds to LF, as indicated by the inferior inhibitory potency of Compounds 2 and 4, which lack them.
It is a reasonable concern that an agent with broad spectrum anti-inflammatory properties could potentially exacerbate the already rapid course of infection and virulence of anthrax through a suppression of the immune response when administered in vivo. Evidence to the contrary for the chemically related non-antimicrobial tetracyclines comes from unpublished studies by C. Rick Lyons and Julie Lovchik at the University of New Mexico, who showed that administration of the chemically modified tetracycline COL-3 to mice simultaneously receiving intratracheally instilled anthrax spores was as effective in preventing death as the classic antibiotic ciprofloxacin and provided more extended post-exposure prophylactic protection. This experiment could be repeated with the curcuminoids in order to rule out the possibility of any undesirable effects developing as a result of treatment.
The implications of this experiment, and of the use of compounds such as curcumin in general, are important in the context of a bioterror attack: if developed further, curcumin and related compounds could potentially be used as post-exposure prophylactic agents, taken immediately after a bioterror attack is initiated, to prevent infection. Furthermore, if the additional metalloproteinase activities of B. anthracis as described by Popov et al. are also inhibited by curcuminoids, the compounds’ broad specificity could be lifesavingCitation15,Citation16.
Several reports have pointed out that the specific cause of death from anthrax is still not fully understood. Although the role of LF in pathogenesis has been defined, and the central role of the MAPK cascade has been established in many cellular functions, inhibition of the MEKs alone does not seem sufficient to account for the systemic organ failure and tissue necrosis that is often seen in animals that have been infected with B. anthracis, or the rapid morbidity seen in Fischer Rats, which typically die within an hour from the time of exposureCitation5,Citation27. Indeed, the more recent discovery that LF also cleaves Nlrp1 and Nlrp1b (in rats and mice, respectively) leading to inflammasome-mediated pyroptosis provides additional information about of the chain of events that contribute to anthrax fatalities in these rodent modelsCitation11,Citation12. Regardless, previous studies have demonstrated that compounds that inhibit LF in vitro have prevented the death of Fischer Rats after intravenous injection of normally lethal amounts of LeTxCitation23, suggesting that inhibition of enzyme activity in vitro may be truly correlated with prevention of morbidity and mortality in vivo. Realistically, treating anthrax effectively may ultimately require some form of combination therapy: antibiotics to combat the bacteria that are the ultimate source of pathogenesis, and small molecule inhibitors specifically directed against the actions of the toxic components produced by the bacteria. The promise of these compounds as therapeutic agents for the treatment of anthrax infection should therefore not be underestimated.
Conclusions
Preliminary data provide strong evidence that curcumin (Compound 1) and Compound 3 act to inhibit LF by both decreasing its catalytic capacity and increasing its substrate affinity. The apparent increase in substrate affinity is mutually exclusive with a mechanism of competitive inhibition, and thus rules out simple occupancy of the substrate-binding site by the inhibitor as an explanation for decreased enzyme activity. This also requires that curcumin and Compound 3 bind to LF at an allosteric site, and therefore may function as uncompetitive or non-competitive inhibitors, though failure to fit the standard model of either of these mechanisms provides evidence for a mixture of inhibitory mechanisms.
Declaration of interest
Dr Francis Johnson and Dr Lorne M. Golub are listed as inventors on patent applications describing some of the compounds in this study. These patent applications have been fully assigned to their institutions, the Research Foundation of Stony Brook University and to Chem-Master International, Inc. This study was conducted with funds from the Stony Brook Foundation.
Supplementary material available online Supplementary Figures 1 and 2
Supplementary Material
Download PDF (401 KB)References
- Mock M, Mignot T. Anthrax toxins and the host: a story of intimacy. Cell Microbiol 2003;5:15–23
- Brittingham KC, Ruthel G, Panchal RG, et al. Dendritic cells endocytose Bacillus anthracis spores: implications for anthrax pathogenesis. J Immunol 2005;174:5545–52
- Rosenberger CM, Finlay BB. Phagocyte sabotage: disruption of macrophage signalling by bacterial pathogens. Nat Rev Mol Cell Biol 2003;4:385–96
- Dixon TC, Meselson M, Guillemin J, et al. Anthrax. N Engl J Med 1999;341:815–26
- Tonello F, Montecucco C. The anthrax lethal factor and its MAPK kinase-specific metalloprotease activity. Mol Aspects Med 2009;30:431–8
- Mastrolorenzo A, Supuran CT. Botulinus toxin, tetanus toxin, and anthrax lethal factor inhibitors. In: Supuran CT, Winum J-Y, eds. Drug design of zinc-enzyme inhibitors. Hoboken (NJ): John Wiley & Sons, Inc.; 2009:705–20
- Mock M, Fouet A. Anthrax. Ann Rev Microbiol 2001;55:647–71
- Brossier F, Mock M. Toxins of Bacillus anthracis. Toxicon 2001;39:1747–55
- Ezzell JW, Jr Abshire TG. Serum protease cleavage of Bacillus anthracis protective antigen. J Gen Microbiol 1992;138:543–9
- Abrami L, Liu S, Cosson P, et al. Anthrax toxin triggers endocytosis of its receptor via a lipid raft-mediated clathrin-dependent process. J Cell Biol 2003;160:321–8
- Hellmich KA, Levinsohn JL, Fattah R, et al. Anthrax lethal factor cleaves mouse nlrp1b in both toxin-sensitive and toxin-resistant macrophages. PLoS ONE 2012;7:e49741
- Levinsohn JL, Newman ZL, Hellmich KA, et al. Anthrax lethal factor cleavage of Nlrp1 is required for activation of the inflammasome. PLoS Pathog 2012;8:e1002638
- Pannifer AD, Wong TY, Schwarzenbacher R, et al. Crystal structure of the anthrax lethal factor. Nature 2001;414:229–33
- Spyroulias GA, Galanis AS, Pairas G, et al. Structural features of angiotensin-I converting enzyme catalytic sites: conformational studies in solution, homology models and comparison with other zinc metallopeptidases. Curr Top Med Chem 2004;4:403–29
- Popov SG, Popova TG, Hopkins S, et al. Effective antiprotease-antibiotic treatment of experimental anthrax. BMC Infect Dis 2005;5:25
- Supuran CT, Scozzafava A, Clare BW. Bacterial protease inhibitors. Med Res Rev 2002;22:329–72
- Gupta SC, Prasad S, Kim JH, et al. Multitargeting by curcumin as revealed by molecular interaction studies. Nat Prod Rep 2011;28:1937–55
- Zhang Y, Golub LM, Johnson F, et al. pKa, zinc- and serum albumin-binding of curcumin and two novel biologically-active chemically-modified curcumins. Curr Med Chem 2012;19:4367–75
- Ammon HP, Wahl MA. Pharmacology of curcuma longa. Planta Med 1991;57:1–7
- Kunwar A, Barik A, Mishra B, et al. Quantitative cellular uptake, localization and cytotoxicity of curcumin in normal and tumor cells. Biochim Biophys Acta 2008;1780:673–9
- Kocer SS, Walker SG, Zerler B, et al. Metalloproteinase inhibitors, nonantimicrobial chemically modified tetracyclines, and ilomastat block Bacillus anthracis lethal factor activity in viable cells. Infect Immun 2005;73:7548–57
- Zhang Y, Gu Y, Lee HM, et al. Design, synthesis and biological activity of new polyenolic inhibitors of matrix metalloproteinases: a focus on chemically-modified curcumins. Curr Med Chem 2012;19:4348–58
- Dell’Aica I, Dona M, Tonello F, et al. Potent inhibitors of anthrax lethal factor from green tea. EMBO Rep 2004;5:418–22
- Gaddis BD, Rubert Perez CM, Chmielewski J. Inhibitors of anthrax lethal factor based upon N-oleoyldopamine. Bioorg Med Chem Lett 2008;18:2467–70
- Numa MM, Lee LV, Hsu CC, et al. Identification of novel anthrax lethal factor inhibitors generated by combinatorial Pictet--Spengler reaction followed by screening in situ. ChemBioChem 2005;6:1002–6
- Kochi SK, Schiavo G, Mock M, et al. Zinc content of the Bacillus anthracis lethal factor. FEMS Microbiol Lett 1994;124:343–8
- Ezzell JW, Ivins BE, Leppla SH. Immunoelectrophoretic analysis, toxicity, and kinetics of in vitro production of the protective antigen and lethal factor components of Bacillus anthracis toxin. Infect Immun 1984;45:761–7