
Abstract
A new series of fluorine containing 1,3,5-triazinyl sulfonamide derivatives obtained from cyanuric fluoride, sulfanilamide/4-aminoethylbenzenesulfonamide followed and incorporating also amin0, amino alcohol and amino acid moieties have been investigated as inhibitors of three β-carbonic anhydrases (CAs, EC 4.2.1.1) from the bacterial pathogen Mycobacterium tuberculosis, mtCA1 (Rv1284), mtCA 2 (Rv3588c) and mtCA 3 (Rv3273). All three enzymes were efficiently inhibited by these sulfonamides with KI values in the nanomolar or submicromolar range, depending on the substitution of one or both fluorine atoms at the 1,3,5-triazine ring. As some of these enzymes are crucial for the life cycle of this bacterium, the class of β-CA inhibitors reported in this study may lead to antimycobacterial agents with a different mechanism of action compared to the clinically used such drugs for which the pathogen developed extensive drug resistance.
Introduction
The genome of the human pathogen Mycobacterium tuberculosis contains at least three carbonic anhydrases (CAs, EC 4.21.1) belonging to the β-class, which have been cloned, purified and characterized in the past. These enzymes, so called mtCA 1, 2 and 3 and encoded, respectively, by the genes Rv1284, Rv3588c and Rv3273Citation1–6. Indeed, the CA superfamily of enzymes comprises the α-, β-, γ-, δ- and ζ-CA classes, all of which are generally efficient catalysts for the reversible interconversion between carbon dioxide and bicarbonate (CO2 + H2O HCO3- + H+)Citation7.
Their catalytic activity and inhibition profiles with many sulfonamides (the main class of CA inhibitors; CAIs) have recently been investigatedCitation3–6. The three mycobacterial CAs were thus shown to be active CAs, with appreciable catalytic activity for the physiological, and to be efficiently inhibited by sulfonamides and related compoundsCitation7–11, with KI in the range of low nanomolar or subnanomolarCitation3–6.
Due to an increase of anti-mycobacterial drugs resistance, which continuously reduces the susceptibility of mycobacteria to the clinically used agents, the inhibition of the β-CAs from this pathogen may represent a new approach for controlling the infections, through a new mechanism of action, as some of these enzymes were shown to be essential for the life cycle of the pathogenCitation12–16. Indeed, many strains of Gram-negative/-positive bacteria (such as among others Staphylococcus aureus, M. tuberculosis, Helicobacter pylori, Brucella suis, Streptococcus pneumoniae, etc.) no longer respond to various classes of antibioticsCitation12–16. Many of these new proteins have been recently explored, revealing CAs very interesting drug targetsCitation11,Citation17. Indeed, it has been reported that sulfonamide derivates are potent inhibitors of bacterial CAs from H. pylori, B. suis, S. pneumoniae and several other similar pathogens, by inhibiting the growth of these bacteria through a mechanism of action not entirely investigated at this momentCitation11,Citation17–21.
The primary sulfonamides, RSO2NH2, are considered the classical CAIs. They have been widely clinically used for more than 50 years as diuretic or systemically acting antiglaucoma drugsCitation7,Citation8,Citation22,Citation23. However, all these drugs target all mammalian CAs (16 such isoforms are known to date in vertebrates), and as thus, they show undesired side effectsCitation7,Citation8,Citation22. Only recently, a large of number of such derivates started to be investigated as anti-infectives, which target bacteria/fungal CAs (generally belonging to β-CA class), but this new field significantly collided with resistance from the scientific community in accepting these enzymes as anti-infective targetsCitation7,Citation11.
However, CAs belonging to the β-CA class are present in pathogenic organism such as fungi and bacteria and they lack from mammals, in which only the α-CAs (as 16 different isoforms, as mentioned above) are ubiquitousCitation7. Therefore, these enzymes started to be seriously considered as possible drug targets for obtaining antibacterial agents devoid of the resistance problems mentioned above, which affect most classes of antibiotics in clinical useCitation12–15.
The 1,3,5-triazine scaffold containing chloride atoms has been used in previous studies from our groupCitation24 to design and synthesised potent sulfonamides CAIs. In this study, we intend to investigate the inhibition profile of 1,3,5-triazine sulphonamide derivatives in which both chlorine atoms have been replaced with fluoride atoms against three β-CAs (mtCA 1–3). We report hereby the inhibition studies against mtCA 1–3 from the bacterial pathogen M. tuberculosis, with a new series of fluorine containing 1,3,5-triazinyl sulfonamides 1–7.
Materials and methods
Chemistry
Compounds 1–7 investigated in this study were reported in a previous study from our groupCitation25.
Enzymology
mtCA1–mtCA 3 were recombinant enzymes obtained in-house as described earlierCitation1–6,Citation26.
CA catalytic and inhibition assay
An Applied Photophysics (Oxford, UK) stopped-flow instrument has been used for assaying the CA catalyzed CO2 hydration activityCitation28. Phenol red (at a concentration of 0.2 mM) has been used as indicator, working at the absorbance maximum of 557 nm, with 20 mM Hepes (pH 8.4) and 20 mM NaBF4 (for maintaining constant the ionic strength), following the initial rates of the CA-catalyzed CO2 hydration reaction for a period of 10–100 s. The CO2 concentrations ranged from 1.7 to 17 mM for the determination of the kinetic parameters and inhibition constants. For each inhibitor, at least six traces of the initial 5–10% of the reaction have been used for determining the initial velocity. The uncatalyzed rates were determined in the same manner and subtracted from the total observed rates. Stock solutions of inhibitor (10 mM) were prepared in distilled-deionized water and dilutions up to 0.01 nM were done thereafter with distilled-deionized water. Inhibitor and enzyme solutions were preincubated together for 15 min at room temperature prior to assay, in order to allow for the formation of the enzyme-inhibitor (E-I) complex. The inhibition constants were obtained by non-linear least-squares methods using PRISM 3, whereas the kinetic parameters for the uninhibited enzymes from Lineweaver–Burk plots, as reported earlierCitation3–6,Citation26,Citation29,Citation30, and represent the mean from at least three different determinations.
Results and discussion
Chemistry
Reported here are novel β-CAI sulfonamide derivates obtained considering chloride containing 1,3,5-triazine scaffold previously studied from our groupCitation24 in which the chlorine atoms were substituted by fluorine. The compounds were obtained by reaction of cyanuric fluoride with sulfanilamide and 4-(2-aminoethyl)-benzenesulfonamide affording, the key intermediates 1 and 2, which were further enacted with one or two equivalents of nitrogen nucleophiles, such as aminoalcohols, α-amino acids and their esters, as well as methylamine (). The compounds were reported and characterized recentlyCitation25.
Scheme 1. Preparation of sulfonamides 3–7 by reaction of the difluoro-triazinyl-benzene sulfonamides 1, 2 with nucleophiles (R1H, R2H) in presence of diisopropylethylamine (DIPEA), in DMF.
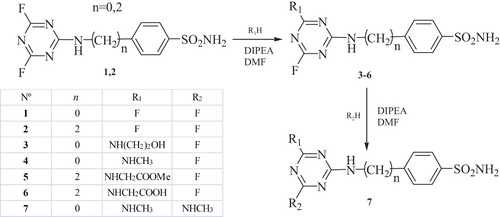
The inhibition studies against all mammalian CA isoforms of this new series of triazinyl-substituted benzenesulfonamides containing fluoride (1–7) have been recently investigated by our groupCitation25. In this study, we extend our earlier inhibition investigations of these triazinyl-containing sulfonamides against the three β-CAs from M. tuberculosis, mtCA1 (Rv1284), mtCA 2 (Rv3588c) and mtCA 3 (Rv3273).
CA inhibition
The inhibition of three β-CAs from the bacterial pathogen M. tuberculosis, i.e. mtCA 1 (Rv1284), mtCA 2 (Rv3588c) and mtCA 3 (Rv3273) has been investigated with derivatives 1–7 reported here, and their inhibition potency compared to those of some sulfonamide inhibitors of mycobacterial CAs such as acetazolamide (AAZ), benzolamide (BZA) (an orphan drugCitation7) and dichlorophenamide, reported earlier by our groupCitation3–6,Citation26.
Data for the inhibition of two of the dominant human CA isoforms (hCA I and II) with these compounds are also included in Citation25 for comparison reasons.
Table 1. Inhibition data of mycobacterial β-CA isoforms mtCA 1–3 with sulfonamides 1–7 reported here, and the standard sulfonamide inhibitors acetazolamide (AAZ), benzolamide (BZA) and dichlorophenamide (DCP) by a stopped flow CO2 hydrase assayCitation27.
The following structure–activity relationship (SAR) can be observed from data of for the inhibition of the β-CAs investigated here with the new group of triazinyl sulfonamides 1–7:
All of the triazinyl-substituted benzenesulfonamides 1–7 behaved as very efficient inhibitors against mtCA I, with inhibition constants in the range of 0.042–0.57 μM, similar or slightly better to those of the best sulfonamide β-CAIs such as AAZ and BZA, which have, respectively, KI values of 0.481 and 0.81 μM (). The best mtCA 1 inhibitor was the difluorotriazine derivative of 4-aminoethyl benzenesulfonamide 2 (KI of 42 nM), which was also a very strong inhibitor of hCA II with KI of 7.1 nM and a moderate inhibitor of the cytosolic isoform hCA I. The difluoro derivative of sulfanilamide (1) was 13.1-fold less effective as an mtCA 1 inhibitor compared to its longer congener (2). The ethanolamine derivative of the sulfanilamide (3) was 1.6 fold more effective mtCA 1 inhibitor compared to its difluoro derivative (1), whereas all the remaining compounds were quite highly effective mtCA 1 inhibitors, with similar inhibition constants in a narrow range of 0.46–0.61 μM. Therefore, SAR is almost impossible to delineate as all substitution patterns lead to highly effective inhibitors of this bacterial CA, mtCA 1.
Against the β-CA mtCA 2, shows that most of these triazine-substituted compounds were weak inhibitors (KI > 0.01 µM). Only derivates 2, 4 and 6 behaved as very strong mtCA 2 inhibitors with KIs in the range of 8.1–9.6 nM, very similar compared to that of AAZ with KI of 9 nM. However, they revealed to be better mtCA 2 inhibitors with the KI values in nanomolar range compared to BZA with KI value only in submicromolar range. Although the active compounds inhibited efficiently mtCA 2, no significant change of KI values has been observed. Therefore, a very flat SAR was noticed in this case.
The third bacterial CA investigated here, mtCA 3, was also efficiently inhibited by almost all derivates of this series of triazinyl-substituted benzenesulfonamides 1–7. Indeed, these derivates showed KIs in the range of 2.1 nM–0.21 µM. The most efficient mtCA 3 inhibitor was the difluorotriazine derivative of 4-aminoethylbenzenesulfonamide 2 (KI of 2.1 nM), which was also a strong inhibitor of mtCA 2, revealing to be 71.4-fold more potent than its shorter congener 1 (difluoro derivative of sulfanilamide) with KI of 0.15 µM. Substitution of one fluorine atom of the difluoro derivate of sulfanilamide 1 with a small moiety such as a methyl amine (4) led to a significant increase (16.8-fold) of the inhibitory potency, whereas substitution of one F atom with more bulky moiety such as ethanolamine (3) or of both F atoms (7) does not significantly change the inhibitory potency as mtCA 3 inhibitor (both with KIs of 0.21 µM).
The mono-glycine methyl ester derivative of 4-aminoethylbenzenesulfonamide (5) (KI of 0.13 μM) was roughly an order of magnitude less inhibitory compared to its difluoro 4-aminoethylbenzenesulfonamide derivative 2 (KI of 2.1 nM). It is also interesting to notice as a small change of the side chain of this mono-glycine methyl ester derivative from an ester to a carboxylic group allows the glycine derivative of 4-aminoethylbenzenesulfonamide (6) to be the least effective mtCA 3 inhibitor of this series of compounds (KI > 1 µM).
An overview of the current inhibition studies shows that among all the three bacterial CAs, mtCA 2 was the most prone to be inhibited by most of these triazinyl-substituted benzenesulfonamides 1–7 with KI in the nanomolar range, followed by mtCA 3, whereas mtCA 1 was the least inhibited bacterial isoform. This inhibition profile is quite similar compared to that of the clinically used drug AAZ. However, it appeared slightly different from the inhibition profile of BZA. For this compound, in fact, the best inhibition has been shown against mtCA 3, followed by mtCA 2 and the least inhibited isoform was mtCA1 like in the series here investigated.
Although the new series of compounds here investigated revealed to be potent bacterial β-CAIs showing a potential for developing new anti-infective drug, the dominant human carbonic anhydrase isoforms (hCA I and II) were also inhibited by compounds 1–7. Indeed, the slow cytosolic isoform hCA I was moderately inhibited by these compounds with inhibition constants in the range of 81.5–250 nM. On the other hand, against the physiologically dominant isoform hCA II, derivatives 1–7 behaved as very strong inhibitors with KIs in the range of 4.9–23 nM.
Therefore, one of the main problems with these mtCA inhibitors investigated so far is that no selectivity for only bacterial CAs was observed since the hCA I and II were also inhibited.
Conclusions
A series of triazinyl-substituted benzenesulfonamides incorporating fluorine and sulfanilamide or 4-aminoethylbenzenesulfonamide scaffolds has been investigated for the inhibition of the three β-CAs from the bacterial pathogen M. tuberculosis, mtCA1 (Rv1284), mtCA 2 (Rv3588c) and mtCA 3 (Rv3273). Some compounds were very efficient inhibitors of mtCA 2 with KI of nanomolar range, also showing excellent inhibition of mtCA 3 and rather efficient inhibitory potency against mtCA 1.
Some of these β-CAIs may have the potential for developing antimycobacterial agents with a different mechanism of action compared to the clinically used drugs for which many strains exhibit multi-drug resistance.
Declaration of interest
The authors report no conflicts of interest. The authors alone are responsible for the content and writing of this article.
This research was financed, in part, by a 7th FP EU grant (METOXIA).
References
- Suarez Covarrubias A, Larsson AM, Hogbom M, et al. Structure and function of carbonic anhydrases from Mycobacterium tuberculosis. J Biol Chem 2005;280:18782–9
- Covarrubias AS, Bergfors T, Jones TA, Hogbom M. Structural mechanics of the pH-dependent activity of beta-carbonic anhydrase from Mycobacterium tuberculosis. J Biol Chem 2006;281:4993–9
- Minakuchi T, Nishimori I, Vullo D, et al. Molecular cloning, characterization, and inhibition studies of the Rv1284 beta-carbonic anhydrase from Mycobacterium tuberculosis with sulfonamides and a sulfamate. J Med Chem 2009;52:2226–32
- Nishimori I, Minakuchi T, Vullo D, et al. Carbonic anhydrase inhibitors. Cloning, characterization, and inhibition studies of a new beta-carbonic anhydrase from Mycobacterium tuberculosis. J Med Chem 2009;52:3116–20
- Guzel O, Maresca A, Scozzafava A, et al. Discovery of low nanomolar and subnanomolar inhibitors of the mycobacterial beta-carbonic anhydrases Rv1284 and Rv3273. J Med Chem 2009;52:4063–7
- Carta F, Maresca A, Covarrubias AS, et al. Carbonic anhydrase inhibitors. Characterization and inhibition studies of the most active beta-carbonic anhydrase from Mycobacterium tuberculosis, Rv3588c. Bioorg Med Chem Lett 2009;19:6649–54
- Supuran CT. Carbonic anhydrases: novel therapeutic applications for inhibitors and activators. Nat Rev Drug Discov 2008;7:168–81
- Neri D, Supuran CT. Interfering with pH regulation in tumours as a therapeutic strategy. Nat Rev Drug Discov 2011;10:767–77
- Pastorekova S, Parkkila S, Pastorek J, Supuran CT. Carbonic anhydrases: current state of the art, therapeutic applications and future prospects. J Enzyme Inhib Med Chem 2004;19:199–229
- Supuran CT. Carbonic anhydrase inhibitors and activators for novel therapeutic applications. Future Med Chem 2011;3:1165–80
- Supuran CT. Bacterial carbonic anhydrases as drug targets: toward novel antibiotics? Front Pharmacol 2011;2:34
- Dye C. Doomsday postponed? Preventing and reversing epidemics of drug-resistant tuberculosis. Nat Rev Microbiol 2009;7:81–7
- Furtado GH, Nicolau DP. Overview perspective of bacterial resistance. Expert Opin Ther Pat 2010;20:1273–6
- Ginsberg AM. Emerging drugs for active tuberculosis. Semin Respir Crit Care Med 2008;29:552–9
- Showalter HD, Denny WA. A roadmap for drug discovery and its translation to small molecule agents in clinical development for tuberculosis treatment. Tuberculosis (Edinb) 2008;88:S3–17
- Tomioka H, Tatano Y, Yasumoto K, Shimizu T. Recent advances in antituberculous drug development and novel drug targets. Expert Rev Respir Med 2008;2:455–71
- Supuran CT, Scozzafava A, Casini A. Carbonic anhydrase inhibitors. Med Res Rev 2003;23:146–89
- Winum JY, Kohler S, Supuran CT. Brucella carbonic anhydrases: new targets for designing anti-infective agents. Curr Pharm Des 2010;16:3310–16
- Vullo D, Nishimori I, Minakuchi T, et al. Inhibition studies with anions and small molecules of two novel beta-carbonic anhydrases from the bacterial pathogen Salmonella enterica serovar Typhimurium. Bioorg Med Chem Lett 2011;21:3591–5
- Nishimori I, Onishi S, Takeuchi H, Supuran CT. The alpha and beta classes carbonic anhydrases from Helicobacter pylori as novel drug targets. Curr Pharm Des 2008;14:622–30
- Supuran CT. Carbonic anhydrase inhibition with natural products: novel chemotypes and inhibition mechanisms. Mol Divers 2011;15:305–16
- Supuran CT. Carbonic anhydrase inhibitors. Bioorg Med Chem Lett 2010;20:3467–74
- Pacchiano F, Carta F, Vullo D, et al. Inhibition of beta-carbonic anhydrases with ureido-substituted benzenesulfonamides. Bioorg Med Chem Lett 2011;21:102–5
- Carta F, Garaj V, Maresca A, et al. Sulfonamides incorporating 1,3,5-triazine moieties selectively and potently inhibit carbonic anhydrase transmembrane isoforms IX, XII and XIV over cytosolic isoforms I and II: solution and X-ray crystallographic studies. Bioorg Med Chem 2011;19:3105--19
- Ceruso M, Vullo D, Scozzafava A, Supuran CT. Inhibition of human carbonic anhydrase isoforms I-XIV with sulfonamides incorporating fluorine and 1,3,5-triazine moieties. Bioorg Med Chem 2013 [Epub ahead of print]. http://dx.doi.org/10.1016/j.bmc.2013.09.031
- Nishimori I, Minakuchi T, Maresca A, et al. The beta-carbonic anhydrases from Mycobacterium tuberculosis as drug targets. Curr Pharm Des 2010;16:3300–9
- Ilies MA, Vullo D, Pastorek J, et al. Carbonic anhydrase inhibitors. Inhibition of tumor-associated isozyme IX by halogenosulfanilamide and halogenophenylaminobenzolamide derivatives. J Med Chem 2003;46:2187–96
- Khalifah RG. The carbon dioxide hydration activity of carbonic anhydrase: I. stop-flow kinetic studies on the native human isoenzymes B and C. J Biol Chem 1971;246:2561–73
- Kolayli S, Karahalil F, Sahin H, et al. Characterization and inhibition studies of an alpha-carbonic anhydrase from the endangered sturgeon species Acipenser gueldenstaedti. J Enzyme Inhib Med Chem 2011;26:895–900
- Schlicker C, Hall RA, Vullo D, et al. Structure and inhibition of the CO2-sensing carbonic anhydrase Can2 from the pathogenic fungus Cryptococcus neoformans. J Mol Biol 2009;385:1207–20