
Abstract
The molecular docking, MD simulation and binding free energy calculation were performed to explore the probable binding modes between PLA and tubulin. Through docking study, three possible binding sites for PLA were speculated as follows: the taxane site, the alternative site and a new site in α-tubulin. Then, 12.0 ns MD simulations show that these binding modes predicted by docking have been changed more or less, whereas the MD simulations offer more reliable binding details. The MM-PBSA binding free-energy calculations reasonably identify that the taxane site is the most favorable binding site of PLA and the alternative site is the secondary one, which can be used to explain some experimental facts. These studies theoretically resolve the priority of binding sites for PLA and offer the reliable binding modes between PLA and tubulin, and thus help to understanding the action mechanism for this kind of inhibitor.
Introduction
Microtubules, major components of the cytoskeleton mainly composed of α- and β-tubulin heterodimers, have a large number of essential cellular functions, such as maintenance of cellular shape, intracellular transport and chromosome segregation. Interference of the tubulin-microtubule dynamics can cause mitotic arrest and eventually trigger the signals for apoptosis. Hence, tubulin is a very attractive therapeutic target for the treatment of cancerCitation1,Citation2.
Taxanes including paclitaxel and docetaxel, microtubule-stabilizing agents binding to the polymerized microtubules and preventing depolymerization of microtubules, are successfully prescribed in anticancer therapiesCitation3. However, these drugs exhibit dose-limiting toxicity and drug resistance to cancer cells that overexpress the P-gpmultiple drug efflux pumpCitation4. Thus, compounds that overcome these limitations may have better curative effect on cancer.
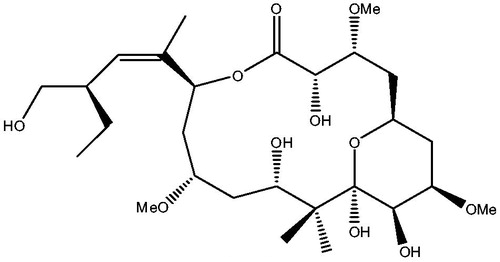
Peloruside A (PLA) (see ), characterized by a polyoxygenated pyranose ring containing 16-membered macrolide, was first isolated in 1999 from New Zealand marine sponge Mycale hentscheli, and it had potent growth inhibition against P388 murine leukemic cellsCitation5. PLA was further found to be a microtubule-stabilizing agent and to have potent antimitotic activity, making cells block in G2/M phase and apoptosisCitation6,Citation7. Interestingly, PLA was likely to overcome the P-gpmultiple-mediated multiple drug resistanceCitation8, and it did not show any obvious toxicity in animal testsCitation9. These results demonstrate the great potential to develop PLA as a promising drug for cancer therapy. Previous experimental studies discovered that PLA did not compete with taxane site drugs for binding to microtubulesCitation8, instead, it could synergize with themCitation10. The initial studies through molecular docking and TR-NOESY nuclear magnetic resonance (NMR) proposed that the binding site of PLA was on α-tubulinCitation11,Citation12. However, Huzil et al.Citation13 proposed a specific PLA-binding site that was adjacent to the taxane-binding site in the β-tubulin by using hydrogen-deuterium exchange mass spectrometry (HDXMS). As a matter of fact, the PLA-binding site proposed by Huzil et al. is also called the alternative site. Recently, Nguyen et al. proposed that PLA was equally likely to bind in the taxane site and the alternative site in β-tubulinCitation14. In the researches for PLA, some pivotal progress has been made through experimental methods; however, so far the exact binding site of PLA has not been accurately identified yet and in particular, the knowledge on how it binds to tubulin at a molecular level is still insufficient.
Figure 1. Structural sketch map of PLA.
Molecular docking is an approach to predict the possible orientations of ligand in active site of receptor to study the interaction mechanisms, and several docking modes of ligands in tubulin have been established through molecular dockingCitation15,Citation16. Molecular dynamics (MD) simulation is a useful methodology providing vivid pictures to depict the fluctuations and conformational changes of molecules, and the dynamic behaviors of some ligand–tubulin complexes have been successfully obtained by performing MD simulationCitation17,Citation18. Therefore, a combined docking and MD simulation study can offer the deep insight into understanding the structural features of ligand–tubulin interactions.
In this work, molecular dockings, MD simulations and binding free-energy calculations were applied to study the probable binding modes of PLA to tubulin. The purpose is to resolve the priority of binding sites and reveal the interaction details between PLA and tubulin. First, the docking calculations offered the possible binding sites. Then, based on the docking results, MD simulations were carried out in explicit solvent model to obtain the insight into the dynamic information of each binding site. Finally, the binding free energy calculation by MM-PBSA method was further used to identify the stability of each binding mode. We expect the obtained results help to understanding the possible stabilization mechanisms of PLA.
Computational methods
Ligand and receptor preparation
The 3D structure of PLA was downloaded from the PubChem Compound Database to serve as docking ligand. The crystallographic structure of tubulin–expothilone complex was taken from the Protein Data Bank (pdb Id code: 1TVK). The water molecules and other unrelated subunits in the complex were removed. Then, the hydrogen atoms were added. They were minimized with a 0.01 kcalċmol−1Å−1 root mean square gradient by adopting the all-atom CHARMm force field and the Adopted Basis Newton--Raphson (NR) Algorithm in Discovery Studio 2.0Citation19. The hydrogen-minimized tubulin was taken as docking receptor.
Molecular docking
To locate the possible binding sites and the appropriate binding orientations of PLA, molecular docking studies were performed using the Lamarckian genetic algorithm local search method with the AutoDock4.2 programCitation20. The α,β subunits interfaces where the microtubule-destabilizing agents’ (vinca alkaloid and colchicine) binding sites are locatedCitation21 were not considered in docking calculations. The tubulin and the macrocycle of PLA were held as rigid and only the torsional bonds of side chains of PLA were taken as free. All used parameters were default except for explained. Then the following two steps, namely global and local dockings, were performed.
First, two global docking runs were carried out to find out possible binding sites. The prepared PLA and tubulin were both assigned for Gasteiger charges. The docking areas defined by grid box were centered on the α-tubulin and β-tubulin, with (x,y,z) = (38.693, −1.735, 3.066) and (−1.349, −1.383, 4.079), respectively. In each docking, the grid map was calculated using 126 × 126 × 126 grid points with 0.375 Å gird spacing, and 100 independent docking processes were calculated with a maximum number of 2.5 × 107 energy evaluations. Sequentially, 100 conformations of PLA were produced, and they were clustered with root-mean square deviation (RMSD) of 2.0 Å and arranged according to the binding energy. The favorable binding sites were identified by judging from the largest number of conformations in the same position or the lowest binding energy. Herein, we selected three possible binding sites 1, 2 and 3.
Because the docking conformations of ligand are very important to influence the results of MD simulations, we further carried out following local docking calculations with a finer grid for each one of the three binding sites to find out the favorable conformation of PLA. The active conformations of PLA were extracted from above docking results with the lowest binding energy in each binding site, and they were also assigned for Gasteiger charges. In each docking, the grid box of 70 × 70 × 70 points was centered on PLA, that is (x,y,z) = (10.32, −19.74, −7.03), (−0.74, −16.70, 12.16) and (45.80, 13.98, −2.58) in binding sites 1, 2 and 3, respectively. One hundred separate dockings were also performed with maximum of energy evaluations to 2.5 × 107. The conformation corresponding to the most cluster members and the lowest binding energy was selected as the most favorable conformation in each binding site. Herein, we selected three favorable binding modes 1, 2 and 3 in corresponding binding sites 1, 2 and 3.
Molecular dynamics simulations
Based on docking results, 12.0 ns MD simulations were carried out with the AMBER10 softwareCitation22 to estimate the dynamic stability of the three docking modes 1, 2 and 3. In each MD simulation, the methods adopted were the same. The electrostatic potential (ESP) of PLA was calculated at the level of Becke’s three-parameter hybrid functional (B3LYP)Citation23 and 6-31 G(d) basis set in the Gaussian 09 programCitation24, and the partial atomic charges for PLA were generated by using the restricted electrostatic potential (RESP)Citation25 method implemented in the Antechamber module. The AMBER ff03 force field and the general AMBER force field (GAFF)Citation26 were respectively used to establish the potential parameters of β-tubulin and PLA. The system was neutralized by adding sodium ions and solvated in a truncated octahedron box with TIP3P water modelCitation27, and the distance between the solute surface and the edge of the box was chosen to be larger than 10 Å. Then, two-stage energy minimizations were carried out to avoid possible steric stress. In first stage, the complex was fixed with restraint constant of 2.0 kcal mol−1 Å−2 and the waters and sodium ions were minimized with steepest descent method for 2000 steps followed by conjugated gradient method for 3000 steps. In second stage, the whole relaxed complex was optimized by 5000 steps steepest descent minimization and 5000 steps conjugated gradient minimization. Next, the complex was gradually heated from 0 to 300 K in 200 ps with a weak constraint of 1.0 kcal mol−1 Å−2 at constant volume and equilibrated for 500 ps at 300 K and 1 atm. Finally, 12 ns production MD simulation was performed in a NPT (constant composition, P = 1 atm and T = 300 K) ensemble. During these steps, the Particle Mesh Ewald (PME)Citation28 method was used to deal with long-range electrostatic interactions with non-bonded cutoff of 10 Å, and SHAKE algorithmCitation29 was turned on to constrain all covalent bonds involving hydrogen atoms with 2 fs time step. The output files and the trajectory files of production MD simulation were saved every 2 ps for following analysis.
Free binding energy calculations
To estimate the binding stability of PLA to tubulin, the binding free energy calculations for the 500 snapshots isolated from the final 1.0 ns simulation of each binding mode were performed by the molecular mechanics Poisson–Boltzmann solvent accessible surface area (MM-PBSA) methodCitation30 enclosed in AMBER10 softwareCitation22.
Results and discussion
Molecular docking
The global dockings were used to find out possible binding sites, and the favorable binding sites were identified by judging from the largest number of conformations in the same position or the lowest binding energy. The results of the global dockings are displayed in and . When the global docking was centered on β-tubulin, the most conformations (25%) were in the site 2, while the conformation with the lowest binding energy (−7.24 kcal/mol) in the first cluster (involving 4% conformations) was in the site 1. Thus, if PLA is activated by binding to β-tubulin, the sites 1 and 2 are possible to be the binding sites. Similarly, when the global docking was centered on α-tubulin, the most conformations (23%) and the conformation with the lowest binding energy (−6.13 kcal/mol) in the first cluster were in the site 3. Thus, if PLA is activated by binding to α-tubulin, the site 3 is also possible to be the binding site. According to these analyses, we selected the three sites 1, 2 and 3 as the probable binding sites of PLA in tubulin.
Figure 2. The three possible binding sites 1, 2 and 3 predicted by global dockings, in which tubulin protein (1TVK) is shown as ribbon and PLA is shown as stick colored by element.
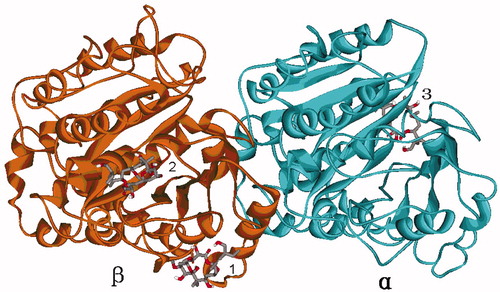
Table 1. The number of conformations and the lowest binding energy in each of the binding sites 1, 2 and 3.
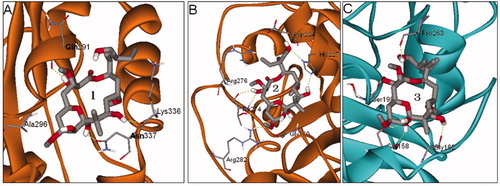
The predicted binding sites 1, 2 and 3 of PLA by global dockings are shown in . The binding sites 1 and 2 are located in β-tubulin, while binding site 3 is in α-tubulin. The binding site 1 consists of β-tubulin residues Gln291, Ala296, Lys336 and Asn337, and it is basically in agreement with the alternative site previously reportedCitation13,Citation14. The binding site 2 is made up of β-tubulin residues Asp224, His227, Thr274, Arg276, Arg282 and Gly360, and it just located in the well-known taxane site. The binding site 3 is distinctly different from the site locating in B9–B10 loop extension of α-tubulinCitation11,Citation12, and it is a new site comprised of α-tubulin residues Ser158, Gly162, Ser198 and Pro263. In short, the taxane site, the alternative site and the new site 3 are possible to be the binding sites of PLA.
Figure 3. The predicted binding sites 1 (A), 2 (B) and 3 (C) of PLA in tubulin by global dockings.
The local dockings with a finer grid were further used to find out the favorable conformation of PLA in each binding site, and the conformation corresponding to the most cluster members and the lowest binding energy was selected as the most probable binding conformation in each binding site. In the binding sites 1, 2 and 3, the conformation from the first cluster including the most docking conformations (71%, 31% and 51%, respectively) with the lowest binding energy (−7.66, −6.65 and −6.24 kcal/mol, respectively) was selected as the favorable conformation of each binding site. Moreover, the different binding sites represent different binding modes; therefore, when the PLA locates respectively in binding sites 1, 2 and 3, the corresponding binding modes are named as modes 1, 2 and 3.
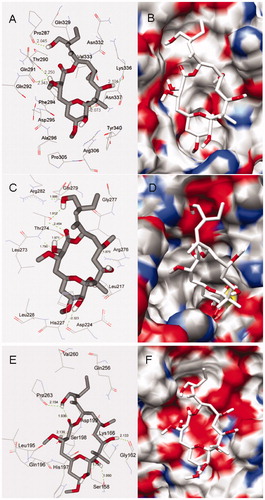
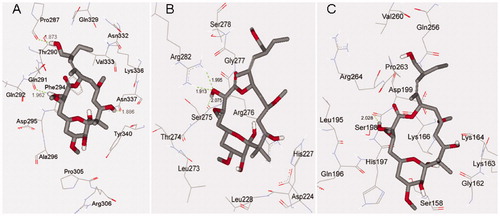
The detailed structures of binding sites in the docking modes 1, 2 and 3 predicted by local dockings are shown in , from which we can see that PLA is suitably situated at each binding site. Because PLA is slightly hydrophilic with LogP = 1.23 calculated by HyperChem softwareCitation31, the hydrophobic characteristic may be important to affect the orientation of PLA in binding site. In docking mode 1, the binding site is composed of β-tubulin residues Pro287, Thr290, Gln291, Gln292, Phe294, Asp295, Ala296, Pro305, Arg306, Gln329, Asn332, Val333, Lys336, Asn337 and Tyr340. It is worth the whistle, the previous experimental study has reported that the candidate region for PLA-binding site, being called the alternative siteCitation14, is composed of peptides β294–301 (H9–H9′ loop), β302–314 (H9′–S8) and β332–340 (H10 loop) detected by HDX-MSCitation13. It is easy to find that the most key residues (Phe294, Asp295, Ala296, Pro305, Arg306, Asn332, Val333, Lys336, Asn337 and Tyr340) of the binding site 1 predicted by the docking appear in the alternative site. Here, the binding site 1 is just the alternative site, therefore, the docking result is basically in agreement with the experimental one. In the binding site, PLA makes hydrophobic interactions with residues Pro305, Phe294, Val333, Lys336 and Tyr340. Moreover, a network of hydrogen bonds is very important for stabilizing the mode. There are two hydrogen bonds between one hydroxyl H atom of PLA and two O atoms of Thr290 and Gln291, with bond lengths of 2.250 and 2.343 Å, respectively. At the same time, other three hydroxyl H atoms of PLA, respectively, form three hydrogen bonds of 2.045, 2.104 and 2.073 Å with carbonyl O atoms of Pro287, Lys336 and Asn337. In docking mode 2, PLA immerges into a pocket constructed by β-tubulin residues Leu217, Asp224, His227, Leu228, Leu273, Thr274, Arg276, Gly277, Gln279 and Arg282. On the one hand, PLA can interact with residues Leu217, Leu228, Leu273 and Arg276 by hydrophilic interaction. On the other hand, it can form seven hydrogen bonds in the binding site as follows: four hydrogen bonds of 1.912, 2.454, 1.971 and 1.790 Å are formed with Thr274, and the other three hydrogen bonds are formed with Asp224, Arg276 and Arg282, with bond distances of 2.323, 1.875, and 1.886 Å, respectively. In docking mode 3, the binding site has less hydrophobic property due to be created by α-tubulin residues Ser158, Gly162, Lys166, Gln196, His197, Ser198, Asp199, Gln256, Val260 and Pro263, and such a property may be the reason for the weakest binding affinity of PLA in the mode, with the highest binding energy (−6.24 kcal/mol). Besides, PLA forms two hydrogen bonds with Ser158 of 1.790 and 1.990 Å, and it also interacts with Gly162, Ser198 and Pro263 by weaker hydrogen bonds with distances of 2.133, 2.139, and 2.154 Å, respectively.
Figure 4. Docking conformations of PLA and corresponding surfaces of tubulin at binding sites 1 (A and B), 2 (C and D) and 3 (E and F), in which the red and blue regions represent oxygen and nitrogen atoms, respectively, whereas gray and white regions represent carbon and hydrogen atoms (as a whole).
Molecular dynamics simulations
To obtain the fluctuation information of these modes, we selected above three docking modes 1, 2 and 3 as initial conformations for following 12.0 ns MD simulations. The physicochemical parameters such as total energy and potential energy in each mode are steady with smooth curves in , which indicate that these systems have reached stable. Compared with corresponding initial conformations, the values of root mean squared deviation (RMSD) of backbone atoms of β-tubulin and PLA in these modes 1, 2 and 3 were also calculated, and the results are outlined in , respectively. Moreover, generally speaking, the studied system theoretically trends to be steady with the increase of simulation time. Therefore, it is easy to understand that the final binding mode after 12.0 ns MD simulations was selected as the desired binding mode between PLA and tubulin in each binding site for following structure-characteristics analyses.
Figure 5. Potential energy (bottom) and total energy (top) evolutions of binding modes 1 (black), 2 (blue) and 3 (red) during the MD simulations (A). RMSD of backbone atoms of β-tubulin (black) and PLA (red) in binding modes 1 (B), 2 (C) and 3 (D), respectively.
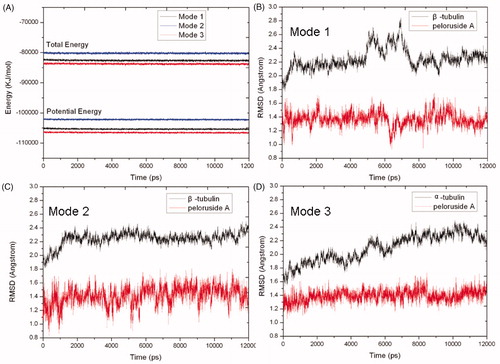
The shows the RMSD values of β-tubulin have some intense fluctuations between 5.0 ns and 7.5 ns up to 2.85 Å, but after 7.5 ns, β-tubulin becomes equilibrated with an average RMSD value of 2.23 Å. Whereas the RMSD values of PLA keep smooth fluctuations around 1.36 Å in the whole simulation. Therefore, the whole system trends to stable after 7.5 ns. Because the RMSD values of β-tubulin and PLA have intense fluctuations at 6.3 ns, we selected a representative snapshot of complex at 6.3 ns to explore the reason why having the big vibrations and study the change of corresponding conformations between 5.0 ns and 7.5 ns. It shows that the conformation of β-tubulin has made a larger change in β210 ∼ β220. This change may be instantaneously incompatible with surrounding circumstance, so it was subsequently pulled back to the original position after 7.5 ns. At 7.5 ns and afterward, the RMSD for PLA is still higher (∼1.37 Å) and the RMSD for β-tubulin is also quite high (∼2.23 Å), it means the ligand and the receptor move simultaneously. Finally, the obtained structure of PLA and the binding site just only have a little change compared with initial docking results (see ). In fact, such a slight change is acceptable due to rigid disposal in docking. In final binding mode 1 (see ), the ligand PLA still interacts with β-tubulin residues Pro305, Phe294, Val333, Lys336 and Tyr340 by hydrophilic interaction. Additionally, the two original hydrogen bonds with Pro287 and Gln291 reappear, but their distances are shorter than original ones, and a new different hydrogen bond of 1.886 Å with Asn337 is also formed. These factors will improve the binding affinity of PLA to tubulin. In a word, the results suggest that the binding mode 1 predicted by docking is basically reasonable and the MD simulation can offer vivid details.
Figure 6. The final (yellow) and initial (gray) structures of binding modes 1(A), 2 (B) and 3 (C) in 12.0 ns MD simulations, respectively.
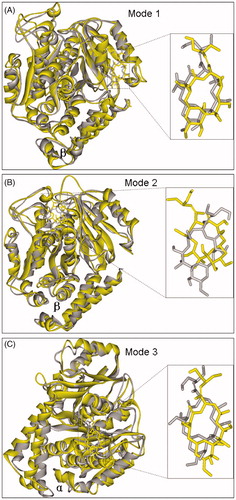
Figure 7. The final binding modes 1(A), 2 (B) and 3 (C) after 12.0 ns MD simulations, respectively.
From , we can see that the fluctuations of β-tubulin and PLA both keep smooth with corresponding mean RMSD values of 2.27 and 1.43 Å after 1.0 ns. It suggests that the system becomes equilibrated after the 1.0 ns. However, via comparing with the initial docking conformation, we find that the final conformation of PLA has already made a change through about 30 degree counterclockwise rotation (see ). The reason for it can be attributed to that the positions of nearby residues β274∼β282 have moved and departed a lot from initial position, resulting in the corresponding change of PLA. The change of PLA induces the harmonious hydrophobic interactions with β-tubulin residues Leu273, Arg276 and Arg282. In addition, we also find that all the hydrogen bonds existing in initial docking mode 2 have disappeared and are substituted by three new hydrogen bonds, including two hydrogen bonds formed with residue Arg282 (1.995 and 1.913 Å) and one hydrogen bond formed with residue Ser275 of 2.075 Å (see ). These results mean that the initial hydrogen bonds are weaker in the mode 2 and thus the mode 2 predicted by docking should not be stable enough.
In , the α-tubulin gradually stabilizes and finally reaches equilibrium after 7.0 ns with a mean RMSD value of 2.28 Å, while PLA keep stable in the whole simulation with a mean RMSD value of 1.39 Å. It indicates that the system becomes equilibrated after 7.0 ns. After 12.0 ns MD simulation, the final structures of PLA and binding site have a slight change compared with initial docking mode (see ). In final binding mode 3 (see ), the α-tubulin residues Lys163, Lys164, Lys166 and Pro263 close to PLA, and it improves the hydrophobic interactions. In addition, although all other hydrogen bonds have vanished, the hydrogen bond formed with Ser198 is much stronger than original one with shorter distance of 2.028 Å. These suggest that the mode 3 predicted by docking is also acceptable.
Binding free-energy analysis
In order to identify the stabilities of the three binding modes 1, 2 and 3, the binding free-energy calculations for the 500 snapshots of the last 1.0 ns MD simulations were also carried out by MM-PBSA method and the corresponding results are listed in . The binding free energies of the three binding modes 1, 2 and 3 are −20.17, −25.21, and −17.09 kcal/mol, respectively. However, the binding energies of the three docking modes 1, 2 and 3 predicted by local dockings are −7.66, −6.65 and −6.24 kcal/mol, respectively. So it is easy to find that the two trends of energies for the docking and MD simulation are somewhat different. In fact, in binding site 2, the equilibrated conformation of PLA has already moved a lot from the initial docking conformation, and the final matching should be the best for the hydrophobic interactions and thus the binding affinity of PLA to tubulin is naturally enhanced. So it is not surprising that the final binding mode 2 becomes the most stable mode instead of the secondary stable mode expected by the docking. Because the MM-PBSA binding free energy is in consideration of the flexibility of β-tubulin by MD simulations but the docking energy is not, the former is certainly more credible than the latter. According to the values of MM-PBSA binding free energies, we can speculate that the binding mode 2 with the lowest binding free energy is the most stable, and the binding mode 1 is secondarily stable, whereas the binding mode 3 is the most instable. As a matter of fact, the binding site of mode 2 is the well-known taxane site, and the binding site of mode 1 is the alternative site, so we can identify that the taxane site is the most favorable binding site of PLA and the alternative site may be the secondary favorable binding site, while the new site 3 may be the weaker candidate binding site of PLA with the highest binding free energy. That is to say, when there is not another competitive taxane site drug with stronger affinity force, PLA will preferentially bind to the taxane site, otherwise it will favorably bind to the alternative site. The result theoretically resolved the priority of binding sites for PLA, and it can be used to reasonably explain the interesting experimental fact that PLA does not compete with taxane-site drugs for binding to microtubulesCitation8, but it can synergize with themCitation10, because PLA will occupy the alternative site while the preferential drugs occupy the taxane site. The components of binding free energy show that, in each binding mode, the van der Waals energy (ΔGvdw) and the electrostatic energy (ΔGele) make major contributions to the binding free energy, and the nonpolar solvation free energy (ΔGnonpolar) is also favorable for the binding, whereas the polar solvation free energy (ΔGpolar) is unfavorable. Usually, the van der Waals energy is closely correlative with the hydrophobic interaction and the electrostatic (ΔGele) energy is correlative with the hydrogen bond energy, so we can further validate that the hydrophobic interactions and hydrogen bonds play very important roles for stabilizing PLA in tubulin.
Table 2. Binding free energies (kcal/mol) obtained by MM-PBSA method for the 500 snapshots of the last 1.0 ns MD simulations.
Conclusions
In this article, a combined method of the molecular docking, MD simulation and binding free-energy calculation was carried out to explore the probable binding modes between PLA and tubulin. The docking studies reveal that there are three possible binding sites: the taxane site and the alternative site, and a new site in α-tubulin. Based on the docking results, 12.0 ns Molecular dynamics (MD) simulations were performed and show that the three binding modes predicted by dockings have been changed more or less, whereas the MD simulations offer more reliable binding details. The binding free-energy calculations for the 500 snapshots of the last 1.0 ns MD simulations by MM-PBSA method identify that the taxane site is the most favorable binding site of PLA and the alternative site in β-tubulin is the secondary one, and it can be used to reasonably explain some experimental facts. Moreover, the study reveals some valuable binding details as follows: (1) The hydrophobic interactions and hydrogen bonds play very important roles for stabilizing PLA in tubulin. (2) In the taxane site, PLA interacts not only with β-tubulin residues Leu273, Arg276 and Arg282 by hydrophilic interaction but also with Arg282 and Ser275 by hydrogen bonds. (3) In the alternative site, PLA interacts not only with β-tubulin residues Pro305, Phe294, Val333, Lys336 and Tyr340 by hydrophilic interaction but also with Pro287, Gln291 and Asn337 by hydrogen bonds. The theoretical results can reasonably resolve the priority of binding sites for PLA and offer the reliable binding modes between PLA and tubulin, and thus help to understanding the action mechanism for this kind of inhibitor.
Declaration of interest
Except for the above support and funding received by the authors to carry out the study, no conflict of interest is required.
We are pleased to thank the financial supports of the Natural Science Foundation of Guangdong Province (S2011040000131), Medical Science and Technology Research Foundation of Guangdong Province (WSTJJ20101231), Science and Technology Project of Guangzhou (2012J2200034), Science research program of Guangzhou municipal colleges (2012C219) and Foundation of Guangzhou medical university (2010C15).
References
- Singh P, Rathinasamy K, Mohan R, Panda D. Microtubule assembly dynamics: an attractive target for anticancer drugs. Iubmb Life 2008;60:368–75
- Jordan MA, Wilson L. Microtubules and actin filaments: dynamic targets for cancer chemotherapy. Curr Opin Cell Biol 1998;10:123–30
- Rowinsky EK. The development and clinical utility of the taxane class of antimicrotubule chemotherapy agents. Annu Rev Med 1997;48:353–74
- Orr GA, Verdier-Pinard P, McDaid H, Horwitz SB. Mechanisms of Taxol resistance related to microtubules. Oncogene 2003;22:7280–95
- West LM, Northcote PT, Battershill CN, Peloruside A. A potent cytotoxic macrolide isolated from the New Zealand marine sponge Mycale sp. J Org Chem 2000;65:445–9
- Miller JH, Rouwé B, Gaitanos TN, et al. Peloruside A enhances apoptosis in H-ras-transformed cells and is cytotoxic to proliferating T cells. Apoptosis 2004;9:785–96
- Hood KA, West LM, Rouwe B, et al. Peloruside A, a novel antimitotic agent with paclitaxel-like microtubule-stabilizing activity. Cancer Res 2002;62:3356–60
- Gaitanos TN, Buey RM, Díaz JF, et al. Peloruside A does not bind to the taxoid site on β-tubulin and retains its activity in multidrug resistant cell lines. Cancer Res 2004;64:5063–7
- Crume KP, O’Sullivan D, Miller JH, et al. Delaying the onset of experimental autoimmune encephalomyelitis with the microtubule-stabilizing compounds, paclitaxel and peloruside A. J Leukocyte Biol 2009;86:949–58
- Hamel E, Day BW, Miller JH, et al. Synergistic effects of peloruside A and laulimalide with taxoid site drugs, but not with each other, on tubulin assembly. Mol Pharmacol 2006;70:1555–64
- Pineda O, Farràs J, Maccari L, et al. Computational comparison of microtubule-stabilizing agents laulimalide and peloruside with taxol and colchicine. Bioorg Med Chem Lett 2004;14:4825–9
- Jiménez-Barbero J, Canales A, Northcote PT, et al. NMR determination of the bioactive conformation of peloruside A bound to microtubules. J Am Chem Soc 2006;128:8757–65
- Huzil JT, Chik JK, Slysz GW, et al. A unique mode of microtubule stabilization induced by peloruside A. J Mol Biol 2008;378:1016–30
- Khrapunovich-Baine M, Menon V, Yang C-PH, et al. Hallmarks of molecular action of microtubule stabilizing agents. J Biol Chem 2011;286:11765–78
- Farce A, Loge C, Gallet S, et al. Docking study of ligands into the colchicine binding site of tubulin. J Enzyme Inhib Med Chem 2004;19:541–7
- Liao SY, Chen JC, Miao TF, et al. Binding conformations and QSAR of CA-4 analogs as tubulin inhibitors. J Enzyme Inhib Med Chem 2010;25:421–9
- Xu S, Chi S, Jin Y, et al. Molecular dynamics simulation and density functional theory studies on the active pocket for the binding of paclitaxel to tubulin. J Mol Model 2012;18:377–91
- Varzhabetyan LR, Glazachev DV, Nazaryan KB. Molecular dynamics simulation study of tubulin dimer interaction with cytostatics. Mol Biol 2012;46:316–21
- Discovery Studio 2.0. Accelrys Software Inc., San Diego, California, USA; 2007
- Morris GM, Huey R, Lindstrom W, et al. AutoDock4 and AutoDockTools4: automated docking with selective receptor flexibility. J Comput Chem 2009;30:2785–91
- Lu Y, Chen J, Xiao M, et al. An Overview of tubulin inhibitors that interact with the colchicine binding site. Pharm Res 2012;29:2943–71
- Case DA, Cheatham TA, Simmerling CL, et al. AMBER 10, University of California, San Francisco, San Francisco, CA; 2008
- Becke AD. A new mixing of Hartree-Fock and local density functional theories. J Chem Phys 1993;98:1372–7
- Frisch MJ, Trucks GW, Schlegel HB, et al. Gaussian 09, Revision A.1. Gaussian Inc., Wallingford, CT; 2009
- Bayly CI, Cieplak P, Cornell WD, Kollman PA. A well-behaved electrostatic potential based method using charge restraints for deriving atomic charges – the RESP model. J Phys Chem 1993;97:10269–80
- Wang J, Wolf RM, Caldwell JW, et al. Development and testing of a general amber force field. J Comput Chem 2004;25:1157–74
- Jorgensen WL, Chandrasekhar J, Madura JD, et al. Comparison of simple potential functions for simulating liquid water. J Chem Phys 1983;79:926–35
- Essman U, Perera L, Berkowitz ML, et al. A smooth particle mesh Ewald method. J Chem Phys 1995;103:8577–93
- Andersen HC. Rattle: a “velocity” version of the shake algorithm for molecular dynamics calculations. J Comput Phys 1983;52:24–34
- Fogolari F, Brigo A, Molinari H. Protocol for MM/PBSA molecular dynamics simulations of proteins. Biophys J 2003;85:159–66
- HyperChem, release 6.03. Hypercube Inc., Waterloo, Canada; 2001