
Abstract
Four series of carbazole derivatives, including N-substituted-hydroxycarbazoles, oxazinocarbazoles, isoxazolocarbazolequinones, and pyridocarbazolequinones, were studied using diverse biological test methods such as a CE-based assay for CK2 activity measurement, a cytotoxicity assay with IPC-81 cell line, determination of MIC of carbazole derivatives as antibacterial agents, a Plasmodium falciparum susceptibility assay, and an ABCG2-mediated mitoxantrone assay. Two oxazinocarbazoles Ib and Ig showed CK2 inhibition with IC50 = 8.7 and 14.0 µM, respectively. Further chemical syntheses were realized and the 7-isopropyl oxazinocarbazole derivative 2 displayed a stronger activity against CK2 (IC50 = 1.40 µM). Oxazinocarbazoles Ib, Ig, and 2 were then tested against IPC-81 leukemia cells and showed the ability to induce leukemia cell death with IC50 values between 57 and 62 μM. Further investigations were also reported on antibacterial and antiplasmodial activities. No significant inhibitory activity on ABCG2 efflux pump was detected.
Introduction
Extensive research in the carbazole chemistry has been reported since the description of the parent 9H-carbazole in 1872, by Graebe and GlaserCitation1. The discovery of the antibacterial activity of murrayanine extracted from the stem bark of Murraya Koenigii (Rutaceae) led to the discovery of new pharmacological activitiesCitation2,Citation3. Wu et al.Citation4 described the antiplatelet and vasorelaxing activities of carbazole derivatives extracted from the Clausena excavata and recently, Peng et al.Citation5 described new derivatives excavatine B and excavatine C with cytotoxic activity and antimicrobial activities. Many examples of compounds such as 5H-benzo[b]carbazolesCitation6, furo[3,4-b]carbazolesCitation7, pyrrolo[2,3-a]carbazolesCitation8,Citation9 as well as indolocarbazolesCitation10 have shown antitumor activities. Nowadays, targeted therapies have become one of the main research activities and kinases have reached the second most important drug target after G-coupled protein receptorsCitation11. Prudent et al.Citation12 have shown the activity of pyridocarbazole as casein kinase 2 (CK2) inhibitors; CK2 is a constitutively active serine/threonine protein kinase that is involved in many diseases including different types of cancerCitation13. Furthermore, the challenges in cancer and infectious diseases are to find active compounds that encounter multidrug resistance frequently associated with overexpression of the ABC transporter family. Indolocarbazole derivatives represent novel classes of ABCG2 inhibitorsCitation14. Miscellaneous activities for carbazole derivatives have been reported such as anti-oxidative, anti-inflammatoryCitation15, and antibacterial activitiesCitation16,Citation17.
To our knowledge, little has been described on the biological activity of oxazinocarbazole derivativesCitation18,Citation19. We have described the synthesis and cytotoxic activities of oxazinocarbazolesCitation20. In this report, we focus on a large panel of pharmaceutical applications ranging from antibacterial, antimalarial activities as well as CK2 and ABCG2 inhibition enriched with the synthesis and activity of new carbazole derivatives.
Materials and methods
Chemistry
Reagents were commercial products of the highest available purity grade. Mitoxantrone was purchased from Sigma Aldrich (Lyon, France). Melting points were determined on an Electrothermal 9200 capillary apparatus (Wolf Laboratories Limited, Pocklington, UK). The IR spectra were recorded on a Perkin Elmer Spectrum Two IR Spectrometer (Perkin Elmer, Waltham, MA). 1H NMR and 13C NMR spectra (broadband decoupling and DEPT-135) were recorded on a Brucker Avance 400 (400 MHz for 1H and 133 MHz for 13C) or a Brucker Avance 500 spectrometer (Perkin Elmer, Waltham, MA) (500 MHz for 1H and 126 MHz for 13C) using CDCl3 or DMSO-d6 as solvents. Chemical shifts (δ) are referred to that of the solvent. Low resolution mass spectra were recorded on an Agilent 1290 Infinity system (Agilent technologies, Palo Alto, CA) equipped with an Agilent 1260 DAD detector and an Agilent 6120 Quadrupole mass detector with an ESI source in a positive mode. HRMS spectra were performed on a Q-TOF Micro Water with an ESI source in the positive mode. Flash chromatography was performed on 230–400-mesh silica.
Synthesis of 9-isopropyl-9H-carbazol-4-ol (1)
In a dried tricol containing 263 mg of NaH 60% (18.75 mmol) under an argon atmosphere, 500 mg of 4-hydroxycarbazole (2.73 mmol) dissolved in 0.4 mL of DMF and 5 mL of anhydrous THF were added dropwise. Within 10 min, 0.3 mL of 2-iodopropane (3 mmol) was added. The mixture was kept at room temperature and under an argon atmosphere for 2 h. The residue was hydrolyzed and neutralized with 15% HCl. After extraction with CH2Cl2, the organic phase was dried over Na2SO4 and concentrated under vacuum. The residue was purified by column chromatography on silica gel (CH2Cl2/Petroleum ether: 1/1). Yield 65%; viscous oil; IR: 3450, 1631, 1599, 1580 cm−1; 1H NMR (δ, CDCl3): 8.36 (m, 1H, Harom.); 7.53 (d, 1H, J = 8.3 Hz, Harom.); 7.44 (m, 1H, Harom.); 7.30–7.24 (m, 2H, Harom.); 7.13 (d, 1H, J = 8.3 Hz, Harom.); 6.56 (dd, 1H, J1 = 7.8 Hz, J2 = 0.6 Hz, Harom.); 5.41 (s, 1H, OH); 4.99 (m, 1H, CHMe2); 1.72 (d, 6H, J = 7.1 Hz, CHMe2); 13C NMR (δ, CDCl3): 152.4 (1Cq); 141.8 (1Cq); 139.2 (1Cq); 126.2 (1CH); 124.9 (1CH); 123.3 (1CH); 122.7 (1Cq); 119.2 (1CH); 111.8 (1Cq); 109.9 (1CH); 104.7 (1CH); 103.2 (1CH); 47.2 (1CH); 21.1 (2CH3); HRMS calculated for C15H15NNaO, MNa+ = 248.1046, found, MNa+ = 248.1046.
Synthesis of 7-isopropyl-3-phenyl-2,3,4,7-tetrahydro[1,3]oxazino[5,6-c]carbazole (2)
A solution containing 1.5 mmol of aniline and 3 mmol of formaldehyde dissolved in 10 mL methanol was stirred for 30 min at 0 °C. A solution containing 1 mmol of hydroxycarbazole 1 dissolved in 10 mL of methanol was added dropwise. The mixture was then stirred at room temperature for 24 h. At the end of the reaction, the solvent was evaporated under vacuum and the residue was purified by column chromatography on silica gel (CH2Cl2/cyclohexane: 1/6). Yield 63%; mp = 132.4 °C; IR: 1627, 1601, 1578 cm−1; 1H NMR (δ, CDCl3): 8.54 (d, 1H, J = 7.7 Hz, Harom.); 7.67 (d, 1H, J = 8.3 Hz, Harom.); 7.59 (m, 1H, Harom.); 7.47–7.37 (m, 7H, Harom.); 7.12 (m, 1H, Harom.); 5.81 (s, 2H, H-2); 5.13 (m, 1H, CHMe2); 5.00 (s, 2H, H-4); 1.87 (d, 6H, J = 7.0 Hz, CHMe2); 13C NMR (δ, CDCl3): 150.9 (1Cq); 149.1 (1Cq); 140.1 (1Cq); 139.2 (1Cq); 129.5 (2CH); 124.8 (1CH); 124.0 (1CH); 123.7 (1CH); 122.5 (1Cq); 121.7 (1CH); 119.0 (3CH); 112.2 (1Cq); 110.0 (1Cq); 109.7 (1CH); 103.1 (1CH); 47.0 (1CH); 80.2 (1CH2); 51.0 (1CH2); 21.1 (2CH3); HRMS calculated for C23H23N2O, MH+ = 343.1805, found, MH+ = 343.1811.
General procedure for the aminomethylation of 9-isopropyl-9H-carbazol-4-ol (1)
A solution containing 2 mmol of primary amine and 2 mmol of formaldehyde dissolved in 10 mL of methanol was stirred for 30 min at 0 °C. A solution containing 1 mmol of hydroxycarbazole 1 dissolved in 10 mL of methanol was added dropwise. The mixture was then stirred at room temperature for 3 h to give a precipitate of the aminometylated compound which was purified by column chromatography on silica gel (CH2Cl2/petroleum ether: 2/1).
3-[(Benzylmethylamino)methyl]-9-isopropyl-9H-carbazol-4-ol (3)
Yield 67%; mp = 150 °C; IR: 2836, 1636, 1601 cm−1; 1H NMR (δ, CDCl3): 8.39 (d, 1H, J = 7.7 Hz, Harom.); 7.39 (d, 1H, J = 8.3 Hz, Harom.); 7.33–7.13 (m, 8H, 7Harom. and OH); 6.96 (d, 1H, J = 8.3 Hz, Harom.); 6.85 (d, 1H, J = 8.3 Hz, Harom.); 4.85 (m, 1H, CHMe2); 3.84 (s, 2H, CH2); 3.58 (s, 2H, CH2); 2.21 (s, 3H, NMe); 1.60 (d, 6H, J = 7.1 Hz, CHMe2); 13C NMR (δ, CDCl3): 154.8 (1Cq); 141.2 (1Cq); 139.4 (1Cq); 137.4 (1Cq); 129.8 (2CH); 128.9 (2CH); 127.9 (1CH); 126.1 (1CH); 124.6 (1CH); 123.7 (1CH); 123.3 (1Cq); 119.0 (1CH); 112.4 (1Cq); 110.9 (1Cq); 109.5 (1CH); 100.9 (1CH); 61.7 (1CH2); 61.4 (1CH2); 47.0 (1CH); 41.5 (1CH3); 21.1 (2CH3); HRMS calculated for C24H27N2O, MH+ = 359.2118, found, MH+ = 359.2120.
3-{[(2-Chlorobenzyl)methylamino]methyl}-9-isopropyl-9H-carbazol-4-ol (4)
Yield 63%; mp = 61 °C; IR: 2925, 1636, 1603 cm−1; 1H NMR (δ, CDCl3): 8.44 (d, 1H, J = 7.7 Hz, Harom.); 7.48–7.19 (m, 8H, 7Harom. and OH); 7.06 (d, 1H, J = 8.3 Hz, Harom.); 6.94 (d, 1H, J = 8.3 Hz, Harom.); 4.93 (m, 1H, CHMe2); 3.98 (s, 2H, CH2); 3.83 (s, 2H, CH2); 2.32 (s, 3H, NMe), 1.69 (d, 7.0 Hz, 6H, CHMe2); 13C NMR (δ, CDCl3): 154.6 (2Cq); 141.2 (1Cq); 139.3 (1Cq); 135.2 (2Cq); 131.9 (1CH); 130.2 (1Cq); 129.3 (1CH); 127.3 (1CH); 126.2 (1CH); 124.6 (1CH); 123.7 (1CH); 123.2 (1Cq); 119.0 (1CH); 110.9 (1Cq); 109.5 (1CH); 100.9 (1CH); 61.5 (1CH2); 59.1 (1CH2); 47.0 (1CH3); 41.4 (1CH); 21.1 (2CH3); HRMS calculated for C24H26ClN2O, MH+ = 393.1728, found, MH+ = 393.1728.
3-{[(3-Chlorobenzyl)methylamino]methyl}-9-isopropyl-9H-carbazol-4-ol (5)
Yield 47%; mp = 182 °C; IR: 2967, 2833, 1633, 1598 cm−1; 1H NMR (δ, CDCl3): 8.46 (d, 1H, J = 7.7 Hz, Harom.); 7.48 (d, 1H, J = 8.3 Hz, Harom.); 7.42–7.21 (m, 7H, 6Harom. and OH); 7.05 (d, 1H, J = 8.3 Hz, Harom.); 6.95 (d, 1H, J = 8.3 Hz, Harom.); 4.99 (m, 1H, CHMe2); 3.94 (s, 2H, CH2); 3.64 (s, 2H, CH2); 2.31 (s, 3H, NMe), 1.69 (d, 6H, J = 7.0 Hz, CHMe2); 13C NMR (δ, CDCl3): 154.6 (1Cq); 141.3 (1Cq); 139.5 (1Cq); 139.4 (1Cq); 134.6 (1Cq); 130.3 (1CH); 129.9 (1CH); 128.1 (1CH); 127.9 (1CH); 126.1 (1CH); 124.7 (1CH); 123.7 (1CH); 123.2 (1Cq); 119.1 (1CH); 112.4 (1Cq); 110.7 (1Cq); 109.6 (1CH); 101.0 (1CH); 61.5 (1CH2); 61.0 (1CH2); 47.0 (1CH3); 41.6 (1CH); 21.1 (2CH3); HRMS calculated for C24H26ClN2O, MH+ = 393.1728, found, MH+ = 393.1726.
3-{[(4-Chlorobenzyl)methylamino]methyl}-9-isopropyl-9H-carbazol-4-ol (6)
Yield 67%; mp = 139 °C; IR: 2985, 2954, 2845, 1633, 1603 cm−1; 1H NMR (δ, CDCl3): 8.46 (d, 1H, J = 7.9 Hz, Harom.); 7.48 (d, 1H, J = 8.1 Hz, Harom.); 7.43–7.21 (m, 7H, 6Harom. and OH); 7.04 (d, 1H, J = 8.1 Hz, Harom.); 6.95 (d, 1H, J = 8.3 Hz, Harom.); 4.94 (m, 1H, CHMe2); 3.93 (s, 2H, CH2); 3.63 (s, 2H, CH2); 2.30 (s, 3H, NMe), 1.69 (d, 6H, J = 7.2 Hz, CHMe2); 13C NMR (δ, CDCl3): 154.6 (1Cq); 141.3 (1Cq); 139.4 (1Cq); 135.9 (1Cq); 133.7 (1Cq); 131.1 (2CH); 129.0 (2CH); 126.1 (1CH); 124.7 (1CH); 123.6 (1CH); 123.2 (1Cq); 119.1 (1CH); 112.4 (1Cq); 110.7 (1Cq); 109.6 (1CH); 101.0 (1CH); 61.4 (1CH2); 60.8 (1CH2); 47.0 (1CH3); 41.5 (1CH); 21.1 (2CH3); HRMS calculated for C24H26ClN2O, MH+ = 393.1728, found, MH+ = 393.1727.
3-{3-[(Benzylmethylamino)methyl]-9-isopropyl-9H-carbazol-4-yloxy}propan-1-ol (7)
A solution containing 100 mg (0.28 mmol) of 3, 38 mg (0.28 mmol) of 3-bromopropan-1-ol, 77 mg (0.56 mmol) of K2CO3 and 10.3 mg (0.028 mmol) of tetrabutylammonium iodide (TBAI) dissolved in 5 mL of CH3CN was stirred at 80 °C for 4 h. The mixture was then filtered on celite and washed with CH3CN. The filtrate was evaporated and the residue was purified by column chromatography on silica gel (EtOAc/petroleum ether: 2/1) to give 7 as a liquid product. Yield 65%; IR: 3381, 2930, 2875, 1622, 1596 cm−1; 1H NMR (δ, CDCl3): 8.19 (d, 1H, J = 7.9 Hz, Harom.); 7.46–7.13 (m, 10H, Harom.); 4.89 (m, 1H, CHMe2); 4.25 (t, 2H, J = 5.8 Hz, CH2); 3.96 (t, 2H, J = 5.8 Hz, CH2); 3.72 (s, 2H, CH2); 3.51 (s, 2H, CH2); 3.05 (bs, 1H, OH); 2.17 (s, 3H, NMe); 2.13 (m, 2H, CH2); 1.62 (d, 6H, J = 7.0 Hz, CHMe2); 13C NMR (δ, CDCl3): 154.0 (1Cq); 141.2 (1Cq); 139.6 (1Cq); 139.0 (1Cq); 129.6 (2CH); 129.5 (1CH); 128.5 (2CH); 127.3 (1CH); 125.3 (1CH); 123.1 (1CH); 121.9 (1Cq); 120.7 (1Cq); 119.3 (1CH); 116.8 (1Cq); 110.1 (1CH); 106.3 (1CH); 71.3 (1CH2); 62.0 (1CH2); 60.6 (1CH2); 57.4 (1CH2); 47.1 (1CH3); 42.8 (1CH); 33.2 (1CH2); 21.1 (2CH3); HRMS calculated for C27H33N2O2, MH+ = 417.2537, found, MH+ = 417.2531.
Preparation of recombinant human CK2 holoenzyme
The preparation of the human recombinant CK2 holoenzyme was performed according to the protocol previously described by Olgen et al.Citation21. The α-subunit (CSNK2A1) and β-subunit (CSNK2B) of the human protein kinase CK2 were expressed in a bacterial pT7-7 system in Escherichia coli BL21 (DE3). Newly transformed starter cultures were grown overnight at 37 °C in LB-medium until they reached the stationary phase. With the separate starter cultures, new medium was inoculated for both subunits and cultivated until an OD500 of 0.6 was reached. Protein expression was induced by addition of IPTG (1 mM final concentration) and carried out at 30 °C for 5–6 h for CSNK2A1 and at 37 °C for 3 h for CSNK2B. Bacterial cells were harvested by centrifugation (6000 × g for 10 min at 4 °C) and disrupted by sonification (three times 30 s on ice). Preparations were then centrifuged to remove the cell debris and the bacterial extracts for both subunits and combined before purification by a three-column procedure. The fractions containing active CK2 holoenzyme were determined by activity measurement using the synthetic peptide substrate RRRDDDSDDD. Finally, fractions exhibiting CK2 activity were combined and analyzed by SDS-PAGE and Western Blot.
Capillary electrophoresis-based assay for the testing of inhibitors of the human CK2
The recently established capillary electrophoresis CK2 activity assay was used for testing the inhibitorsCitation22. Therefore, 2 µL of the dissolved inhibitors (stock solution in DMSO) were mixed with 78 µL of CK2 supplemented kinase buffer which was composed of 1 µg CK2 holoenzyme, 50 mM Tris/HCl (pH 7.5), 100 mM NaCl, 10 mM MgCl2, and 1 mM DTT. The reaction was initiated by the addition of 120 µL assay buffer, which was composed of 25 mM Tris/HCl (pH 8.5), 150 mM NaCl, 5 mM MgCl2, 1 mM DTT, 100 µM ATP, and 0.19 mM of the substrate peptide RRRDDDSDDD. The reaction was carried out for 15 min at 37 °C and stopped by the addition of 4 µL EDTA (0.5 M). Subsequently the reaction mixture was analyzed by a PA800 capillary electrophoresis from Beckman Coulter (Krefeld, Germany). Acetic acid (2 M, adjusted with conc. HCl to a pH of 2.0) was used as the electrolyte for electrophoretic separation. The separated substrate and product peptide were detected at 214 nm using a DAD-detector. Pure solvent was used as a negative control (0% inhibition) and assays devoid of CK2 were used as a positive control (100% inhibition). For primary testing an inhibitor concentration of 10 µM was used. Compounds that revealed at least 50% inhibition at 10 µM were used for IC50 determinations. The IC50 inhibition was determined using nine inhibitor concentrations ranging from 0.001 µM to 100 µM. IC50 values were calculated from the resulting dose–response curves.
Cytotoxicity assay
IPC-81 acute myelogenic leukemia rat cellsCitation23 were maintained at logarithmic growth in the medium (DMEM) supplemented with 10% horse serum. For cytotoxicity testing, the IPC-81 cells were seeded in 96 well tissue culture plates at 150 000 cells/mL and exposed to various concentrations of CK2 inhibitors or vehicles for 24 h. The cells were next fixed in 2% buffered formaldehyde (pH 7.4) with the DNA-specific dye Hoechst 33342 (Polysciences Inc., Eppelheim, Germany) and scored for death by fluorescence microscopic evaluation of apoptotic chromatin condensation. At least 100 cells in three different microscopic fields were counted. IPC-81 cells were also analyzed for intactness of metabolic activity (formazan oxidation) by the WST-1 assay (Roche Diagnostics, Basel, Switzerland) following the supplier’s instructions.
Determination of antibacterial activity
The following reference strains were used for testing the antibacterial activity: E. coli DSM 1103 and Pseudomonas aeruginosa DSM 1117 for Gram negative bacteria (Deutsche Sammlung für Mikroorganismen, Braunschweig, Germany) and Staphylococcus aureus CIP 103.429 and Enterococcus faecalis CIP 103.214 (Collection de l’Institut Pasteur, Paris, France) for Gram-positive bacteria. Bacteria were grown overnight at 35 °C in Tryptic Soy Broth and streaked on Tryptic Soy Agar (AES, Bruz, France). From these isolation plates, inocula were prepared according to CLSI recommendationsCitation24. The broth microdilution technique was carried out as advised using selected carbazole derivatives dissolved in DMSO at concentrations ranging from 0.0625 to 128 µg/mL. Ciprofloxacin and DMSO were used in each series of experiments as a positive and a negative control, respectively. The MIC was determined as the lowest drug concentration at which wells remained visually clear.
Plasmodium falciparum susceptibility assay
The in vitro activities of oxazinocarbazole derivatives were tested over a concentration range of 0.78–10 000 nM against the susceptibility to chloroquine (CQ) strain P. falciparum 3D7. The traditional labeled hypoxanthine method was used to assess antimalarial activity, as described by Desjardins and coworkersCitation25. CQ was routinely included as positive controls as well as negative controls using a solvent. The resulting IC50s were calculated using Pk-Fit softwareCitation26 (Simulations Plus, Inc., Lancaster, CA). Inhibition growth (I) and corresponding drug concentration (C) were fitted according a sigmoid model, described as
where Imax is the maximum inhibition growth and gamma the sigmoid factor of the curb.
Inhibitory activity on human ABCG2-transfected cells
All compounds were dissolved in DMSO and then diluted in the DMEM high glucose medium. The stock solution was stored at −20 °C and warmed to 25 °C just before use. The human fibroblast HEK293 cell line transfected with ABCG2 was obtained as previously describedCitation27. Cells were maintained in Dulbecco’s modified Eagle’s medium (DMEM high glucose), supplemented with 10% fetal bovine serum (FBS), 1% penicillin/streptomycin, and drug supplemented with 0.75 mg/mL G418. To perform the ABCG2-mediated mitoxantrone assay, HEK293-ABCG2 cells were seeded at a density of 1 × 105 cells/well into 24-well culture plates. After 48 h incubation, the cells were exposed to 5 µM mitoxantrone for 30 min at 37 °C, in the presence or absence of compounds at various concentrations. The cells were then washed with phosphate buffer saline (PBS) and after being trypsinized and subsequently resuspended in ice-cold PBS (0.2 mL), they were kept on ice until analysis by flow cytometry. The data of intracellular drug fluorescence were monitored with a FACS Calibur cytometer (Becton Dickinson, Franklin Lakes, NJ) using a FL4-H channel. At least 10 000 events were collected, for which the maximal fluorescence (100%) was the difference between geometric mean fluorescence of cells incubated with 5 µM of GF120918 as a control (100% inhibition) and without a inhibitor.
Results and discussion
Chemistry
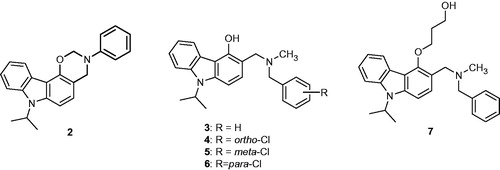
Among the 43 carbazole derivatives described in this study, the synthesis of only 2–7 has never been reported until now ().
Figure 1. Structures of oxazinocarbazole 2 and aminomethylated carbazoles 3–7.
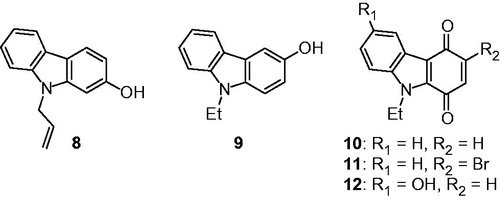
For the other series, experimental works are previously described. The N-substituted hydroxycarbazoles 8 and 9 were prepared in one step by a chemoselective N-alkylation of 2- or 3-hydroxycarbazole with 2.5 eq of NaH in DMF/THF mixture under argon atmosphere at room temperature and subsequent treatment with the alkylating agentCitation20,Citation28. The tricyclic carbazolequinones 10–12 were obtained starting from the corresponding hydroxycarbazoles by oxidation with Frémy’s salt ()Citation29,Citation30.
Figure 2. Structures of tricyclic hydroxycarbazole and carbazolequinone derivatives 8–12.
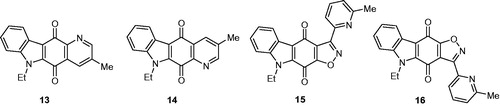
Syntheses of pyridocarbazolediones 13 and 14 have been achieved using regiospecific hetero-Diels–Alder reaction between an azadiene and an appropriate bromocarbazolequinoneCitation29. Syntheses of isoxazolocarbazolediones 15 and 16 have been accomplished through a regioselective 1,3-dipolar cycloaddition of nitrile oxide with an appropriate bromocarbazolequinone ()Citation31.
Figure 3. Structures of pyrido and isoxazolocarbazolediones 13–16.
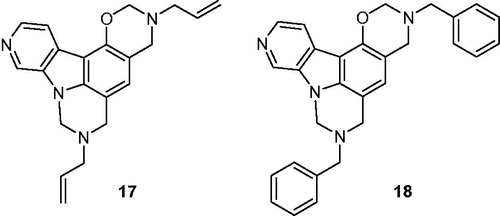
The oxazinopyrimidocarboline derivatives 17 and 18 were synthesized through a Mannich reaction employing 5-hydroxy-β-carboline, 4 eq of formaldehyde and 2 eq of a primary amine in MeOH at room temperature ()Citation32.
Figure 4. Structures of oxazinopyrimidocarboline derivatives 17 and 18.
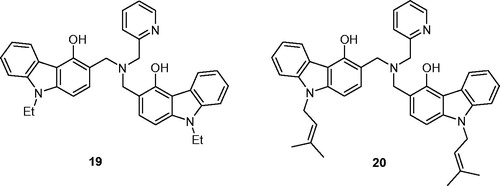
Dimers 19 and 20 were obtained as by-products through a dimerization process in the course of the Mannich condensation among N-substituted 4-hydroxycarbazole, formaldehyde, and 2-(aminomethyl)pyridine as the primary amine ()Citation20.
Figure 5. Structures of N,N-bis(carbazolylmethyl)amines 19 and 20.
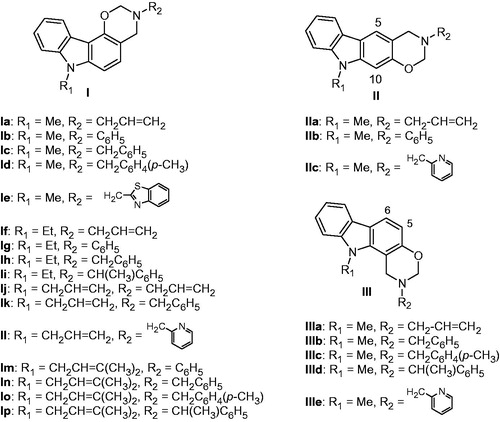
The oxazino[5,6-c]carbazoles Ia–p were prepared by a Mannich type condensation of N-substituted 4-hydroxycarbazole with 1.5 eq of various primary amines and 3 eq of formaldehyde. The Mannich reaction of N-substituted 2-hydroxycarbazole with the primary amines and formaldehyde led to a mixture of the two regioisomers, the oxazino[6,5-b]carbazoles IIa–c and the oxazino[5,6-a]carbazoles IIIa–e which were isolated by chromatography. Identification of the regioisomeric oxazinocarbazoles was unambiguously established from their 1H NMR 300 MHz spectra ()Citation20. Thus, for oxazinocarbazoles II, the 1H NMR spectra shows two singlets at 7.63 and 6.78 ppm for the aromatic protons H-5 and H-10 while in the regioisomers III, it exhibits two doublets at 7.83 and 6.78 ppm for the aromatic protons H-5 and H-6 with 8.5 Hz as a coupling constant value.
Figure 6. Structures of oxazinocarbazoles Ia–p, IIa–c, and IIIa–e.
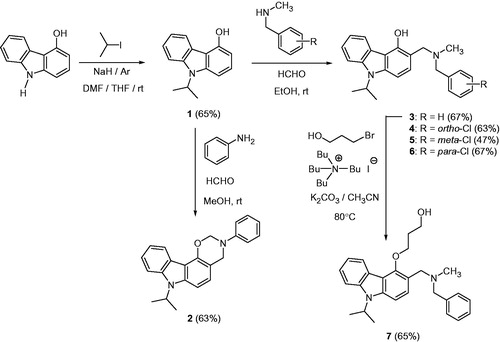
The N-isopropylated compound 1 was chemoselectively prepared by treatment of 4-hydroxycarbazole with sodium hydride in the presence of isopropyl iodide in the solvent system THF-DMF. Then, compound 2 was prepared by a Mannich reaction of 1 with 1.5 eq of aniline and 3 eq of formaldehyde in MeOH at room temperature. In contrast, syntheses of 3–6 have been carried out using a Mannich reaction of 1 with 2.5 eq of the corresponding secondary amines and 2.5 eq of formaldehyde in methanol at room temperature. The O-alkylation of 3 with 1 eq of 3-bromopropan-1-ol in the presence of 0.1 eq of tetrabutylammonium iodide (TBAI) and 2 eq of anhydrous K2CO3 in CH3CN at 80 °C gave the compound 7 (Scheme 1).
Scheme 1. Synthesis of compounds 1–7.
Biological results
Carbazoles derivatives as potent inhibitors of human protein kinase CK2
Thirty-seven compounds were evaluated by the capillary electrophoresis (CE)-based assayCitation22 for human protein kinase CK2 activity measurement and inhibitor screening (). With this recently developed assay, the produced and purified recombinant human CK2 holoenzyme was used. The synthetic peptide RRRDDDSDDD was used as the substrate, which is reported to be most efficiently phosphorylated by CK2. The purity of the CK2 holoenzyme was higher than 99% ()Citation33. We can directly monitor the degree of CK2 substrate phosphorylation without pretreatment of the kinase reaction before capillary electrophoresis. This enabled us to reliably determine CK2 activity/inhibition.
Figure 7. SDS-PAGE of purified human protein kinase CK2 holoenzyme. About 15 µL of purified protein solution (0.25 µg/µL) were separated on a 12.5% acrylamide gel and stained with Coomassie Brilliant Blue G250. At the left, the apparent molecular mass of the marker proteins (lane 1) is given. Lane 2 shows the purified human CK2 enzyme holoenzyme (3.75 µg). The band below the CK2α belongs to the well-known degradation product of the α-subunit CK2α (amino acids 1–335), which is supposed to be enzymatically active and using occurs during purificationCitation33.
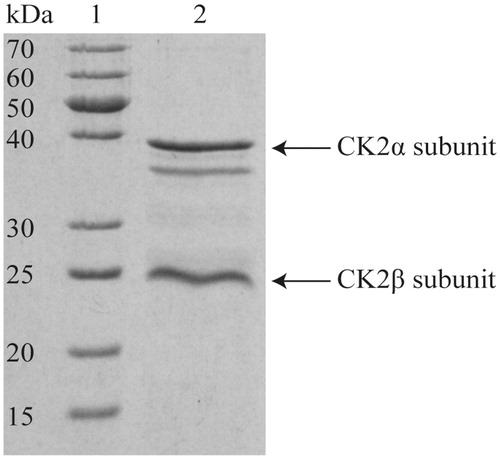
Table 1. Inhibition of human protein kinase CK2 by carbazole derivatives 10–16, Ia–p, IIa–c, and IIIa–e.
For initial testing, inhibition was determined relative to the control at inhibition concentration of 10 μM of compounds dissolved in DMSO. Negative control cells were cultured with pure DMSO that never exceeded 1% as a final concentration and set to 0% inhibition. Reactions without CK2 were used as the positive control and set to 100% inhibition. Compounds with more than 50% inhibition at a concentration of 10 μM were subjected to an IC50 determination. For this purpose, inhibition was measured at final concentrations ranging from 0.001 to 100 μM in appropriate intervals. IC50 were calculated from the resulting dose–response curves. Each value was determined at least in triplicate in independent experimentsCitation22.
Tricyclic carbazolequinones 10–12 showed weak CK2 inhibition (% inhibition values <26%). Series of pyridocarbazolequinones 13,14 and isoxazolocarbazolequinones 15,16 were not very soluble at 10 μM in pure DMSO. Nevertheless, the saturated solutions of derivatives 13, 15, and 16 were tested and showed weak CK2 inhibitory activity (% inhibition values <29%), having in mind that these compounds could have stronger inhibitory activities without the problem of solubility. In the series of oxazinocarbazoles I–III, all compounds were soluble and the assay was performed in triplicate. Two oxazino[5,6-c]carbazoles Ib and Ig appeared to be potential CK2 inhibitors (% inhibition values >50%). They are structurally characterized by a short alkyl chain in R1 (methyl for Ib and ethyl for Ig) and a common phenyl group in R2. The IC50 values were calculated: 8.7 and 14.0 μM, respectively.
Further pharmacomodulation works were realized to optimize these first results. In a recent SAR studyCitation34 on another tetracyclic scaffold (indeno[1,2-b]indole), the best inhibitory activity against CK2 was observed by introducing an isopropyl group on indolic nitrogen. Then the oxazinocarbazole 2 exerted the best CK2 inhibition with an IC50 value of 1.40 μM () and could be used as lead compound for further structural exploration. In contrast, the opening of the oxazine ring did not improve the activity. In fact, aminomethylated carbazoles 3–7 showed a weak level of inhibition (<32%).
Table 2. Inhibition of human protein kinase CK2 by oxazinocarbazole 2 and aminomethylated carbazoles 3–7.
Oxazinocarbazoles as antileukemic agents
We tested the oxazinocarbazoles potency against IPC-81 leukemia cells. The IPC-81 cell line is derived from the transplantable BNML model of acute myelogenic leukemia (AML), known to be a reliable predictor of the clinical efficiency of antileukemic agents, like the first-line AML anthracycline drug daunorubicin (DNR)Citation35. The oxazinocarbazoles had IC50 values between 57 and 62 μM (). All the tested compounds had a similarly steep dose dependency, and were capable of inducing 100% IPC cell death at concentrations above 80 μM (). The cell death morphology had apoptotic features, with nuclear condensation and surface membrane blebs (not shown), which are typical for such cells treated with various apoptosis inducers and anti-leukemic drugsCitation36. We also tested the cell’s mitochondrial formazan conversion (WST-assay, see Methods section). This assay agreed with the morphological assessment of cell death for long incubation times. For short incubation periods, the microscopic assay was superior, presumably because the already formed mitochondrial formazan converting enzymes only lose their activity when the cell undergoes secondary necrosis with loss of ATP and general proteolysis.
Figure 8. The ability of oxazinocarbazoles to induce IPC-81 leukemia cell death. IPC-81 cells were incubated with various concentrations of the compounds Ib, Ig, and 2, and apoptosis assessed by microscopic evaluation of leukemia cell morphology after 24 h.
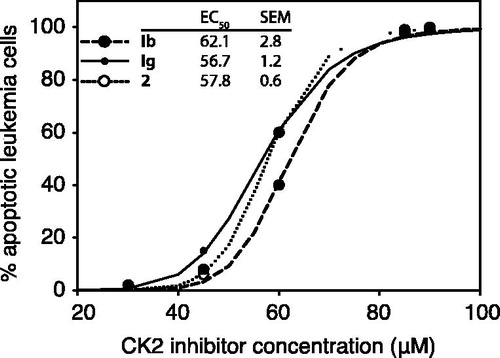
We conclude that the oxazinocarbazole derivatives were indeed able to induce leukemia cell death at concentrations well below 80 µM.
Oxazinocarbazoles as antibacterial agents
Most of the tested compounds did not display any significant antibacterial activity at the tested concentrations (). Only hydroxycarbazoles 8,9 and non-substituted carbazolequinone 10 were found to be somewhat effective, the latter being the most efficient, but only on Gram-positive bacteria (i.e. S. aureus and E. faecalis). These molecules had in common a low molecular weight, as compared to the ineffective ones. Surprisingly, compound 12, which only differs from compound 10 by the addition of a hydroxyl substituent, lost all activity at concentrations up to 128 µg/mL despite a rather low molecular weight. The overall lack of efficiency on Gram-negative bacteria can be hypothesized to be linked with an impermeability of the Gram-negative cell wall to the tested compounds.
Table 3. Antimicrobial activities (MIC µg/mL) of carbazole derivatives 8–10, 12, 17–20, Ic, Ik,l, Ip, and IIc.
Oxazinocarbazoles as antimalarial agents
Sixteen compounds were evaluated in vitro for their antimalarial activity against P. falciparum 3D7 strain according with the IC50 values as shown in . As the strain used in this assay was well described, chloroquinosensible and chloroquine were used for the validation of the assay. The susceptibility to available antimalarial drugs for this strain following the used isotopic method is close to the range 2–200 nM with end points corresponding to dihydroartemisinin and quinine, respectively. The susceptibility to doxycycline which is only used for malaria prophylaxis is closed to 10 µM. Four compounds showed interesting results with IC50 under 1 µM: hydroxycarbazole derivative 8 (IC50 = 675 nM), oxazinopyrimidocarboline derivative 18 (IC50 = 551 nM) and two oxazinocarbazole derivatives Ip (IC50 = 659 nM) and IIc (IC50 = 430 nM).
Table 4. Antiplasmodial activities (IC50 nM) of carbazole derivatives 2, 8–10, 12, 17–20, Ib,c, Ig, Ik,l, Ip, and IIc.
Oxazinocarbazoles and ABCG2-mediated drug transport
To investigate the activity of carbazole derivatives in multidrug resistance, oxazinocarbazoles 2, Ia–c, Ig, Ij,k, In, IIa,c, IIIa,e, and dimers 19,20 were screened for their inhibition of mitoxantrone efflux in ABCG2-transfected cells. Ko143 was used as other control and showed an inhibition of 115%. Only very low inhibition (20% for compound IIIe) was observed at 10 µM for the tested compounds.
Conclusions
Five different biological assays were used to explore new pharmacological potencies of carbazole derivatives: inhibition of CK2, IPC-81 cell line cytotoxicity, antibacterial activity, antimalarial activity, and inhibitory activity on HEK293-ABCG2 cells. The best results were obtained on CK2 assay with the oxazinocarbazoles Ib (IC50 = 8.7 μM), Ig (IC50 = 14.0 μM), and 2 (IC50 = 1.4 μM). Cytotoxicity activity of these CK2 inhibitors was determined on IPC-81 leukemia cells (IC50 lower than 80 µM). From the point of view of the structure, oxazinocarbazoles Ib, Ig, and 2 have a short indolic N-substituted side chain (e.g. methyl, ethyl, and isopropyl) and a phenyl group in the oxazine ring. New pharmacomodulations on oxazinocarbazole template are currently studied and will later be published. In contrast, investigation concerning the antiplasmodial activities showed encouraging results with carbazole derivatives 8 (IC50 = 675 nM), 18 (IC50 = 551 nM), Ip (IC50 = 659 nM), and IIc (IC50 = 430 nM). Further structural explorations should be envisaged to determine a new pharmacophore model for antimalarial agents. No carbazole derivatives showed significative activities on bacteria and ABCG2 efflux pump for further investigations. Finally carbazole and its congeners (e.g. oxazinocarbazoles) continue to be central templates for drug design of new small molecules.
Declaration of interest
The authors report no declarations of interest. We acknowledge financial support to Zouhair Bouaziz and Marc Le Borgne by the “Bonus Qualité Recherche” (BQR) of the University Claude Bernard Lyon 1, by the “Cluster 5 Chimie Durable et Chimie pour la Santé” of the Region Rhône-Alpes and by the “ARC 1 Santé” of the Region Rhône-Alpes. The “Institut des Sciences Pharmaceutiques et Biologiques” (ISPB) is also gratefully acknowledged for the funding of an uHPLC/DAD/MS system.
The present work was also supported by the “Partenariat Hubert Curien” (PHC) (Campus France, Programme Aurora, Grant Agreement No. 27460VC). Pr. Marc Le Borgne also thanks the “Institut français d’Oslo” for their support via the Åsgard Programme 2010.
Dr. M. Baitiche is very grateful to the Ministry of Higher Education and Research and to the University of Setif for sponsoring the trainings carried out at EA 4446 “Biomolécules Cancer et Chimiorésistances”.
We acknowledge financial support given to Samar Issa by the “Agence Universitaire de la Francophonie” (AUF) and the Région Rhône-Alpes (MIRA).
Acknowledgements
Dr. Z. Bouaziz and Pr. M. Le Borgne would like to gratefully thank Christine Ranquet and Dr. L. Ettouati for their assistance in the stock management of the small molecule library of EA 4446 B2C. We also would like to thank ChemAxon for providing us license to their cheminformatics software.
References
- Schmidt AW, Reddy KR, Knölker HJ. Occurrence, biogenesis, and synthesis of biologically active carbazole alkaloids. Chem Rev 2012;112:3193–328
- Nagappan T, Ramasamy P, Wahid ME, et al. Biological activity of carbazole alkaloids and essential oil of Murraya koenigii against antibiotic resistant microbes and cancer cell lines. Molecules 2011;16:9651–64
- Ma Q, Tian J, Yang J, et al. Bioactive carbazole alkaloids from Murraya koenigii (L.) Spreng. Fitoterapia 2013;87:1–6
- Wu TS, Huang SC, Wu PL, Teng CM. Carbazole alkaloids from Clausena excavata and their biological activities. Phytochemistry 1996;43:133–40
- Peng WW, Zeng GZ, Song WW, Tan NH. A new cytotoxic carbazole alkaloid and two new other alkaloids from Clausena excavata. Chem Biodivers 2013;10:1317–21
- Asche C, Frank W, Albert A, Kucklaender U. Synthesis, antitumour activity and structure-activity relationships of 5H-benzo[b]carbazoles. Bioorg Med Chem 2005;13:819–37
- Hajbi Y, Neagoie C, Biannic B, et al. Synthesis and biological activities of new furo[3,4-b]carbazoles: potential topoisomerase II inhibitors. Eur J Med Chem 2010;45:5428–37
- Giraud F, Akué-Gédu R, Nauton L, et al. Synthesis and biological activities of 4-substituted pyrrolo[2,3-a]carbazole Pim kinase inhibitors. Eur J Med Chem 2012;56:225–36
- Akué-Gédu R, Letribot B, Saugues E, et al. Kinase inhibitory potencies and in vitro antiproliferative activities of N-10 substituted pyrrolo[2,3-a]carbazoles derivatives. Bioorg Med Chem Lett 2012;22:3807–9
- Lee HJ, Schaefer G, Heffron TP, et al. Noncovalent wild-type-sparing inhibitors of EGFR T790M. Cancer Discov 2013;3:168–81
- Indumathi T, Muthusankar A, Shanmughavel P, Rajendra Prasad KJ. Synthesis of hetero annulated carbazoles: exploration of in vitro cytotoxicity and molecular docking studies. Med Chem Commun 2013;4:450–5
- Prudent R, Moucadel V, Nguyen CH, et al. Antitumor activity of pyridocarbazole and benzopyridoindole derivatives that inhibit protein kinase CK2. Cancer Res 2010;70:9865–74
- Liu S, Hsieh D, Yang YL, et al. Coumestrol from the national cancer Institute's natural product library is a novel inhibitor of protein kinase CK2. BMC Pharmacol Toxicol 2013;14:36
- Robey RW, Shukla S, Steadman K, et al. Inhibition of ABCG2-mediated transport by protein kinase inhibitors with a bisindolylmaleimide or indolocarbazole structure. Mol Cancer Ther 2007;6:1877–85
- Bandgar BP, Adsul LK, Chavan HV, et al. Synthesis, biological evaluation, and docking studies of 3-(substituted)-aryl-5-(9-methyl-3-carbazole)-1H-2-pyrazolines as potent anti-inflammatory and antioxidant agents. Bioorg Med Chem Lett 2012;22:5839–44
- Zhang FF, Gan LL, Zhou CH. Synthesis, antibacterial and antifungal activities of some carbazoles derivatives. Bioorg Med Chem Lett 2010;20:1881–4
- Biamonte MA, Wanner J, Le Roch KG. Recent advances in malaria drug discovery. Bioorg Med Chem Lett 2013;23:2829–43
- Tenbrink RE. Oxazinocarbazoles for the treatment of CNS diseases, European Patent 1200447 A1; 2002
- McCarthy TJ, Merchant KM, Ten Brink RE. Labeled oxazinocarbazoles as diagnostic agents European Patent 1495031 A1; 2005
- Issa S, Walchshofer N, Kassab I, et al. Synthesis and antiproliferative activity of oxazinocarbazole and N,N-bis(carbazolylmethyl)amine derivatives. Eur J Med Chem 2010;45:2567–77
- Olgen S, Götz C, Jose J. Synthesis and biological evaluation of 3-(substituted-benzylidene)-1,3-dihydro-indolin derivatives as human protein kinase CK2 and p60(c-Src) tyrosine kinase inhibitors. Biol Pharm Bull 2007;30:715–18
- Gratz A, Götz C, Jose J. A CE-based assay for human protein kinase CK2 activity measurement and inhibitors screening. Electrophoresis 2010;31:634–40
- Lacaze N, Gombaud-Saintonge G, Lanotte M. Conditions controlling long-term proliferation of Brown Norway rat promyelocytic leukemia in vitro: primary growth stimulation by microenvironment and establishment of an autonomous Brown Norway ‘leukemic stem cell line'. Leuk Res 1983;8:145–54
- Clinical and Laboratory Standards Institute. Methods for dilution antimicrobial susceptibility tests for bacteria that grow aerobically; approved standard. 8th ed. Document M07-A8. Wayne (PA): CLSI; 2009
- Desjardin RE, Canfield CJ, Haynes JD, Chulay JD. Quantitative assessment of antimalarial activity in vitro by a semiautomated microdilution technique. Antimicrob Agents Chemother 1979;16:710–18
- Farenc C, Fabreguette JR, Bressolle F. Pk-fit: a pharmacokinetic/pharmacodynamic and statistical data analysis software. Comput Biomed Res 2000;33:315–30
- Arnaud O, Boumendjel A, Géze A, et al. The acridone derivative MBLI-87 sensitizes breast cancer resistance protein-expressing xenografts to irinotecan. Eur J Cancer 2011;47:640–8
- Compain-Batissou M, Latreche D, Gentili J, et al. Synthesis and Diels-Alder reactivity of ortho-carbazolequinones. Chem Pharm Bull 2004;52:1114–16
- Poumaroux A, Bouaziz Z, Fillion H. Regiospecific hetero Diels-Alder synthesis of pyrido[2,3-b] and pyrido (3,2-b]carbazole-5,11-diones. Heterocycles 1997;45:585–96
- Bouaziz Z, Ghérardi A, Régnier F, et al. Synthesis of carbazolequinone derivatives as inhibitors of Toxoplasma gondii purine nucleoside phosphorylase. Eur J Org Chem 2002;2002:1834–8
- Compain-Batissou M, Gentili J, Walchshofer N, et al. Regioselectivity in the 1,3-dipolar cycloaddition reactions of nitrile oxides and organic azides with carbazole-1,4-diones. Heterocycles 2007;71:27–38
- Issa S, Walchshofer N, Kassab I, et al. Mannich reaction of 5-hydroxy-β-carboline. Application to the synthesis of novel oxazinopyrimidocarboline derivatives. Heterocycles 2008;75:2761–7
- Ermakova I, Boldyreff B, Issinger OG, Niefind K. Crystal structure of a C-terminal deletion mutant of human protein kinase CK2 catalytic subunit. J Mol Biol 2003;330:925–34
- Hundsdörfer C, Hemmerling HJ, Götz C, et al. Indeno[1,2-b]indole derivatives as a novel class of potent human protein kinase CK2 inhibitors. Bioorg Med Chem 2012;20:2282–9
- McCormack E, Bruserud O, Gjertsen BT. Animal models of acute myelogenous leukaemia – development, application and future perspectives. Leukemia 2005;19:687–706
- Huseby S, Gausdal G, Keen TJ, et al. Cyclic AMP induces IPC leukemia cell apoptosis via CRE- and CDK-dependent Bim transcription. Cell Death Dis 2011;2:e237