
Abstract
An anti-inflammatory complex of Ag(I), namely [Ag(tpp)3(asp)](dmf) [tpp = triphenylphosphine, aspH = aspirin, dmf = N,N-dimethylformamide], was synthesized in an attempt to develop novel metallotherapeutic molecules. STD 1H NMR experiments were used to examine if this complex binds to LOX-1. The 1H NMR spectra in buffer Tris/D2O betrayed the existence of two complexes: the complex of aspirin and the complex of salicylic acid produced after deacetylation of aspirin. Nevertheless, the STD spectra showed that only the complex of salicylic acid is bound to the enzyme. Molecular docking and dynamics were used to complement our study. The complexes were stabilized inside a large LOX-1 cavity by establishing a network of hydrogen bonds and steric interactions. The complex formation with salicylic acid was more favorable. The in silico results provide a plausible explanation of the experimental results, which showed that only the complex with salicylic acid enters the binding cavity.
Introduction
Non-steroidal anti-inflammatory drugs (NSAID’s) is a class of pharmaceutical molecules, which inhibit cyclooxygenase (COX) and thus the prostaglandin synthesisCitation1. Prostaglandins belong to postanoids that constitute potent substances, which trigger a variety of reactions in cells of many tissues; the most typical reaction is the activation of inflammatory cells (leukocytes). On the other hand, lipoxygenase (LOX) on the other hand converts arachidonic acid into 5-hydroperoxyeicosatetraenoic acid, which is the precursor of leukotrienes. Leukotrienes are formed mainly in leukocytes and they are potent pro-inflammatory mediators. Recent reports have shown that NSAID’s may be used as anticancer drugsCitation2–5. They are found to activate the mechanism of apoptosis and inhibit angiogenesis. These two mechanisms help to suppress both the formation of malignant tumor and the malignant transformationCitation5.
Aspirin (aspH, ) is a well-known NSAID that differs from the rest homologs drugs in the mechanism of its actionCitation6,Citation7. Among NSAID’s, only aspirin inhibits with an irreversible manner and affects more the COX-1 variant than the COX-2 of the enzymeCitation5,Citation8. In contrast, and as it is shown in our previous study, aspirin alone does not interact with LOX-1Citation9.
Figure 1. (left) O-acetylsalicylic acid (aspirin) and (right) its complex Ag(tpp)3(asp).
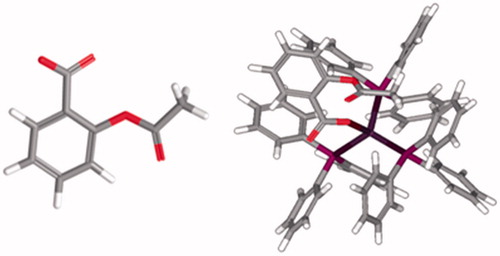
Most drugs (as aspirin) that are used today are purely organic compounds. The success of metallo-organic complexes, e.g. cisplatin in the treatment of many tumor types, initiated the interest in using metal complexes for therapeutic purposes. Within cells, metal complexes participate in reactions that cannot be achieved with conventional organic substances. Silver complexes have found biomedical uses in antibacterial actionCitation10. Recently, silver(I) complexes were synthesized and tested for their antitumor activityCitation11–14.
In order to investigate the relationship between inflammation and carcinogenesis in the development of new metallotherapeutics, the synthesis of silver(I) complexes with anti-inflammatory drugs, such as aspirin of formulae [Ag(tpp)3(asp)](dmf) (tpp = triphenylphosphine, aspH = o-acetylsalicylic acid and dmf = N,N-dimethylformamide) () is reported in previous workCitation15,Citation16. Three phosphorus atoms from tpp ligands and one deprotonated carboxylic oxygen atom from aspirin form a tetrahedral geometry around the Ag(I) ion. The complex was characterized by elemental analyses, spectroscopic techniques and X-ray crystallography at ambient conditionsCitation15,Citation16. Moreover, the complex was tested for its in vitro cytotoxic activity against leiomyosarcoma cancer cells (LMS), human breast adenocarcinoma cells (MCF-7) and normal human fetal lung fibroblasts cells (MRC-5). The results of biological tests showed that the complex is more active than cisplatin and less active against MRC-5 cells proliferationCitation15,Citation16. Also, the complex shows significantly stronger activity than its ligands (tpp and aspNa) and stronger activity (2-fold) than that of Ag(I) nitrate, a known antimicrobial agentCitation16. Due to the morphology of the cells after incubation with the complex, apoptosis was suggested. Indeed, the cells had undergone programmed cell death as it was confirmed by flow cytometry and DNA fragmentation. DNA-binding tests indicated that the ability of the complex might modify the activity of the cells by interacting strongly with their DNACitation15. The influence of the complex on the oxidation of linoleic acid by the enzyme LOX was studied in a wide concentration range. Specifically, the IC50 value was found for the complex to be 7.2 μM. Thus, the complex exhibits significantly higher inhibitory activity against LOX than cisplatin that has IC50 value equal to 65.9 μΜCitation15,Citation16. In these studies the congener complex Ag(tpp)2(sal) was evaluated showing IC50 = 2.3 μM.
In this study, the binding affinity of the [Ag(tpp)3(asp)](dmf) to enzyme LOX-1 is studied using STD 1H-NMR experiment (saturation transfer difference 1H NMR). The experimental results are complemented by in silico studies using docking and molecular dynamics (MD) calculations in an attempt to provide a plausible explanation.
Methods
NMR spectroscopy
NMR samples for STD experiments were prepared in 99.9% D2O buffer containing 20 mM Tris (98% D11), 7 mM (ND4)2SO4 (98% D8), 3.5 mM MgCl2 and 0.3 mM DTT (98% D10), pD 7.2. Ligands concentration was 0.4 mM and the protein concentration was 0.004 mM resulting in protein–ligand ratio of 1:100 [actual concentration of complex Ag(tpp)3(asp) was lower due to poor solubility]. Samples were subjected to STD experiments directly or after bath sonication for 30 min at 37 °C.
STD NMR experimentsCitation17 were recorded on Varian 800 MHz spectrometer (Palo Alto, CA) with spectral width of 8223 Hz, 8192 complex data points. Pulse sequences provided in Varian libraries of pulse programs were used. Relaxation delay was set to 10 s. Selective on-resonance irradiation frequency was set to 0.32 parts per million (ppm) with saturation time of 0.4 s. Selective saturation was achieved by a train of 50-ms Gauss-shaped pulses separated by a 1-ms delay. Off-resonance irradiation frequency for the reference spectrum was applied at 30 ppm. Water suppression was achieved with excitation sculptingCitation18,Citation19. Spectra were zero filled twice and line broadening function of 1 Hz was applied.
For the STD experiments on Varian NMR spectrometer, firstly, we obtain reference spectrum (off-resonance spectrum). The frequency of saturation is moved far away from the signals of the protein so it has no effect. Secondly, we obtain STD spectrum. This pulse sequence actually records two different spectra: off-resonance spectrum, as described above and on-resonance spectrum where the protein is saturated. These two spectra are automatically subtracted (this is done by the pulse sequence), so we can’t see individual spectra, but only STD spectrum, which is result of the subtraction.
Docking experiments
The computational studies were performed using the Molegro Virtual Docker software (MVD; version 5, Aarhus, Denmark) provided by Molegro ApSCitation20. The X-ray crystal structure of soybean LOX-1 (PDB entry code: 1F8N) was obtained from RCSB Protein Data Bank and it was in the apo form. All structures were prepared using Molegro’s Molecules and Protein Preparation Wizard. Proper bond assignments, bond orders, hybridization and charges were calculated by Molegro Virtual Docker softwareCitation21. Explicit hydrogen atoms were added, and their hydrogen bonding (HB) patterns were also determined by MVD.
In order to predict the potentially active regions of LOX-1, in which an inhibitor may bind, the grid-based cavity detection algorithmCitation22 was employed using the molecular surface feature. At least five potential cavities were detected, but the largest one has been selected because of the bulky complexes, and the corresponding volume was 558.592 ÅCitation3. Docking calculations were performed using the heuristic search algorithm MolDock SE (simplex evolution) in combination with the grid-based version of the MolDock Score [GRID]Citation22.
For each ligand, five poses were generated. The MolDock Score [Grid] is a grid-based scoring function which pre-calculates potential energy values on an evenly spaced cubic grid, in order to speed up calculations. A grid resolution of 0.30 Å was set to initiate the docking process, and the binding site on the protein was defined as extending in x, y and z directions around the selected cavity with a radius of 15 Å. For the pose generation, the default setting was applied (MolDock SE), namely a maximum of 1500 iterations combined with a population size of 50. These poses were built incrementally from their rigid root point. The pose generator tests a number of different torsion angles, rotations and translations, evaluates the affected part of the molecule and chooses the value, which results in the lowest energy contributionCitation20. If the generated pose has an energy below the predefined “energy threshold” (100.0, expressed in arbitrary units) it is accepted into the initial population for the “simplex evolution” algorithm. The “simplex evolution” algorithm performs a combined local/global search on the poses generated by the pose generator. The number of the maximum iterations of the simplex evolution algorithm (Nelder–Mead simplex minimization) was set to 300, while the neighbor distance factor (the factor, which determines how close the point of the initial simplex will be to the other randomly selected individuals in the population) was set to 1.0 (causes the initial simplex to span the neighbor points exactly)Citation20.
MD simulations
The structures of the two Ag complexes (aspirin and salicylic) bound to LOX-1 as obtained after docking were considered for further MD calculations. All-atom, unrestrained MD simulations in explicit water have been performed for the protein complexes using the SANDER program under the AMBER 11 software suiteCitation23. Another MD simulation for the apo form of LOX-1 has been carried out to distinguish differences in structural features from the bound proteins. Missing residues 1–5, 19–30 and 117–120 have been included in the crystal structure with the Maestro program of SchrödingerCitation24. Crystal water molecules were removed from the structure before adding all missing hydrogen atoms with the tLEaP module of AMBER. Atomic partial charges, bond lengths, bond angles, dihedral angles, force constants and van der Waals parameters for LOX-1 were represented by the AMBER ff99SB force fieldCitation25. Partial charges and force field parameters for each Ag complex were assigned as follows: first, missing hydrogen atoms were added with the program ReduceCitation26. Next, the geometry of Ag complexes was optimized with Gaussian 09 using the HF/6-31G* method for C, P, O and H atomsCitation27, while for Ag(I) an effective core potential of 28 electrons was usedCitation28. The non-bonded parameters for Ag(I), namely the van der Waals radius and the well-depth have been assigned the values rvdw = 1.72 Å and ɛvdw = 0.050 kcal mol−1, respectivelyCitation29. Finally, the RESP atomic partial chargesCitation30 for Ag complexes were derived with the ANTECHAMBER module, and the general AMBER GAFF force field was employed to obtain the force field parametersCitation31. Equilibrium distances, angles and dihedrals in Ag complexes have been defined according to crystal structure parameters provided by Poyraz et al.Citation16. Force constants have been estimated according to GAFF definitions and relevant work by the Merz groupCitation32. The atom of iron was modeled in a +2 oxidation state (rvdw = 1.20 Å, ɛvdw = 0.050 kcal mol−1), without bond restraints involving Fe and its ligand residues. Each system (Ag complexes: LOX-1 and apo LOX-1) was neutralized with tLEaP by adding 11 Na+ counterions, and was solvated with ∼29 000–30 000 water molecules. Explicit solvation has been represented by the TIP3P water modelCitation33 in truncated octahedral periodic boundary conditions, with a cutoff distance of 10 Å. The long-range electrostatic interactions have been calculated using the particle mesh Ewald (PME) methodCitation34. A four-step, energy minimization process with the combination of steepest descent and conjugate gradient methods was used to relieve undesired steric interactions that may appear after adding the missing residues, and to direct the systems toward an energetically favorable state. The first step kept the solute (either Ag complex: LOX-1 or apo LOX-1) constrained with a harmonic force constant of 500 kcal mol−1 Å−2, while the water molecules were allowed to relax. Next, the strength of the constraint was gradually reduced in two steps to 2 kcal mol−1 Å−2. Finally, the restraint was removed, to allow all atoms to move freely. Each of the four steps was realized in 5000 cycles with a cutoff of 20 Å. The next procedure involved the gentle heating of the complex under constant volume, over 100 ps with the gradual increase of the temperature from 0 to 300 K. The SHAKE algorithmCitation35 was applied to constrain all bond lengths involving hydrogen atoms to their equilibrium distance, thus a 2 fs time step was used. The Langevin thermostat with a collision frequency of 2.0 ps−1 kept the average temperature constantCitation36, and a restraint of 10 kcal mol−1 Å−2 was also applied to the solute. The same restraint was employed for the next 100 ps of equilibration in the NPT ensemble. A final equilibration stage of 100 ps was performed with all atoms of the system unrestrained. The subsequent MD calculations lasted for 60 ns. The SHAKE method and the Langevin thermostat, along with a 10 Å non-bonded cutoff were applied during the heating, equilibration and production MD periods. Further analysis (RMSD, atomic fluctuations and HB calculations) was performed on the resulting trajectories with the ptraj program under AMBER. Cutoffs of 3.5 Å for the donor–acceptor distance and of 120° for the donor–hydrogen–acceptor angle have been used to define HB interactions. Free energy calculations were performed for every frame (6000 in total) of the two Ag complex–LOX-1 trajectories with the molecular mechanics Poisson–Boltzmann surface area (MM-PBSA) method. Due to the extreme computational cost for the entropy calculation in large systems (such as LOX-1), we have calculated only the enthalpy contributions in order to estimate relative binding affinities. Details on the MM-PBSA methodology are provided in the “Supplementary material” section.
Atoms-in-molecules analysis of HB interactions
The ab initio analysis of a selected hydrogen bond was based on the theory of atoms-in-molecules (AIM), developed by BaderCitation37. The main concept of AIM involves the changes of the electron density distribution resulting from either bond or complex formation, and allows the study of the nature of the chemical bond and its strengthCitation37,Citation38. The topological features of the electron density are analyzed, thus providing useful information for the characterization of the type of bonding, through the determination of bond path and bond critical points (BCP)Citation39. A BCP is the point, where the gradient of density function, vanishesCitation39. The electron density, ρ(r), and its Laplacian [
] at the BCP can be used to characterize the type of interactions as covalent, ionic and hydrogen bondingCitation39. The ρ(r) is small (∼10−2) for both weak and strong HBsCitation40. For weak and medium HBs, the Laplacian is positive (0.024–0.139 a.u.)Citation40, while for a strong HB is negative. An increase in the HB strength causes an increase in the absolute value of the Laplacian of the electron density. The energy of the HB (EHB) is computed by using the following relationshipCitation41:
where, V(r)BCP is the local potential energy density at the BCP. The V(r)BCP is computed by employing the following formula:
where, Τ(r)BCP is the local kinetic energy density at the BCP.
The computation of the density function ρ(r) was done by employing the Gaussian quantum chemistry codeCitation27 at the HF level, by using the 6-31 + G* basis set for C, P, H, O and N, while for Ag an effective core potential of 28 electrons was usedCitation28. All the BCPs and the Laplacians were computed with the AIM2000 software (Büro für Innovative Software, Bielefeld, Germany)Citation42.
Results and discussion
1H NMR spectroscopy studies
The synthetic complex of [Ag(tpp)3(asp)](dmf) is soluble in DMSO and its stability was tested by UV–Vis spectra and conductivity measurements in the same solventCitation16. The 1H NMR spectrum of the complex is shown in . As it can be observed, all the peaks of the different components are visible. A small deacetylation of aspirin is attributed to the acidic character of silver (<3%). In Supplementary Table S1, the chemical shifts of the identified peaks are also shown.
Figure 2. 1H NMR spectrum of the complex Ag(tpp)3(asp) in DMSO-d6: (top) aliphatic region and (bottom) aromatic region.
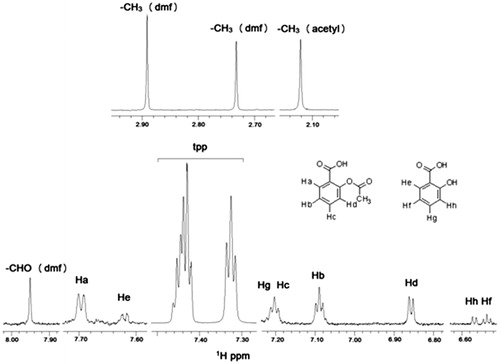
The stability of the complex of Ag with aspirin was also tested using HRMS in methanol (Supplementary Figure S1). Attempts to obtain decent spectra in DMSO and buffer Tris/D2O were not successful as the ions of the solvents overlapped the ions of the molecule.
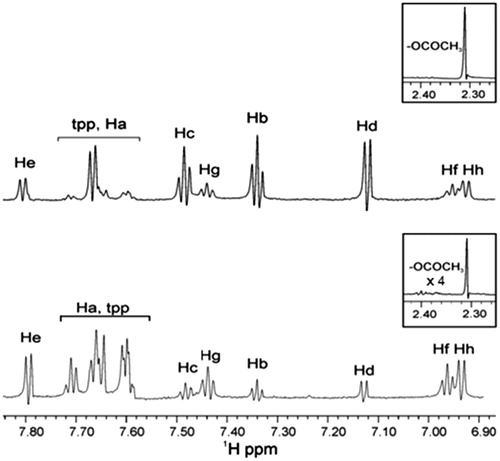
The 1H NMR experiments of complex Ag(tpp)3(asp) with LOX-1 in buffer Tris/D2O were performed without or after sonication (). The spectra clearly show the existence of two complexes due to the presence of pairs of peaks in the aromatic region. The second complex is with the salicylic acid that is produced after the hydrolysis of aspirin. Without sonication, the peaks attributed to tpp segment are barely shown in the spectrum, as they are buried into Ha peak. This is an awkward result and we attributed it to the low solubility of the complex in the Tris buffer containing LOX-1. To improve the solubility of the complex, we applied sonication. As it can be observed, the tpp peaks are higher in intensity but, the approximate ratio of the two complexes Ag(tpp)3(asp):Ag(tpp)3(salicylic acid) is lower ∼1:3 (∼3:1 without sonication). This difference in the ratio proves that sonication accelerates the deacetylation of aspirin complex to salicylic complex. The binding of the two complexes of Ag with LOX was investigated by STD 1H NMR experiment. In Table S2, the chemical shifts of the identified peaks are also shown.
Figure 3. 1H NMR spectrum of soybean LOX-1 with complex Ag(tpp)3(asp) in Tris/D2O: (top) without sonication and (bottom) after sonication.
STD 1H NMR experiments
Without sonication
From the aromatic region, it can be observed that only peaks of the aromatic ring attributed to the complex of salicylic acid are seen in the STD 1H NMR experiment, thus indicating that this is localized in the active site the soybean LOX-1 (), while in the reference spectrum, two complexes of Ag are observed [ratio based on the integration of the aromatic peaks is Ag(tpp)3(asp):Ag(tpp)3(salicylic acid) ∼1:3]. Although the two reference spectra [ (top) and (top)] show identical peaks, the ratio of the complexes Ag(tpp)3(asp):Ag(tpp)3(salicylic acid) differs. The reference spectrum of is obtained using a longer time with respect to the sample preparation shown in . The ratio of the complexes Ag(tpp)3(asp):Ag(tpp)3(salicylic acid) is lower in signifying that even in the non-sonicated samples there is a dynamic interconversion of the complex Ag(tpp)3(asp) into Ag(tpp)3(salicylic acid).
Figure 4. STD experiment of 1H NMR of soybean LOX-1 with complex Ag(tpp)3(asp) in Tris/D2O (without sonication): the reference NMR spectrum of aliphatic and aromatic region (top), and the STD NMR spectrum of aromatic region (bottom).
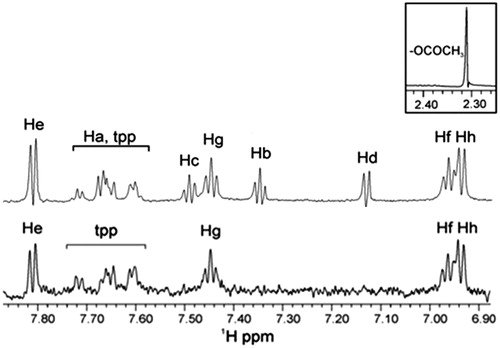
The measured intensities of the peaks of the STD spectrum were calculated and the relative degree of saturation of protons was estimated. The Hf, Hg and Hh protons of the aromatic ring have almost the same STD intensities, between 86 and 100%. The proton He STD intensity was lower, ∼70%, while lower intensities corresponding to the protons of tpp (30–60%) were observed.
These results are in agreement with the ones published in previous workCitation16. However, in this publicationCitation16 we have not interpreted the results in terms of the existence of the two complexes Ag(tpp)3(asp) and Ag(tpp)3(salicylic acid).
STD 1H NMR experiments
After sonication
Similarly, the spectroscopic data show that only the peaks of aromatic protons of complex of salicylic acid are barely appeared (). In the reference spectrum, two complexes of Ag are observed. The ratio of the two complexes as revealed by the integration of peaks was found to be aspirin:salicylic acid ∼1:10. Although the two reference spectra [ (bottom) and (top)] show identical peaks, the ratio of the complexes Ag(tpp)3(asp):Ag(tpp)3(salicylic acid) differs. The reference spectrum of is obtained using a longer time in comparison to the sample preparation shown in . The ratio of the complexes Ag(tpp)3(asp):Ag(tpp)3(salicylic acid) is lower in due to the dynamic interconversion of the complex Ag(tpp)3(asp) into Ag(tpp)3(salicylic acid) with time.
Figure 5. STD 1H NMR of soybean LOX-1 with complex Ag(tpp)3(asp) in Tris/D2O (after sonication): the reference NMR spectrum of aliphatic and aromatic regions (top), and on-resonance STD NMR spectrum of aromatic region (bottom).
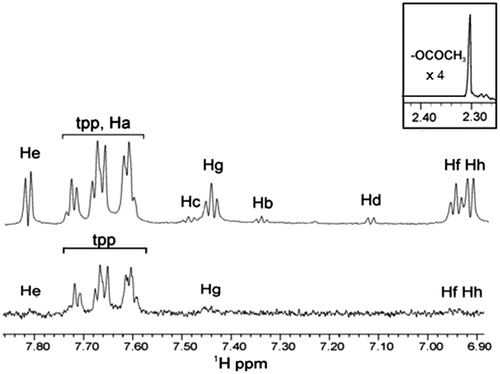
The peaks attributed to tpp gave much stronger signals compared with the STD aromatic protons of salicylic acid. Additionally, the multiplicity of peaks in the signals of salicylic acid is unclear. The measured intensities of the peaks of the spectrum are calculated and the relative degree of saturation of protons is estimated. The protons of tpp have STD intensities between 50 and 100%. The lowest intensities corresponding to He, Hg, Hf and Hh protons of the aromatic ring have almost the same STD intensities, ranging between 4 and 15% (the largest percentage is for Hf).
The obtained results show that when sonication is applied, the formed complex Ag(tpp)3(salicylic acid) expells the (lower in ratio) complex Ag(tpp)3(aspirin) from the active site cavity.
Docking studies
In order to further investigate the binding of Ag complexes at the active site of LOX, in silico docking studies were performed. Docking experiments at LOX-1 were performed by MVD software according to the protocol described in materials and methods.
In this study, the crystallographic structure of the soybean LOX-1 in the apo form, published by Tomchick et al.Citation43 (PDB ID: 1F8N) was used. The high resolution of the X-ray structure was 1.40 Å.
It is reported in the literature that the active center of LOX-1 consists of an iron atom with its four or six ligandsCitation44,Citation45. Boyington et al.Citation44 published the crystallographic data of LOX-1 in high resolution (2.60 Å), and suggested that in the active center iron is attached to four ligands, namely three histidines (His499, His504 and His690) and the carboxyl end of Ile839Citation44. Three years later, Minor et al.Citation45 concluded that the center of the iron has six ligands: the four were identical to those previously reported and Fe was also attached to a water molecule and Asn694Citation45.
The oxygen atom of the carboxamide group of Asn694 is properly oriented toward the iron atom, but in relatively long distance (Fe–O694 = 3.05 Å); therefore, it was concluded that asparagine serves as a rather weak ligandCitation45. In the recently reported crystallographic structure with entry code 1F8N, the Fe–O694 distance was measured more accurately to be 2.87 ÅCitation43.
Therefore, the recent crystallographic studies of LOX-1 converge on the view that the center of iron consists of six ligands, with the amino acid of Asn694 not tightly attached. It is also noted that the atom of iron is in the oxidation state +2, contrary to other LOXs where Fe is in oxidation state +3.
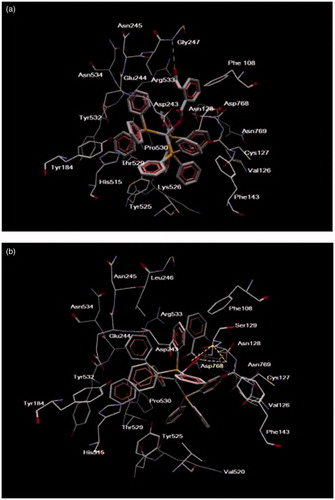
The Ag complexes were docked in the observed cavity of LOX-1. The silver complexes are accommodated in the cavity, as they are depicted by their MolDock score. In the case of the complex of aspirin, the MolDock score was −140.894 (expressed in arbitrary units) and it is characterized by HB with Gly247, and numerous steric interactions with amino acids Asn128, Val126, Asn769, Arg533, Thr529, Lys526, His515, Tyr532, Asn534, Leu246, Phe108, Cys127, Ser129, Asp243, Glu244, Val520, Tyr525 and Phe143 (). In the case of the complex of salicylic acid, the MolDock Score was −153.648 (expressed in arbitrary units) and there are three hydrogen bonds with Asn128 and Asp768, and numerous steric interactions with several amino acids, such as Val126, Asn769, Arg533, Thr529, His515 and Tyr184 (). The docking results suggested that the complex that is preferred to bind to LOX-1 is the complex of salicylic acid. In addition, the aromatic ring of aspirin points toward the solvent, thus providing a plausible explanation for the STD results. More details on the stability of the complexes and their dynamic properties are given in the session of MD.
Figure 6. Ligand map (dotted lines: hydrogen bonds) of the complex of (a) aspirin and (b) salicylic acid in the cavity of LOX-1 (1F8N).
MD analysis
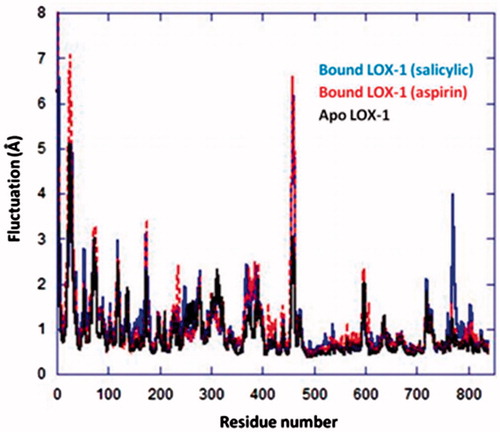
The MD simulations of each Ag complex bound to LOX-1 () started from the conformations obtained by the docking calculations on 1F8N. Initial conformational changes of the proteins were observed during the first 10 ns of each simulation, which eventually resulted in relatively stable trajectories. The stabilization of the systems is indicated by the structural deviations during the last part of the simulations (, red curves). A Cα-based RMSD calculation with respect to the initial structure of LOX-1 yielded an average value of 2.3 Å (for LOX-1 bound to aspirin complex), thus suggesting that the simulated protein resembles the crystal structure (); however, it is noted that the protein acquired a stable conformation during the last 10 ns of the run. A similar behavior was observed for the apo form of LOX-1 (average RMSD = 2.5 Å), although it presented higher structural deviations than the bound protein, especially during the first half of the simulation. This is not surprising, since it is well expected for the unbound form to be relatively flexible due to the absence of a ligand. The salicylic complex initially induced greater conformational changes and flexibility to the structure of LOX-1 than the aspirin complex, with RMSD values reaching up to 3.3 Å (); it was however observed that during the last 15 ns of the simulation, the protein was eventually stabilized to conformations resembling its structure with aspirin. The above findings are further supported by the atomic fluctuation calculations on the three systems (): the overall greater flexibility of LOX-1 when bound to salicylic complex (compared to aspirin complex) may be attributed to the increased mobility of residues, such as His53, and especially Pro770 (, blue curve). Despite differences in flexibility of individual residues, it is noted that both bound systems may be considered stable overall, with the exceptions of the flexible termini and Leu459. Additionally, it was observed that the two Ag complexes were stabilized inside the cavity of LOX-1 after ∼35 ns (, blue) and remained practically unchangeable throughout the second part of the simulations.
Figure 7. The Ag complex inside binding cavity of LOX-1. The atom of Fe(II) in the active site of cavity 1 is also displayed.
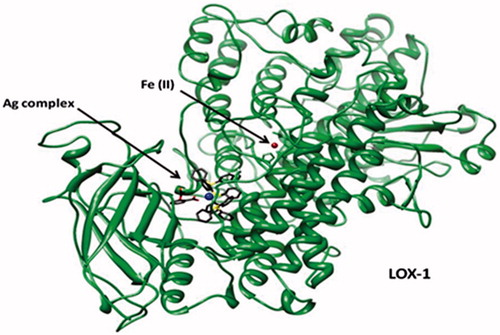
Figure 8. RMSD of LOX-1 bound to (a) Ag complex (salicylic) and (b) Ag complex (aspirin). RMSD values for the apo form of LOX-1 and for the two Ag complexes in the bound-protein systems are also shown. Calculations were initiated from each structure obtained after docking and overlapped on the same structure. For LOX-1 and Ag complexes, superposition involved Cα atoms and all heavy atoms, respectively.
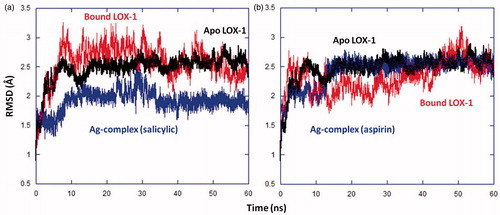
Figure 9. Cα atomic fluctuations for all residues of LOX-1 in its apo, and Ag complex-bound forms.
Hydrogen bonding analysis on the MD trajectory of the protein complex with salicylic revealed the existence of a broad HB network, which associates the Ag complex with residues Arg767, Asp768 and Asn769 in LOX-1. Multiple HB interactions involving oxygen atoms (of salicylic) stabilize the Ag complex inside LOX-1 in a compact structure. In particular, it was indicated that an HB between the salicylic group of Ag complex and Arg767 is prevalent during the first half of the MD run. Interestingly, after the first 30 ns of the simulation, additional interactions between the Ag complex and Asp768 (also observed in the docking scheme)/Asn769 were developed. Therefore, the stabilization of the system after the first 35 ns as depicted by the RMS plots in may be justified by this HB enhancement. Hydrogen bonds as percentage occurrence are summarized in , and the aforementioned interactions of the silver complex are highlighted in . In the case of aspirin complex, the HB calculations indicated that only one major interaction exists between the complex and LOX-1, namely, between O3 (aspirin) and Asn128 ( and ). The Ag complex–Asn128 hydrogen bond was also identified by the docking analysis. Interestingly, this interaction was prevalent only during the second half of the simulation, while in the first 30 ns the aspirin complex formed several weak interactions with Asn128 and Cys127 ().
Figure 10. Hydrogen bonds between Ag complex and residues in binding cavity of LOX-1 for (a) the aspirin complex and (b) the salicylic complex. Interactions are represented by dotted lines, and involve oxygen atoms (red) in the aspirin/salicylic moiety of Ag complex. Ag(I) and P atoms are shown in blue and yellow, respectively; for simplicity, the hydrogen atoms of Ag complex are not shown.
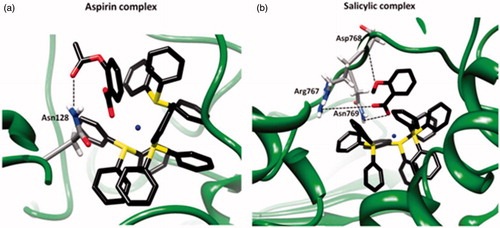
Table 1. Hydrogen bonding interactions between Ag complexes and LOX-1.
Previous studies on the performance of the MM-PBSA method have suggested that for the estimation of relative binding affinities concerning structurally similar compounds, one may omit the computationally expensive entropy calculation without worsening the qualitative predictionsCitation46–48. Additionally, it has been shown that the use of normal mode analysis tends to overestimate significantly the entropy loss for ligand bindingCitation49. Taking into account the above, and since LOX-1 is a particularly large system to perform entropy calculations in a time-efficient way, we have decided to focus our analysis only on the relative binding energies as expressed by the enthalpy calculations.
Thus, MM-PBSA calculations were performed for the two Ag complex–LOX-1 systems, and provided further support to the experimental observations and the theoretical (docking and HB) calculations, since they showed that the binding of salicylic complex is over 9 kcal mol−1 more favorable than the Ag-aspirin–LOX-1 complex formation (). Enthalpy decomposition indicated that the major contributions to binding for both complexes originate from van der Waals and non-polar interactions. Importantly, the results of also suggested that differences in the van der Waals and (total) electrostatic contributions between the two complexes are the main reasons for the reduced binding enthalpy in aspirin complex.
Table 2. MM-PBSA energy calculations* for the two Ag complexes in LOX-1.
Analysis of the prevalent HB interaction in the salicylic–LOX-1 complex
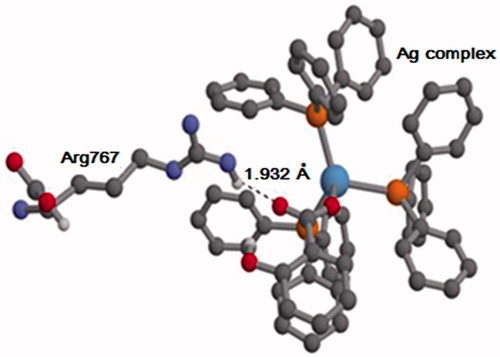
Employing the AIM method, we calculated the strength of the HB interaction between Arg767 and Ag complex (salicylic), which was observed in the MD simulations (). The average geometry, as obtained from the MD trajectory, was used for the AIM calculations. The AIM analysis showed that the energy of the HB is −7.59 kcal/mol, which is a typical value for HB interactions. At the BCP, the Laplacian [] of the electron density is 0.025 a.u. and the energy density (HBCP) is 0.0004 a.u. According to referenceCitation50, the sign of
and HBCP indicates the strength of the HB. Since both
and HBCP are positive, the HB is relatively weak. It is however noted that since the AIM calculation was performed only on one (average) snapshot of the trajectory, the strength of the interaction may vary during the simulation. Furthermore, the HB analysis () strongly suggested that the Ag complex–Arg767 interaction is prevalent throughout the MD run ().
Figure 11. The geometry of Arg767 and the Ag complex (salicylic), as obtained from the MD trajectory. For clarity, only some of the hydrogen atoms are shown. O, C, N, P, Ag and H atoms are shown in red, gray, violet, orange, blue and white, respectively.
Conclusions
As a continuation of our previous work on metallotherapeutics, we have in the present study examined the molecular interactions of the synthetic anti-inflammatory complex of Ag(I), [Ag(tpp)3(asp)](dmf) [tpp = triphenylphosphine, aspH = aspirin and dmf = N,N-dimethylformamide], with LOX-1. To provide direct evidence for the binding of the above complex to LOX-1, we have used STD 1H NMR experiments. The low solubility of the complex has driven us to complement our experiments using sonication, in an attempt to increase the solubility of Ag(I) complex in the enzyme environment. As it is shown in our previous study, aspirin alone does not interact with LOX-1Citation9. Interestingly, Ag(tpp)3(asp) complex was not stable, and gradually the aspirin segment of the complex was converted to salicylic acid. This intriguing result allowed us to study the details of the interaction of both complexes in equilibrium with LOX-1. The complex of salicylic acid appeared to govern the interactions with LOX-1. These experimental results motivated us to study the detailed interactions of the two complexes with LOX-1 using MD calculations. MD results confirmed the binding of both complexes to LOX-1, and in agreement with experimental data, the binding with salicylic acid was stronger in comparison to the one with aspirin. Ab initio calculations were further applied to confirm the stability of hydrogen bonds. The obtained results may be related to the different biological activities of Ag(tpp)2(sal) (IC50 = 2.3 μM) and Ag(tpp)3(asp) (IC50 = 7.2 μM)Citation16.
Declaration of interest
The authors report no conflicts of interest.
G.L. acknowledges the funding provided by the European Commission for the FP7-REGPOT-2009-1 Project “ARCADE” (Grant Agreement No. 245866) and for the Project “NanoPUZZLES” (Grant Agreement No. NMP-SL-2012-309837). The NMR studies were supported by EN-FIST Center of Excellence (Dunajska 156, SI-1000 Ljubljana, Slovenia) and Ministry of Higher Education, Science and Technology of Slovenia.
Supplemental Material.pdf
Download PDF (60.5 KB)Acknowledgements
The authors would like to thank Thomsen R. for providing Molegro software.
References
- Vane JR, Flower RJ, Botting RM. History of aspirin and its mechanism of action. Stroke 1990;21:12–23
- Taketo M. Cyclooxygenase-2 inhibitors in tumorigenesis (Part I). J Natl Cancer Inst 1998;90:1529–36
- Taketo M. Cyclooxygenase-2 inhibitors in tumorigenesis (Part II). J Natl Cancer Inst 1998;90:1609–20
- Janne PA, Mayer RJ. Chemoprevention of colorectal cancer. N Engl J Med 2000;342:1960–8
- Zha S, Yegnasubramanian V, Nelsona WG, et al. Cyclooxygenases in cancer: progress and perspective. Cancer Lett 2004;215:1–20
- Siegel MI, McConnell RT, Porter NA, et al. Aspirin-like drugs inhibit arachidonic acid metabolism via lipoxygenase and cyclo-oxygenase in rat neutrophils from carrageenan pleural exudates. Biochem Biophys Res Commun 1980;92:688–95
- Paajanen H, Männistö J, Uotila P. Aspirin inhibits arachidonic acid metabolism via lipoxygenase and cyclo-oxygenase in hamster isolated lungs. Prostaglandins 1982;23:731–41
- Ingo O. Modulation of the biological properties of aspirin by formation of bioorganometallic derivative. Angew Chem Int Ed 2009;48:1160–3
- Vrontaki E, Leonis G, Papadopoulos MG, et al. Comparative binding effects of aspirin and anti-inflammatory Cu complex in the active site of LOX-1. J Chem Inf Model 2012;52:3293–301
- Rosenkranz HS, Rosenkranz S. Silver sulfadiazine: interaction with isolated deoxyribonucleic acid. Antimicrob Agents Chemother 1972;2:373–9
- Kyros L, Kourkoumelis N, Kubicki M, et al. Structural properties, cytotoxicity, and anti-inflammatory activity of silver(I) complexes with Tris(p-tolyl)phosphine and 5-chloro-2-mercaptobenzothiazole. Bioinorg Chem Appl 2010;2010:386860. doi: 10.1155/2010/386860
- Zartilas S, Hadjikakou SK, Hadjiliadis N, et al. Tetrameric 1:1 and monomeric 1:3 complexes of silver(I) halides with tri(p-tolyl)-phosphine: a structural and biological study. Inorg Chim Acta 2009;362:1003–10
- Zachariadis PC, Hadjikakou SK, Hadjiliadis N, et al. Synthesis, characterization and in vitro study of the cytostatic and antiviral activity of new polymeric silver (I) complexes with ribbon structures derived from the conjugated heterocyclic thioamide 2-mercapto-3,4,5,6-tetrahydropyrimidine. Eur J Inorg Chem 2004;7:1420–6
- Hadjikakou SK, Ozturk II, Xanthopoulou MN, et al. Synthesis, structural characterization and biological study of new organotin(IV), silver(I) and antimony(III) complexes with thioamides. J Inorg Biochem 2008;102:1007–15
- Banti CN, Giannoulis AD, Kourkoumelis N, et al. Mixed ligand–silver(I) complexes with anti-inflammatory agents which can bind to lipoxygenase and calf-thymus DNA, modulating their function and inducing apoptosis. Metallomics 2012;4:545–60
- Poyraz M, Banti CN, Kourkoumelis N, et al. Synthesis, structural characterization and biological studies of novel mixed ligand Ag(I) complexes with triphenylphosphine and aspirin or salicylic acid. Inorg Chim Acta 2011;375:114–21
- Mayer M, Meyer B. Group epitope mapping by saturation transfer difference NMR to identify segments of a ligand in direct contact with a protein receptor. J Am Chem Soc 2001;123:6108–17
- Hwang TL, Shaka AJ. Water suppression that works. Excitation sculpting using arbitrary wave-forms and pulsed-field gradients. J Magn Reson Ser A 1995;112:275–9
- Dalvit C. Efficient multiple-solvent suppression for the study of the interactions of organic solvents with biomolecules. J Biomol NMR 1998;11:437–44
- Molegro. Molegro virtual docker: user manual, MVD 2011.5.0. Aarhus, Denmark: Molegro ApS; 2012
- Thomsen R, Christensen MH. MolDock: a new technique for high-accuracy molecular docking. J Med Chem 2006;49:3315–21
- Ul-Haq Z, Khan W. Molecular and structural determinants of adamantly susceptibility to HLA-DRs allelic variants: an in silico approach to understand the mechanism of MLEs. J Comput Aided Mol Des 2011;25:81–101
- (a) Case DA, Cheatham T, Darden T, et al. The amber biomolecular simulation programs. J Comput Chem 2005;26:1668–88. (b) Case DA, Darden TA, Cheatham III TE, et al. Amber 11: user manual. University of California, San Francisco, CA; 2010
- Maestro. Maestro version 9.1: user manual. Schrödinger, LLC: New York, NY; 2008
- Hornak V, Abel R, Okur A, et al. Comparison of multiple Amber force fields and development of improved protein backbone parameters. Proteins 2006;65:712–25
- Word JM, Lovell SC, Richardson JS, Richardson DC. Asparagine and glutamine: using hydrogen atom contacts in the choice of side-chain amide orientation. J Mol Biol 1999;285:1735–47
- Frisch MJ, Trucks GW, Schlegel HB, et al. Gaussian 09, revision A.02. Gaussian, Inc., Wallingford, CT; 2009
- (a) Bergner A, Dolg M, Küchle W, et al. Ab-initio energy-adjusted pseudopotentials for elements of groups 13–17. Mol Phys 1993;80:1431–41. (b) Kaupp M, Schleyer PvR, Stoll H, Preuss H. Pseudopotential approaches to Ca, Sr, and Ba hydrides. Why are some alkaline earth MX2 compounds bent? J Chem Phys 1991;94:1360–6. (c) Dolg M, Stoll H, Preuss H, Pitzer RM. Relativistic and correlation effects for element 105 (hahnium, Ha): a comparative study of M and MO (M = Nb, Ta, Ha) using energy-adjusted ab initio pseudopotentials. J Phys Chem 1993;97:5852–9
- Bondi A. Van der Waals volumes and radii. J Phys Chem 1964;68:441–51
- Woods RJ, Khalil M, Pell W, et al. Derivation of net atomic charges from molecular electrostatic potentials. J Comput Chem 1990;11:297–310
- Wang J, Wolf RM, Caldwell JW, et al. Development and testing of a general amber force field. J Comput Chem 2004;25:1157–74
- Op’t Holt B, Merz KM Jr. Insights into Cu(I) exchange in HAH1 using quantum mechanical and molecular simulations. Biochemistry 2007;46:8816–26
- Jorgensen WL, Madura JD, Impey RW, Klein ML. Comparison of simple potential functions for simulationg liquid water. J Chem Phys 1983;79:926–35
- Darden T, York D, Pedersen L. Particle mesh Ewald: an N.Log(N) method for Ewald sums in large systems. J Chem Phys 1993;98:10089–92
- Ryckaert JP, Ciccotti G, Berendsen HJC. Numerical integration of the cartesian equations of motion of a system with constraints: molecular dynamics of n-alkanes. J Comput Phys 1977;23:327–41
- Izaguirre JA, Catarello DP, Wozniak JM, Skeel RD. Langevin stabilization of molecular dynamics. J Chem Phys 2001;114:2090–8
- Bader RFW. Atoms in molecules: a quantum theory. Oxford: Clarendon Press; 1990
- Bader RFW. A bond path: a universal indicator of bonded interactions. J Phys Chem A 1998;102:7314–23
- Parthasarathi R, Subramanian V. Characterization of hydrogen bonding: from van der Waals interactios to covalency. Unfied picture of hydrogen bonding based on electron density topography analysis (Chapter 1). In: Grabowski SJ, ed. Hydrogen bonding: new insights. Dordrecht, the Netherlands: Springer; 2006:1–50
- Grabowski SJ. Hydrogen bonding strength – measures based on geometric and topological parameters. J Phys Org Chem 2004;17:18–31
- Espinosa E, Molins E, Lecomte C. Hydrogen bond strengths revealed by topological analyses of experimentally observed electron densities. Chem Phys Lett 1998;285:170–3
- Biegler-König F, Schönbohm J, Bayles D. AIM2000 – a program to analyze and visualize atoms in molecules. J Comp Chem 2001; 22:545–59
- Tomchick DR, Phan P, Cymborowski M, et al. Structural and functional characterization of second-coordination sphere mutants of soybean lipoxygenase-1. Biochemistry 2001;40:7509–17
- Boyington JC, Gaffney BJ, Amzel LM. The three-dimensional structure of an arachidonic acid 15-lipoxygenase. Science 1993;260:1482–6
- Minor W, Steczko J, Stec B, et al. Crystal structure of soybean lipoxygenase L-1 at 1.4 Å resolution. Biochemistry 1996;35:10687–701
- Wang JM, Morin P, Wang W, Kollman PA. Use of MMPBSA in reproducing the binding free energies to HIV-1 RT of TIBO derivatives and predicting the binding mode to HIV-1 RT of efavirenz by docking and MM-PBSA. J Am Chem Soc 2001;123:5221–30
- Zhuang S, Zou J, Jiang Y, et al. Some insights into the stereochemistry of inhibition of macrophage migration inhibitory factor with 2-fluoro-p-hydroxycinnamate and its analogues from molecular dynamics simulations. J Med Chem 2005;48:7208–14
- Rafi SB, Cui G, Song K, et al. Insight through molecular mechanics Poisson-Boltzmann surface area calculations into the binding affinity of triclosan and three analogues for FabI, the E. coli enoyl reductase. J Med Chem 2006;49:4574–80
- Steinbrecher T, Labahn A. Towards accurate free energy calculations in ligand protein-binding studies. Curr Med Chem 2010;17:767–85
- Rozas I, Alkorta I, Elguero J. Behavior of ylides containing N, O, and C atoms as hydrogen-bond acceptors. J Am Chem Soc 2000;122:11154–61
Supplementary material available online Supplemental material