
Abstract
Inosine 5′-monophosphate dehydrogenase (IMPDH) is important molecular target for potential anticancer, antiviral, antibacterial and immunosuppressive agents. A lot of compounds were obtained to establish their activity toward this enzyme, and to improve therapeutic properties of IMPDH inhibitors used as the drugs. Some of the recently reported analogs exhibited promising results during in vitro and in vivo examinations in comparison to substances applied in clinic. In this review, we describe synthesis and biological activity evaluations of the newly designed IMPDH inhibitors.
Introduction
Inosine 5′-monophosphate dehydrogenase [IMPDH; IMP-NAD oxidoreductase, Enzyme Classification (EC) 1.2.1.14] catalyzes a metabolic branch point reaction in purine synthesis and has been an area of intellectual convergence in biological and medicinal chemistryCitation1. This enzyme is responsible for the catalysis of NAD-dependent oxidation of inosine monophosphate (IMP) as a substrate to xanthosine 5′-monophosphate (XMP), which is used in the de novo biosynthesis of guanine. XMP is converted to guanosine monophosphate (GMP) with glutamine as an amino donor. The de novo pathway of purine synthesis is considered as a more significant source of nucleotides for B and T cells than the salvage pathway, in order that IMPDH inhibitors are potential immunosuppressive agentsCitation2,Citation3. In this review, we focus mainly on synthesis and biological activity of human IMPDH inhibitors.
The inhibition of IMPDH induces a reduction in guanine nucleotide pools that produce an interruption of DNA and RNA synthesisCitation4, a decline in intracellular signalingCitation5, and down-regulation of c-myc and Ki-ras oncogenes in vitroCitation6–8 and in leukemic cells of patients treated with inhibitorsCitation8. IMPDH inhibition results in apoptosis in both neoplastic cell lines and activated T-lymphocytesCitation6,Citation9.
The role of IMPDH as a chemotherapeutic target was further advanced by the discovery, in 1990 by Natsumeda et al.Citation10, that the enzyme exists as two isoforms: labeled type I and type IICitation10. These isoforms are of identical size and share 84% sequence identity. However, the type I “housekeeping” isoform is constitutively expressed in both normal and neoplastic cellsCitation11, while type II expression is preferentially up-regulated in human neoplastic cell linesCitation10,Citation12.
The type II isoform is also a target for immunosuppression. The role of this isoform in immunosuppression has been elucidated by a series of mice knockout models described by Gu et al.Citation13. While the type II enzyme is the major isoform in normal human T-lymphocytes, these cells appear to induce both type I and type II enzymes when stimulated by mitogenCitation13. Recently, hIMPDH-I has been identified as anti-angiogenic drug target and mycophenolic acid (MPA) was found to block tumor-induced angiogenesis in vivoCitation14. Therefore, in this review newly designed compounds are also discussed, which indicated interesting selectivity between both isofoms of hIMPDH.
Enzymes of purine nucleotide biosynthesis pathways are attractive targets for the design of potential anticancer, immunosuppressant, antiviral and antibacterial agentsCitation14–20. There were obtained many of the compounds that inhibit IMPDH activity, but only a few have been used in medicine.
IMPDH inhibitors used in clinic
IMPDH is recognized as a validated target for several major therapeutic areasCitation21. A number of potent and selective inhibitors of IMPDH are used in clinic (). Ribavirin (1) is a prodrug of corresponding 5′-monophosphate, which as a competitive inhibitor interacts with the IMP domain of IMPDHCitation22,Citation23.
Figure 1. Currently used drugs that target IMPDH.
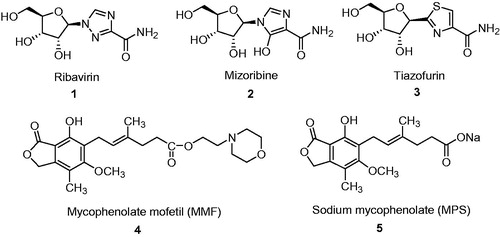
Ribavirin (1) (1-β-D-ribofuranosyl-1,2,4-triazole-3-carboxamide, Virazole®, Valeant Pharmaceuticals, Laval, Quebec, Canada) is a broad spectrum antiviral agent inhibiting the replication of a wide range of DNA and RNA viruses in vitro and in vivo, the most sensitive being HSV-1, HSV-2, vaccinia, influenza, parainfluenza, measles, rhino, respiratory syncytial and some tumor virusesCitation24–26. It is also active against various retroviruses including HIV and is used for treatment of AIDS patientsCitation27. The combination of interferon-α and ribavirin was proved to be successful in therapy for patients with chronic hepatitis C infectionCitation28.
Mizoribine (2) (MZB) is an imidazole nucleoside and an immunosuppressive agent. The immunosuppressive effect of MZB has been reported to be due to the inhibition of DNA synthesis in the S-phase of the cell cycle. After phosphorylation, mizoribine-5′-monophosphate (MZB-P) inhibits GMP synthesis by antagonistic blocking of IMPDH and GMP synthetase in the pathway from IMP to GMP in the purine synthesis system. MZB was found to suppress both humoral and cellular immunity by selective inhibition of lymphocyte proliferation, which led to its development as an immunosuppressive agentCitation29. MZB (Bredinin®, Asahi Kasei Pharma Corporation, Tokyo, Japan) is used in Japan as an immunosuppressant similarly to mycophenolate mofetil (MMF). Noteworthy, MZB-P (Ki value of 10 nM) inhibits stronger IMPDH than ribavirin monophosphate (Ki value of 250 nM), which can be due to additional hydrogen bond from hydroxyl group in imidazole ringCitation30.
The third, tiazofurin (3) (TR) is metabolically converted into thiazole-4-carboxamide adenine dinucleotide (TAD) and interacts with NAD site of enzyme. Noteworthy, TR is the only IMPDH inhibitor applied clinically in cancer treatment. It was approved as an orphan drug in case of patients in blast crisis of chronic myelogenous leukemia (CML)Citation31.
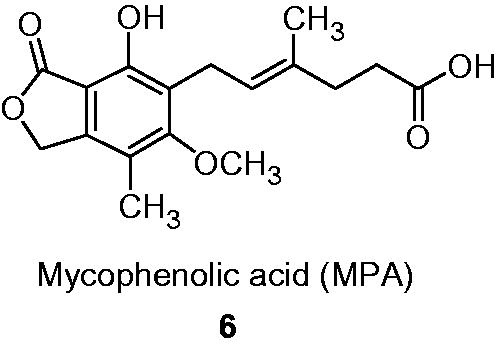
The last ones: mycophenolate mofetil (4) (2-morfolinoethyl, MMF, CellCept, F. Hoffmann-La Roche Ltd, Basel, Switzerland) and sodium mycophenolate (5) (MPS, Myfortic, Novartis Pharmaceuticals Corporation) are used in clinic as immunosuppressants in prophylaxis of organ transplant rejection. The former is a prodrug of MPA 6 (), which binds to the N sub-site of IMPDH (an uncompetitive inhibitor) and is one of the most potent inhibitors of human IMPDH (hIMPDH), with a slight higher potency against isoform II (Ki in the range of 7–14 nM) over isoform I of the enzyme (Ki in the range of 11–33 nM)Citation32. The both proteins consist of 514 amino acids and possess molecular mass 56 kDaCitation1,Citation30,Citation32–34. According to molecular modeling studies, functional groups in MPA interact in binding site with adequate amino acids in IMPDH: carboxylic group—Ser 276, phenol group—Gln 441, Thr 333, lactone—Thr 333 and Gly 326. Trans configuration of the double bond in side chain is important for the proper arrangement of the interacting with Ser 276 carboxylic group and maintenance of biological activity. In van der Waals contacts are involved: Asn 303, Arg 322, Asp 274, Gly 415 and Met 414Citation30,Citation32. However, none of the mentioned agents shows significant selectivity against IMPDH type II isoform.
Figure 2. Structure of MPA 6.
Despite of the progress in development of clinically applied IMPDH inhibitors, new drugs with improved therapeutic properties are still desired. As a result, new compounds are designed to evaluate their potential activity. It includes structural modifications of known IMPDH inhibitors and investigations of new substances, which were synthetically obtained or isolated from natural sourcesCitation30–32,Citation35,Citation36.
New potent IMPDH inhibitors derived from MPA
During last years derivatives of MPA are one of the most interesting group of IMPDH inhibitors. In literature we can find a lot of new analogs, unfortunately only a few of them exhibit interesting activityCitation3,Citation35,Citation36. Moreover, immunosuppressive agents cause side effect like dose-limiting gastrointestinal (GI) toxicity upon MMF or MPA oral administration. Yang et al.Citation37 synthesized 35 isobenzofuran derivatives and assessed their structure–activity relationship within T-cell proliferation and IMPDH type II inhibition. The synthesis of the three series of isobenzofuran analogs 8, 10 and 11 is depicted in Scheme 1. The starting material for all target compounds 8, 10 and 11 was 5-aminoisobenzofuran-1(3H)-one 7.
Scheme 1. The synthesis of isobenzofuran analogs 8, 10 and 11Citation37.
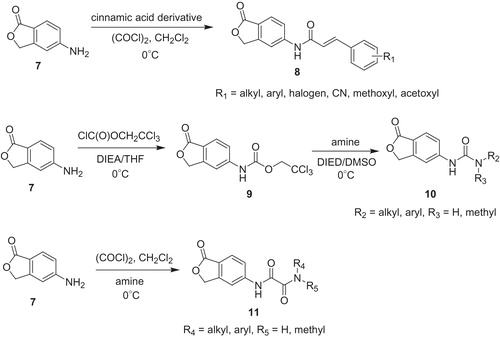
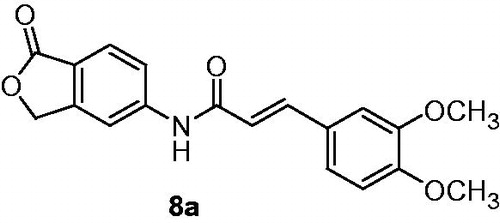
Treatment of 7 with generated in situ derivative of cinnamic acid chloride provided α,β-unsaturated amides 8. Among these compounds, the highest activity toward T-cells (the proliferative response of mouse splenocytes to concanavalin A) revealed analogs possessing fluor, methoxyl and acetoxyl as R1 substituent at position 3 (in case of methoxyl also disubstituted in 3 and 4 and 8a; ) of phenyl ring of cinnamic acid moiety. T-cells proliferation test gave results IC50 ca. 0.03 µM, whereas MPA occurred to be less active (IC50 value of 0.28 µM). In contrast to that, electron-withdrawing CN group at 4th position, similarly to bulky substituents at 2nd position, decreased observed activity.
Figure 3. Isobenzofuran derivative 8a active in vitro toward both T-cells proliferation and IMPDH type inhibitionCitation37.
The first stage of urea analogs 10 synthesis involved reaction of (7) with 2,2,2-trichloroethylchloroformate. Then, carbamate 9 underwent nuclephilic substitution at acyl carbon atom with appropriate amine. In general, urea analogs 10 were less active than α,β-unsaturated amides 8, and secondary amine derivative of 10 (R2 = phenyl, R3 = methyl) showed loss of activity (IC50 > 100 µM) probably due to hydrogen bonding donor role of amide group. On the other hand, primary amine derivatives, especially bearing substituted phenyl ring (R2 = 2-methylphenyl, R3 = H) exhibited considerable activity (IC50 value of 0.90 µM). Interestingly, size of the halogen substituent at para position improved inhibitory properties too (R2 = 4-fluorophenyl, R3 = H, IC50 value of 4.19 µM and R2 = 4-iodophenyl, R3 = H, IC50 value of 1.37 µM). Furthermore, aromatic amine derivatives (R2 = aryl, R3 = H) indicated better activity if compared with alkyl ones (R2 = cyclohexyl or n-propyl, R3 = H). On the basis of these results it was concluded, that benzene-ring conjugated system is crucial for retain or increase activity of urea analogs 10.
Different structure–activity relationship was observed in case of diamide analogs 10. These compounds were obtained in the reaction of 7 with oxalyl chloride and adequate amine. Analog hold aliphatic amine (R4 = tert-butyl, R5 = H, IC50 value of 3.40 µM) occurred to be more active like derivative having aromatic amine (R4 = phenyl, R5 = H, IC50 value of 36.07 µM). However, presence of substituent being hydrogen bond donor in aromatic ring enhanced potency significantly (R4 = 4-hydroxyphenyl, R5 = H, IC50 value of 5.03 µM, R4 = 4-methoxyphenyl, R5 = H, IC50 value of 153.33 µM). The highest activity was observed in case p-fluorophenyl derivative (R4 = 4-fluorophenyl, R5 = H, IC50 value of 0.57 µM).
Subsequently, isobenzofuran analogs 8, 10 and 11 were evaluated as IMPDH type II inhibitors. The highest potency was observed in case of 3,4-dimethoxyphenyl cinnamic acid derivative 8a (). Its inhibition toward IMPDH type II (IC50 value of 2.1 µM) was slightly lower than MPA (IC50 value of 1.5 µM).
Among IMPDH inhibitors can be found also acridone derivativesCitation31. Nitroacridines are known as a high cytotoxic compounds and in search of potent MPA derivatives, Malachowska-Ugarte et al.Citation38 designed conjugates of MPA and nitroacridine/acridone 13 and 15 (Scheme 2). The amide bond was formed between free carboxylic group of MPA 6 and primary amine group of 9-(ω-aminoalkyl)amino-1-nitroacridines 12, and 1-[(ω-aminoalkyl)-4-nitro-9(10H)]acridones 14 in the presence of optimized coupling reagent.
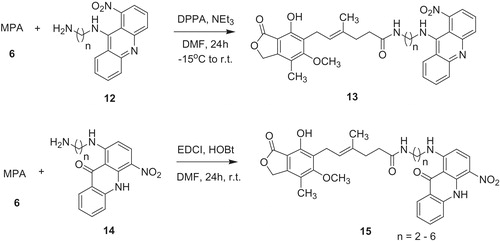
Conjugates 13 and 15 were evaluated as inhibitors of leukemia cell lines (Jurkat, Molt-4, HL-60, CCRF and L1210) and human peripheral blood mononuclear cells (PBMC). These compounds occurred to be IMPDH inhibitors in the experiments based on addition of GMP to Jurkat cell lineCitation2. Their activity depend both on length of the diamine linker and type of heterocycle. Basically, acridine analogs 13 indicated higher potency than acridones 15 and parent MPA 6. However, Yurkat cells were surprisingly more sensitive toward acridones 15 (IC50 in MTT test value of <0.0008 µM), than acridines 13 (IC50 in the range of 0.17–0.86 µM) or MPA 6 (IC50 value of 0.193 µM). Conjugates 13 and 15 exhibited intermediate activities between respective starting acridine 12 or acridone 14 and MPA 6, so that the observed effects were rather additive, not synergistic. On the other hand, it could be advantageous for optimization cytotoxic properties for in vivo investigations.
In literature are reported some structural modifications of MPA based on polar groups at the end of its side chain. They provided some derivatives indicating significant activity if comparison with parent MPA, which is in good agreement with molecular modeling studiesCitation30,Citation32,Citation35.
Iwaszkiewicz-Grzes et al.Citation39 synthesized amino acids derivatives of MPA 17 and 18 (Scheme 3). First, MPA 6 produced with amino acid methyl esters 16 in the presence of EDCI as coupling reagent analogs 17. Subsequently, esters 17 were hydrolyzed to amino acid derivatives 18 possessing free carboxylic groups. Compounds 17 and 18 occurred to be IMPDH inhibitors and their activity was evaluated against lymphoid cell line Jurkat and activated PBMC as in vitro model of immunosuppression.
Scheme 3. Synthesis of amino acid MPA derivative 17 and 18Citation39.
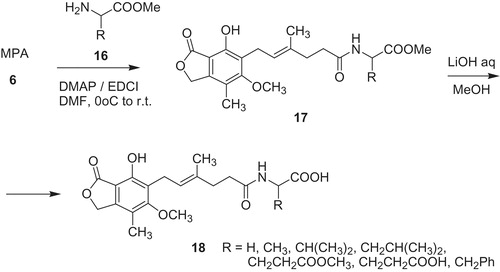
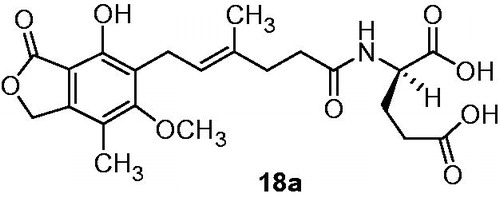
According to obtained results, recovering of free carboxylic group-enhanced activity. Furthermore, inhibitory properties depended on amino acid attached (R substituent) and configuration at chiral center. The most promising outcome in this series of compounds was received in case of d-glutamic acid analog 18a (). Antiproliferation activity toward Jurkat cell line of 18a expressed as EC50 (antiproliferation test based on incorporation [3H]thymidine) was 0.45 µM and selectivity index SI defined as SI = EC50/IC50 (MTT test) value of 47.1, whereas MPA 6 gave EC50 value of 9.45 µM, and SI value of 2.99.
Figure 4. N-mycophenoyl-d-glutamic acid 18aCitation39.
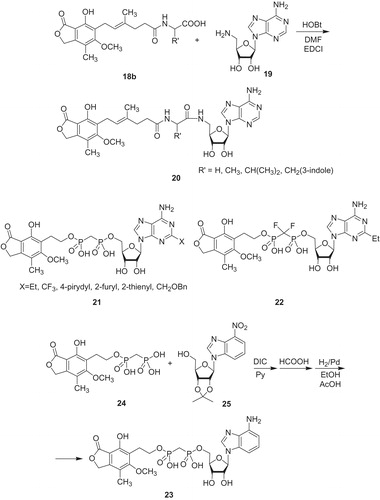
Taking the fact, that IMPDH is a nicotinamide adenine dinucleotide (NAD)-dependent enzyme, Pankiewicz et al.Citation40 designed several classes of NAD-based IMPDH inhibitors. For instance, MPA amino acid derivatives 18b were coupled with 5′-aminoadenosine 19 to respective diamides 20 (Scheme 4). In this series D-valine derivative of 20 revealed nanomolar potency toward IMPDH-I (Ki value of 3 nM) and IMPDH-II (Ki value of 88 nM). In the same communicate were reported also new mycophenolic adenine dinucleotide (MAD) analogs 21 being substituted at position 2. High activities, both toward IMPDH-I (Ki value of 0.6 nM), IMPDH-II (Ki value of 14 nM), and several cancer cell lines were observed in case of 2-(4-pyridyl) MAD derivative of 21. The authors also modified linker between adenosine and MPA derived units. For instance, 2-ethyl substituted difluoromethylene MAD analog 22 gave Ki value of 0.6 nM with IMPDH-I, Ki value of 24 nM with IMPDH-II and IC50 value of 0.4 nM against K562. In search of selective IMPDH-II inhibitor, the same scientific group designed 4-aminobenzimidazole analog 23. This compound was prepared via coupling of bis(phosponate) 24 with O-isopropylidene-4-nitrobenzimidazole 25, followed by deprotection of 2′and 3′-hydroxyl groups and hydrogenolysis. The inhibitory toward IMPDH-I reached Ki = 491 nM, and was considerably better in case of IMPDH-II: Ki = 165 nM. These results are coincident with analysis of the crystal structure of IMPDH-II–NAD complex, whereas adenine ring interacts with two amino acids: His 253 and Phe 282 possessing aromatic systems. In contrast to that, IMPDH-I provides only Tyr 282. As a result, replacement of adenine moiety by 4-aminobenzimidazole enables stronger stacking interactions and better selectivity to IMPDH-II.
Scheme 4. NAD-based IMPDH inhibitorsCitation40.
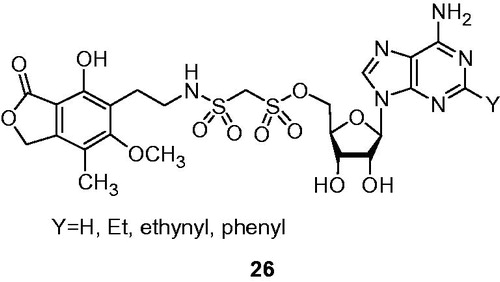
Mycophenolic adenine bis(sulfonoamides)Citation41 (MABSs) () 26 possess isosteric linker to the methylenebis(phosphonates) 21. The tetrahedral sulfur atoms provide similar geometry and charge delocalization if compared with the naturally occurring pyrophosphate linkage. These series of compounds revealed comparable to MAD analogs 21 IMPDH inhibition, together with an observable selectivity to isoform II of the enzyme.
Figure 5. Methylenebis(sulfonamide) analogs 21 of mycophenolic adenine methylenebis(phosphonate)s 26Citation41.
Dual inhibitor of histone deacetylase (HDAC, zinc-dependent enzyme) and IMPDH, e.g. hydroxamic acid derivative of MPA 27 (), are considered as potential anticancer agents, which could provide also the selectivity toward IMPDH-II over IMPDH-ICitation29,Citation40. Sunohara et al.Citation42 focused on synthesis of 10 new derivatives of MPA 28, 29 possessing efficient zinc-binding moieties, as potential dual inhibitors for HDAC and IMPDH-II that would improve transplantation therapy and chemotherapy.
Figure 6. MPA derivatives as dual IMPDH and HDAC inhibitors.
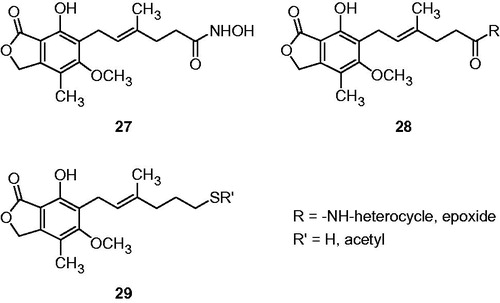
Authors proposed to combine MPA at C-6′ position with groups of heterocyclic amide, thiols, and investigate their structure–activity relationships for inhibitory activity against human IMPDH (in vitro), inductive activity on erythroid differentiation (K562 cells), and inhibitory effect on K562 cell proliferation. Base assumption in modeling of the new structure of derivatives was that zinc-dependent enzyme can be molecular target in cancer treatment. Another designed zinc-binding moiety was epoxide similarly to potential anticancer agent Trapoxin.
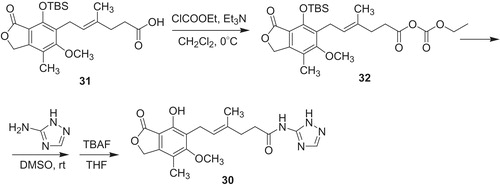
The most potent compound against K562 cells occurred to be N-(2,3,5-triazoyl)mycophenolic amide 30 (IC50 = 0.48 μM, Scheme 5), and was comparable to MPA 6 (IC50 = 0.19 μM). Its inhibitory activities were IC50 = 0.105 μM toward IMPDH-I, IC50 = 0.098 μM against IMPDH-II, which gave selectivity index IMPDH-I/IMPDH-II value of 1.1. To compare, MPA 6 indicated IC50 = 0.019 μM toward IMPDH-I, IC50 = 0.012 μM against IMPDH-II and selectivity index IMPDH-I/IMPDH-II was 1.6. Since amine group of 3-amino-1,2,4-triazole is a poor nucleophile, synthesis of N-(2,3,5-triazoyl)mycophenolic amide 30 required use of MPA possessing protected phenol group. Tert-butyldimethylsilyl ether of MPA 31 was treated with ethyl chloroformate to produce mixed anhydride 32, followed by reaction with 3-amino-1,2,4-triazole. Finally, deprotection of phenol group with tetrabutylamonium fluoride yielded amide 30.
Scheme 5. Synthesis of N-(2,3,5-triazoyl)mycophenolic amide 30Citation42.
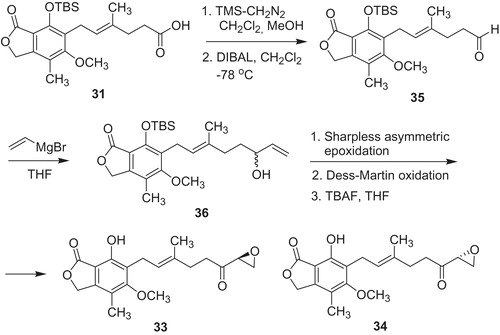
Tert-butyldimethylsilyl ether of MPA 31 was also used in synthetic pathway of epoxyketones 33 and 34 (Scheme 6). Carboxylic group of 30 was methylated, subsequently respective methyl ester underwent reduction by DIBAL to aldehyde 35. Then, nucleophilic addition of vinyl magnesium bromide to 35 produced racemic alcohol 36. Alcohol (R)-36 provided epoxyketone 33 and (S)-36 led to epoxyketone 34. The both products 33 and 34 revealed similar to MPA selectivity index IMPDH-I/IMPDH-II, 1.7 and 1.8, respectivelyCitation42.
Scheme 6. Synthesis of enantiomeric mycophenolic epoxyketones 33 and 34Citation42.
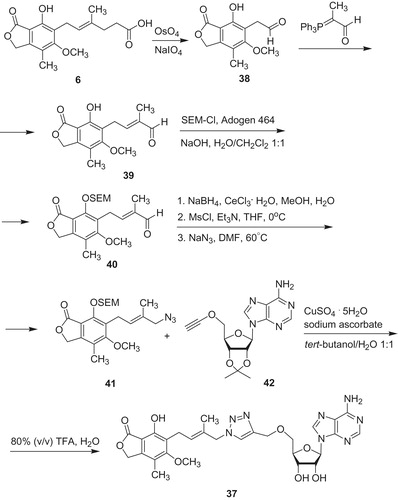
Chen et al.Citation43 designed triazole-linked MPA derivatives, which revealed high activity in vitro toward IMPDH. Moreover, compound 37 (Scheme 7) occurred to be not only IMPDH inhibitor (Ki value of 0.070 µM against type I and Ki value of 0.044 µM against type II), but also toward Mycobacterium tuberculosis IMPDH (Ki value of 1.5–2.2 µM) whereas MPA 6 gave 0.033 µM (Type I), 0.007 µM (Type II) and 62 µM (mtIMPDH), respectively. The target compound 37 was obtained from MPA 6. First, MPA 6 was oxidized to aldehyde 38, which reacted with 2-(triphenylphosphoranylidene)propionaldehyde to produce 39. In the next step, phenol group was protected under phase transfer catalysis with 2-(trimethylsilyl)etoxymethyl chloride (SEM-Cl). Subsequently, aldehyde 40 underwent reduction followed by mesylation and convertion to adequate azide 41. Then, terminal alkyne 42 participated in catalyzed by Cu(I) 1,3-dipolar cycloaddition to azide 41. Both 2-(trimethylsilyl)etoxymethyl (SEM) ether and isopropylidene protecting groups were removed with aqueous TFA to yield 37.
Scheme 7. Synthesis of triazole-linked IMPDH inhibitor 37Citation43.
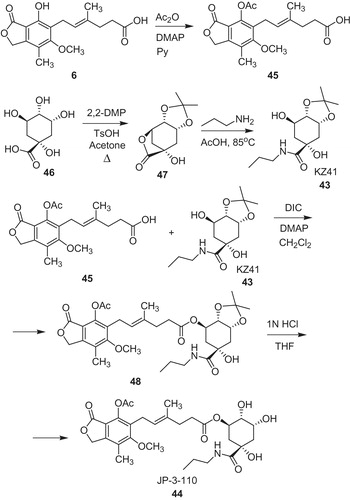
The other types of conjugates were developed by Wu H. and co-workersCitation44. In search of less toxic immunosuppressant to prevent rejection in human islet transplantation, MPA was coupled with quinic acid derivative 43 (KZ41) as anti-inflammatory component (Scheme 8). The obtained ester 44 (JP-3-110) revealed similar immunosuppressive activity in vitro to MPA 6; however, it can provide safer drug for human islet transplantation due to diminishing of pro-apoptotic effect of 6. The synthesis of conjugate 45 required use of MPA with protected phenol group (45). Quinic acid (46) was converted to 3,4-O-isopropylidene-1,5-quinic lactone (47) in the reaction with 2,2-dimethoxypropane (2,2-DMP), p-toluenesulfonic acid in boiling acetone. Then, lactone (47) provided amide 43 upon treatment with n-propyl amine in acetic acid. Subsequently, MPA derivative 45 was coupled with quinic acid analog 43 (KZ41) in the presence of N,N′-diisopropylcarbodiimide (DIC) to ester 48, followed by removing of protecting acetonide group. The structure of the obtained conjugate 44 (JP-3-110) will be optimized toward better solubility and stability to improve its pharmacological properties.
Scheme 8. Synthesis of conjugate of MPA and quinic acid derivative 44Citation44.
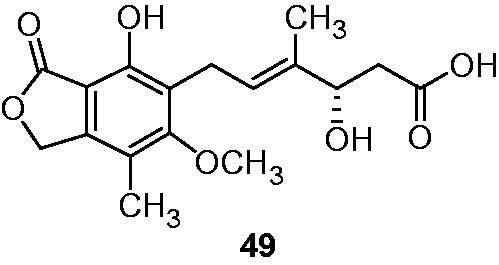
Three new derivatives of MPA were isolated and identified from marine-derived Penilillium fungus (sp. SOF07, from South China Sea) by Chen et al.Citation45. These compounds exhibited moderate activity in the range IC50 value of 6.43–73.24 µM toward IMPDH-II and IC50 value of 2.46 µM to >30 µM against mouse splenocyte proliferation. MPA 6 and 4′-hydroxymycophenolic acid (49) () were also isolated from mentioned sample. Their inhibitions were IC50 value of 0.63 µM, 1.79 µM toward IMPDH-II, and IC50 value of 0.32 µM, 1.10 µM against mouse splenocyte proliferation, respectively.
Figure 7. Structure of 4′-hydroxymycophenolic acid 49 isolated from Penilillium fungus (sp. SOF07)Citation45.
IMPDH inhibitors including oxazol moiety
According to molecular modeling studies, the C7 phenol group in MPA 6 provides very significant interaction between MPA 6 molecule and IMPDH and is crucial for drug activityCitation30. On the other hand, the C7 phenol group undergoes glucuronidation in vivo, which considerably limits therapeutic properties. To overcome this metabolic drawback, derivatives possessing other polar group or heterocyclic moieties were designedCitation35,Citation46,Citation47. Chen et al.Citation48 developed suberoylanilide hydroxamic acid (50) (SAHA, potent HDACs inhibitor) derivative 51 (Scheme 9), as dual HDAC and IMPDH inhibitor. Apart from zinc-binding hydroxamic acid group, compound 51 hold also phenyloxazol moiety possessing ortho methoxy substituent as a part interacting with IMPDH.
Scheme 9. Synthesis of dual HDAC and IMPDH inhibitor 51Citation48.
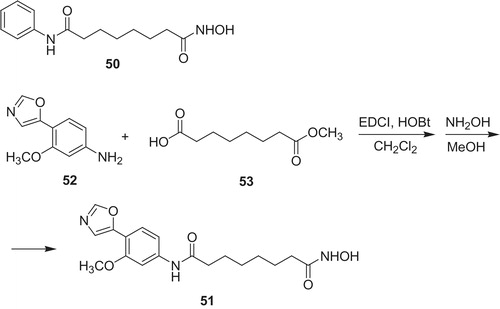
Synthesis of 51 included coupling of 3-methoxy-4-(5-oxazolyl)phenylamine (52) with suberic acid monomethyl ester (53), followed by selective nucleophilic substitution at acyl carbon atom of methyl ester with hydroxylamine. The product 51 revealed promising activity. Its inhibition toward IMPDH was Ki value of 5.0 µM (Type I) and 1.7 µM (Type II) in comparison to MPA: 0.04 µM (Type I) and 0.01 µM (Type II), respectively. Noteworthy, potency of 51 against HDAC (IC50 value of 0.06 µM) was higher than in case of MAHA (19), SAHA (50): 5.0 and 0.41 µM, respectively. Additionally, activity of compound 50 toward K562 (IC50 value of 0.29 µM) occurred to be better like of MPA 6, MAHA (27) and SAHA (50): 7.7, 4.8 and 0.75 µM, respectively.
Merimepodib (54) (Scheme 10) is a potent IMPDH inhibitor (Ki in the range of 6–10 nM for the both hIMPDHs) and drug candidate for hepatitis C developed by Vertex Pharmaceuticals (South Boston, MA)Citation31,Citation49,Citation50. There was completed a Phase 2b clinical trial and synthesis of 54 optimized to commercial route. The final step depicted in Scheme 10 was coupling of 3-methoxy-4-(5-oxazolyl)aniline 52 with carbamate 55 in the presence of diisopropylethylamine as a base.
Scheme 10. Synthesis of merimepodib 54Citation49.
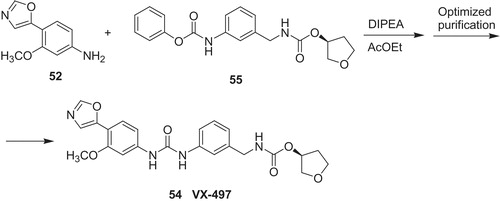
Several synthetic pathways toward 3-methoxy-4-(5-oxazolyl)aniline 52 were reported in the chemical literatureCitation51,Citation52. The starting material, 2-methoxy-4-nitrotoluene 56 was oxidized to respective 1,1-diacetate 57, followed by hydrolysis to aldehyde 58 (Scheme 11). Then, 58 was converted with TosMIC (toluenesulfonylmethyl isocyanide) to oxazolyl derivative 59. Subsequently, hydrogenolysisCitation52 of 59 provided amine 52.
Scheme 11. Synthesis of 3-methoxy-4-(5-oxazolyl)aniline 52Citation52.
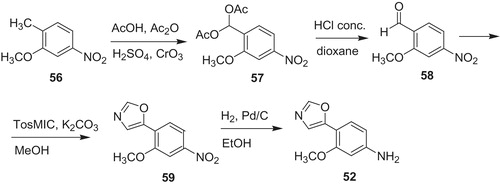
Carbamate 55 was obtained from (S)-3-hydroxytetrahydrofuran (60) (Scheme 12). The reaction of the alcohol 60 with phosgene (or triphosgene) gave crude chloroformate 61, which underwent nucleophilic substitution at acyl carbon atom with amine generated from hydrochloride 62. Next, hydrogenolysis of nitro compound 63 to amine 64, followed by treatment with phenyl chloroformateCitation53 led to carbamate 55.
Scheme 12. Synthesis of carbamate 55Citation53.
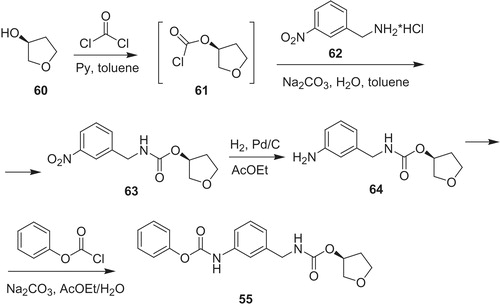
Research group from Bristol–Myers Squibb developed IMPDH inhibitors, in which urea linker in VX-497 (54) was replaced by heterocyclesCitation53,Citation54. In the series of 2-aminooxazoles, BMS-337197 (65) (Scheme 13) revealed excellent immunosuppressive activity in mouse model of arthritis. In the key synthetic stage isothiocyanate (66) reacts with β-keto azide (67) in the presence to triphenylphosphine to form 2-(N-aryl)-1,3-oxazole (68). Then, acetate in 68 was hydrolyzed, and obtained hydroxyl group was converted to adequate methanesulfonate. Next, nucleophilic substitution with morpholine provided desired product 65.
Scheme 13. Synthesis of BMS-337197 65Citation51,Citation52.
In search of new chemotherapeutics against drug resistance, Pankiewicz et al. developed dual inhibitors of IMPDH (isoform I and II), and HDAC based on cinnamic hydroxamic acid (CHA) and N-(3-metoxy-4-(5-oxazolyl)phenyl]amino (MOA) scaffold possessing urea (69) or diamide (70) linkerCitation55 (). MOA structural unit was derived from potent IMPDH inhibitors [VX-497 (54), BMS-337197], whereas CHA moieties are known as zinc-dependent HDAC inhibitors. To compare with VX-148 [IMPDH inhibitor derived from N-(4-cyano-3-methoxyphenyl)amine (CMA)], Pankiewicz et al. reported also urea analogs, in which 5-oxazolyl ring was replaced by –CN substituent.
Figure 8. Urea and diamide derivatives of cinnamic hydroxamic acid and N-(3-metoxy-4-(5-oxazolyl)phenyl]amine 69 and 70Citation55.
![Figure 8. Urea and diamide derivatives of cinnamic hydroxamic acid and N-(3-metoxy-4-(5-oxazolyl)phenyl]amine 69 and 70Citation55.](/cms/asset/c05853e8-5760-4b3d-91a6-caafc4f5d7c9/ienz_a_951349_f0008_b.jpg)
One of the most potent compounds occurred to be urea derivative 69a (Scheme 14). This compound was active toward both isofoms IMPDH (Ki value of 0.30 µM against Type I and Ki value of 0.25 µM against Type II), HDAC (IC50 value of 0.55 µM) and also K562 cell line.
Scheme 14. Synthesis of urea derivative 69aCitation53.
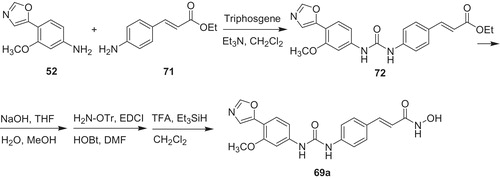
The synthetic pathway includes reaction of 3-methoxy-4-(5-oxazolyl)aniline (52) with ethyl 4-aminocinnamate (71) in the presence of triphosgene to form urea (72). Then, ethyl ester in 72 underwent alkaline hydrolysis, followed by coupling of respective carboxylic acid with O-tritylhydroxylamine. Subsequently, deprotection with triethylsilane gave the product 69a.
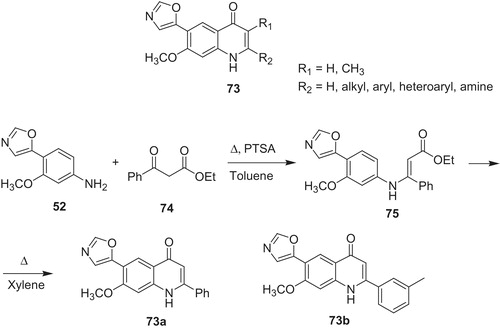
Scientists from Bristol–Myers Squibb developed series of quinolone-based IMPDH inhibitors 73 (Scheme 15) to establish their structure–activity relationship (SAR). In case of derivatives 73, urea or diamide linkage is replaced by bicyclic quinolone scaffoldCitation56. The authors rationalized, that quinoline NH group would provide hydrogen bond with the carboxylate of Asp 274 of IMPDH. Additionally, quinolone carbonyl could be involved as hydrogen bond acceptor in interaction with Gln 441 of IMPDH. Some of the developed compounds revealed comparable or better activity in vitro than MPA 6. For instance, analog 73a gave IC50 = 0.099 μM toward IMPDH-I and IC50 = 0.008 μM against IMPDH-II, (selectivity IMPDH-I/IMPDH-II value of 12), where MPA 6 exhibited 0.055, 0.014 and 3.9, respectively. The synthesis of 73a included reaction of amine 52 with β-ketoester (74) in the presence of p-toluenesulfonic acid. In the next stage, ethyl ester 75 underwent cyclization under heating to product 73a. The best selectivity IMPDH-I/IMPDH-II (value of 30) was obtained in case 3-methyl derivative 73b, which showed IC50 = 0.15 μM toward IMPDH-I and IC50 = 0.005 μM against IMPDH-IICitation54. However, this high selectivity was neither explained nor confirmed at higher concentrations of the enzymeCitation32.
Scheme 15. Structure and synthesis of quinolone derivatives 73Citation56.
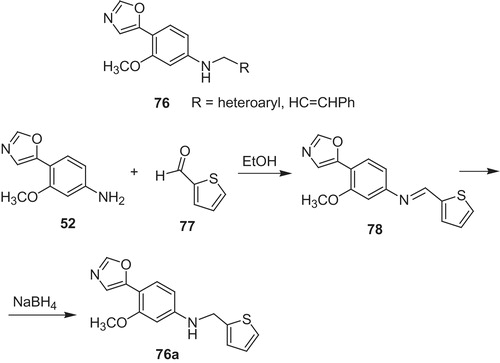
Zhong et al.Citation57 obtained and characterized novel (5-oxazolyl)phenyl amines (76) (Scheme 16) as potential antiviral agents for HCV and CVB. Since, there is currently no vaccine against hepatitis C virus compounds being active toward HCV are highly desired. One of the most active derivative occurred to be N-[3-methoxy-4-(5-oxazolyl)phenyl]-(2-thienylmethyl) amine (76a), which gave CC50 (50% cytotoxic concentration) 51.32 µM, IC50 = 0.28 µM (SI = 183.3) against HCV. The product 76a was received in the reaction of amine 52 with 2-thiophenecarboxaldehyde (77), followed by reduction of imine 78.
Scheme 16. Synthesis of (5-oxazolyl)phenyl amines 76Citation57.
Miscellaneous
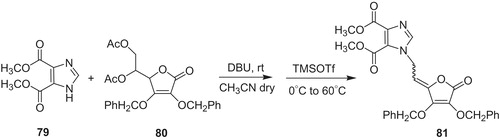
Wittine et al.Citation58 developed 1,2,4-triazole and imidazole derivatives of l-ascorbic acid as potential agents toward tumor and hepatitis C virus. The synthesis of target compounds was based on Vorbrüggen condensation and biological results were referred to ribavirin (1) (). The most promising derivative occurred to be imidazole analog 81 (Scheme 17), which was obtained from 4,5-dimethylimidazole carboxylate (79) and 2,3-di-O-benzyl-5,6-di-O-acetyl-l-ascorbic acid (80) in the presence of 1,8-diazobicyclo[5.4.0]undec-7-en (DBU) and trimethylsilyl trifluoromethanesulfonate (TMSOTf). The product 81 revealed the highest activity against hepatitis C virus replication in the Huh 5-2 replication system and comparable to ribavirin (1) in vitro antitumor activity. For example, cytostatic activity of 81 against human T-cell acute lymphoblastic leukemia (CEM) reached IC50 value of 10 µM, whereas ribavirin (1) gave IC50 value of 63 µM. Moreover, imidazole derivative (81) exhibited non-toxic effect on human diploid fibroblasts and initial experiments with L1210 and CEM cell lines proved, which its mechanism of action includes IMPDH inhibition.
Scheme 17. Synthesis of imidazole derivative of l-ascorbic acid 81Citation58.
Nakanishi et al.Citation59 optimized lead compound from Astellas library to novel and highly potent IMPDH inhibitor () named N-((4-fluorophenyl)(1-methyl-1H-imidazol-2-yl)methyl)-2-methyl-3-(1,2,4-thiadiazol-5-yl)-1H-indole-6-carboxamide, AS2643361 (82). Inhibitory in vitro activities of 82 and MPA 6 against two types of human IMPDH were similar. However, IC50 received in case of 82 was 71 nM toward IMPDH-II in the presence of 50% human serum and was eight times better than for MPA 6 (540 nM). These results suggested, that AS2643361 (82) binds weaker than MPA 6 to serum proteins.
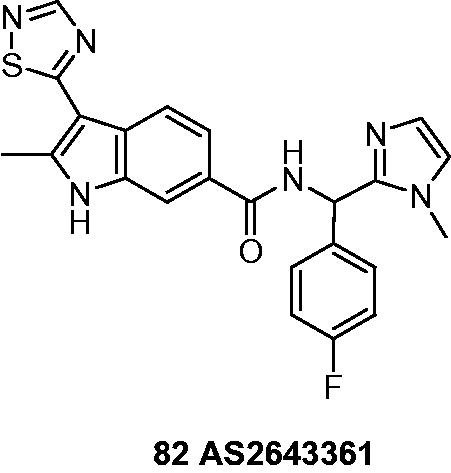
Further tests concentrated on cellular activity and included concanavalin A-stimulated T cell proliferation, lipopolysaccharide-stimulated B cell proliferation, mixed lymphocyte reaction and in vitro IgM production. On this field AS2643361 showed five- to six-fold higher potency over MPA 6. Evaluations in vivo were performed on rats’ models of cardiac transplantation. Important fact is that AS2643361 (82) shows four- to eight-fold lower GI toxicity than MMF 4 and is promising candidate for acute and chronic rejection in transplant medicine.
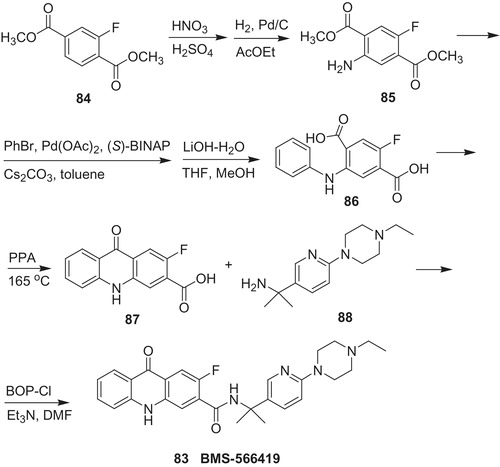
In the course of SAR of acridone derivatives, Watterson et al.Citation60 discovered potent IMPDH-II inhibitor BMS-566419 (83) (Scheme 18). Compound 83 gave ca. three-fold better result than MMF 4 () in its therapeutic index with respect to GI toxicity in rat adjuvant arthritis model. The synthesis started from dimethyl 2-fluoroterephtalate 84, which underwent nitration followed by reduction to amine 85. Then, amine 85 was used in Buchwald–Hartwig condensation with phenyl bromide in the presence of palladium(II) acetate, (S)-(-)-2,2′-bis(diphenylphosphino)-1,1′-binaphtyl, cesium carbonate. Subsequently, ester groups in 2-fluoro-5-phenylaminoterephtalic acid dimethyl ester were hydrolized to 2-fluoro-5-phenylaminoterephtalic acid (86). In the next stage, cyclization of 86 under heating with polyphosphoric acid provided 2-fluoro-9-oxo-9,10-dihydroacridine-3-carboxylic acid (87), which was coupled in the presence of N,N-bis[2-oxo-3-oxazolidinyl]phosphorodiamidic chloride (BOP-Cl) as a coupling reagent with 2-(6-(4-ethylpiperazin-1-yl)pyridine-3-yl)propan-2-amine (87) to produce BMS-566419 (83).
Scheme 18. Synthesis of BMS-566419 84Citation60.
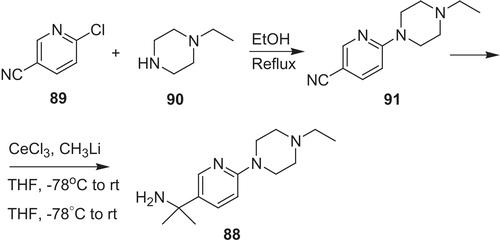
The amine 88 was obtained via nucleophilic aromatic substitution of 2-chloropyridine-5-carbonitrile (89) by N-ethylpiperazine (90) (Scheme 19). Next, 6-(4-ethylpiperazin-1-yl)nicotinonitrile (91) was converted to amine 88 upon dimethylation with generated in situ CH3CeCl2Citation60.
Scheme 19. Synthesis of amine 88Citation60.
Conclusions
The important progress was made recently in the development of new IMPDH inhibitors. Considerable part of designed structures included isobenzofuran moiety, which were derived from MPA as conjugates or analogs possessing modified functional groups. However, data received from molecular modeling studies enabled also to develop other classes of compounds giving interesting results in the course of in vitro or in vivo biological activity evaluations. Selectiveness toward IMPDH-II over IMPDH-I remains still challenging, but among the recently reported compounds can be found drug candidates having promising antiviral, anticancer, immunosuppressive and antibacterial properties.
Declaration of interest
The authors report no conflicts of interest. The authors alone are responsible for the content and writing of this article.
References
- Pimkin M, Markham GD. Inosine 5′-monophosphate dehydrogenase. Adv Enzymol Relat Areas Mol Biol 2009;76:1–53
- Mitsuhashi S, Takenaka J, Iwamori K, et al. Structure-activity relationships for inhibition of inosine monophosphate dehydrogenase and differentiation induction of K562 cells among the mycophenolic acid derivatives. Bioorg Med Chem Lett 2010;18:8106–11
- Bentley R. Mycophenolic acid: a one hundred year odyssey from antibiotic to immunosuppressant. Chem Rev 2000;100:3801–25
- Jayaram HN, Dion RL, Glazer RI, et al. Initial studies on the mechanism of action of a new oncolytic thiazole nucleoside, 2-β-D-ribofuranosylthiazole-4-carboxamide (NSC 286193). Biochem Pharmacol 1982;31:2371–80
- Manzoli L, Billi AM, Gilmour RS, et al. Phosphoinositide signaling in nuclei of Friend cells: tiazofurin down-regulates phospholipase C beta 1. Cancer Res 1995;55:2978–80
- Vitale M, Zamai L, Falcieri E, et al. IMP dehydrogenase inhibitor, tiazofurin, induces apoptosis in K562 human erythroleukemia cells. Cytometry 1997;30:61–6
- Olah E, Csokay B, Prajda N, et al. Molecular mechanisms in the antiproliferative action of taxol and tiazofurin. Anticancer Res 1996;16:2469–77
- Weber G, Prajda N, Abonyi M, et al. Tiazofurin: molecular and clinical action. Anticancer Res 1996;16:3313–22
- Allison AC, Eugui EM. Mycophenolate mofetil and its mechanisms of action. Immunopharmacology 2000;47:85–118
- Natsumeda Y, Ohno S, Kawasaki H, et al. Two distinct cDNAs from human IMP dehydrogenase. J Biol Chem 1990;265:5292–5
- Nagai M, Natsumeda Y, Weber G. Selective up-regulation of type II inosine 5′-monophosphate dehydrogenase messanger RNA expression in human leukemias. Cancer Res 1992;52:258–61
- Konno Y, Natsumeda Y, Nagai M, et al. Expression of human IMP dehydrogenase types I and II in Escherichia coli and distribution in human normal lymphocytes and leukemic cell lines. J Biol Chem 1991;266:506–9
- Gu JJ, Spychala J, Mitchell BS. Regulation of the human inosine monophosphate dehydrogenase type I gene. Utilization of alternative promoters. J Biol Chem 1997;272:4458–66
- Nair V, Shu Q. Inosine monophosphate dehydrogenase (IMPDH) as a probe in antiviral drug discovery. Antiviral Chem Chemother 2007;18:245–58
- Chong CR, Qian DZ, Pan F, et al. Identification of type I inosine monophosphate dehydrogenase as an antiangiogenic drug target. J Med Chem 2006;49:2677–80
- Felczak K, Pankiewicz KW. Rehab of NAD(P)-dependent enzymes with NAD(P)-based inhibitors. Curr Med Chem 2011;18:1891–908
- Chen L, Petrelli R, Felczak K, et al. Nicotinamide adenine dinucleotide based therapeutics. Curr Med Chem 2008;15:650–70
- Hedstrom L. The dynamic determinants of reaction specificity in the IMPDH/GMPR family of (β/α)8 barrel enzymes. Crit Rev Biochem Mol Biol 2012;47:250–63
- Dunkern T, Prabhu A, Kharkar PS, et al. Virtual and experimental high-throughput screening (HTS) in search of novel inosine 5′-monophosphate dehydrogenase II (IMPDH II) inhibitors. J Comput Aided Mol Des 2012;26:1277–92
- Shu Q, Nair V. Inosine monophosphate dehydrogenase (IMPDH) as a target in drug discovery. Med Res Rev 2008;28:219–32
- Barnes BJ, Izydore RA, Eakin AE, Hall IH. Mechanism of action of the antitumor agents 6-benzoyl-3,3-disubstituted-1,5-diazabicyclo[3.1.0]hexane-2,4-diones: potent inhibitors of human type II inosine 5′-monophosphate dehydrogenase. Int J Cancer 2001;94:275–81
- Felczak K, Chen L, Wilson D, et al. Cofactor-type inhibitors of inosine monophosphate dehydrogenase via modular approach: targeting the pyrophosphate binding sub-domain. Bioorg Med Chem 2011;19:1594–605
- Gish RG. Treating HCV with ribavirin analogues and ribavirin-like molecules purine riboside. J Antimicrob Chemother 2006;57:8–13
- Wingard JR, Hess AD, Stuart RK, et al. Effect of several antiviral agents on human lymphocyte functions and marrow progenitor cell proliferation. Antimicrob Agents Chemother 1983;23:593–7
- Sidwell RW, Robins RK, Hillyard IW. Ribavirin: an antiviral agent. Pharmacol Ther 1979;6:123–46
- Wyde PR. Respiratory syncytial virus (RSV) disease and prospects for its control. Antiviral Res 1998;39:63–79
- Fernandez-Larson R, Patterson JL. Ribavirin is an inhibitor of human immunodeficiency virus reverse transcriptase. Mol Pharmacol 1990;36:766–70
- Main J, McCarron B, Thomas HC. Treatment of chronic viral hepatitis. Antiviral Chem Chemother 1998;9:449–60
- Kawasaki Y. Mizoribine: a new approach in the treatment of renal disease. Clin Develop Immunol 2009;2009:681482 (1–10)
- Sintchak MD, Nimmesgern E. The structure of inosine 5′-monophosphate dehydrogenase and the design of novel inhibitors. Immunopharmacology 2000;47:163–84
- Petrelli R, Vita P, Torquati I, et al. Novel inhibitors of inosine monophospate dehydrogenase in patent literature of the last decade. Recent Pat Anticancer Drug Discov 2013;8:103–25
- Hedstrom L. IMP dehydrogenase: structure, mechanism and inhibition. Chem Rev 2009;109:2903–28
- Chen L, Wilson DJ, Xu Y, et al. Triazole-linked inhibitors of inosine monophosphate dehydrogenase from human and Mycobacterium tuberculosis. J Med Chem 2010;53:4768–78
- Risal D, Strickler MD, Goldstein BM. Ternary complex of the human type II inosine monophosphate dehydrogenase with ribavirin monophosphate and C2-mycophenolic adenine dinucleotide. Protein Data Bank 2004. Available from: http://www.rcsb.org/pdb/explore.do?structureId=1NFB [last accessed 19 Aug 2014]
- Cholewinski G, Malachowska-Ugarte M, Dzierzbicka K. The chemistry of mycophenolic acid – synthesis and modifications towards desired biological activity. Curr Med Chem 2010;17:1926–41
- Nelson PH, Carr SF, Devens BH, et al. Structure-activity relationships for inhibition of inosine monophosphatedehydrogenase by nuclear variants of mycophenolic acid. J Med Chem 1996;39:4181–96
- Yang N, Wang Q, Wang W, et al. The design, synthesis and in vitro immunosuppressive evaluation of novel isobenzofuran derivatives. Bioorg Med Chem Lett 2012;22:53–6
- Malachowska-Ugarte M, Cholewinski G, Dzierzbicka K, Trzonkowski P. Synthesis and biological activity of novel mycophenolic acid conjugates containing nitro-acridine/acridone derivatives. Eur J Med Chem 2012;54:197–201
- Iwaszkiewicz-Grzes D, Cholewinski G, Kot-Wasik A, et al. Synthesis and biological activity of mycophenolic acid-amino acid derivatives. Eur J Med Chem 2013;69:863–71
- Felczak K, Vince R, Pankiewicz KW. NAD-based inhibitors with anticancer potential. Bioorg Med Chem Lett 2014;24:332–6
- Chen L, Petrelli R, Olesiak M, et al. Bis(sulfonamide) isosters of mycophenolic adenine dinucleotide analogues: inhibition of inosine monophosphate dehydrogenase. Bioorg Med Chem 2008;16:7462–9
- Sunohara K, Mitsuhashi S, Shigetomi K, Ubukata M. Discovery of N-(2,3,5-triazoyl)mycophenolic amide and mycophenolic epoxyketone as novel inhibitors of human IMPDH. Bioorg Med Chem Lett 2013;23:5140–4
- Chen L, Wilson DJ, Xu Y, et al. Triazole-linked inhibitors of inosine monophosphate dehydrogenase from human and Mycobacterium tuberculosis. J Med Chem 2010;53:4768–78
- Wu H, Pagadala J, Yates Ch R, et al. Synthesis and characterization of an anti-apoptotic immunosuppressive compound for improving the outcome of islet transplantation. Bioconjugate Chem 2013;24:2036–44
- Chen Z, Zheng Z, Huang H, et al. Penicacids A-C, three new mycophenolic acid derivatives and immunosuppressive activities from the marine-derived fungus Penicillium sp. SOF07. Bioorg Med Chem Lett 2012;22:3332–5
- Chen L, Wilson DJ, Labello NP, et al. Mycophenolic acid analogues with a modified metabolic profile. Bioorg Med Chem 2008;16:9340–5
- El-Araby ME, Bernacki RJ, Makara GM, et al. Synthesis, molecular modeling, and evaluation of nonphenolic indole analogs of mycophenolic acid. Bioorg Med Chem 2004;12:2867–79
- Chen L, Wilson D, Jayaram HN, Pankiewicz KW. Dual inhibitors of IMP-dehydrogenase and histone deacetylases for cancer treatment. J Med Chem 2007;50:6685–91
- Looker AR, Littler BJ, Blythe TA, et al. Development and manufacture of the inosine monophosphate dehydrogenase inhibitor merimepodid, VX-497. Org Process Res Dev 2008;12:666–73
- Jain J, Almquist SJ, Shlyakhter D, Harding MW. VX-497: a novel, selective IMPDH inhibitor and immunosuppressive agent. J Pharm Sci 2001;90:625–37
- Herr RJ, Fairfax DJ, Meckler H, Wilson JD. Preparation of 5-(2-methoxy-4-nitrophenyl)oxazole: a key intermediate for the construction of VX-497. Org Process Res Dev 2002;6:677–81
- Gu HH, Iwanowicz EJ, Guo J, et al. Novel diamide-based inhibitors of IMPDH. Bioorg Med Chem Lett 2002;12:1323–6
- Dhar TGM, Shen Z, Guo J, et al. Discovery of N-[2-[2-[[3-methoxy-4-(5-oxazolyl)phenyl]-amino]-5-oxazolyl]phenyl]-N-methyl-4-morpholineacetamide as a novel and potent inhibitor of inosine monophosphate dehydrogenase with excellent in vivo activity. J Med Chem 2002;45:2127–30
- Dhar TGM, Guo J, Shen Z, et al. A modified approach to 2-(N-aryl)-1,3-oxazoles: application to the synthesis of the IMPDH inhibitor BMS-337197 and analogues. Org Lett 2002;4:2091–3
- Chen L, Petrelli R, Gao G, et al. Dual inhibitors of inosine monophosphate dehydrogenase and histone deacetylase based on a cinnamic hydroxamic acid core structure. Bioorg Med Chem 2010;18:5950–64
- Watterson SH, Carlsen M, Dhar TGM, et al. Novel inhibitors of IMPDH: a highly potent and selective quinolone-based series. Bioorg Med Chem Lett 2003;13:543–6
- Zhong Z-J, Zhang D-J, Peng Z-G, et al. Synthesis and antiviral activity of a novel class of (5-oxazolyl)phenyl amines. Eur J Med Chem 2013;69:32–43
- Wittine K, Stepkowić Babić M, Makuc D, et al. The novel 1,2,4-triazole-3-carboxamide and imidazole derivatives of l-ascorbic acid: synthesis, anti-HCV and antitumor activity evaluations. Bioorg Med Chem 2012;20:3675–85
- Nakanishi T, Kozuki Y, Eikyu Y, et al. In vitro and in vivo characterization of AS2643361, a novel and highly potent inosine 5′-monophosphate dehydrogenase inhibitor. Eur J Pharm 2012;674:58–63
- Watterson SH, Chen P, Zhao Y, et al. Acridone-based inhibitors of inosine 5′-monophosphate dehydrogenase: discovery and SAR leading to the identification of N-(2-(6-(4-ethylpiperazin-1-yl)pyridin-3-yl)propan-2-yl)-2-fluoro-9-oxo-9,10-dihydroacridine-3-carboxamide (BMS-566419). J Med Chem 2007;50:3730–42