
Abstract
Mycobacterial enoyl-ACP-reductase, an enzyme contributing in mycolic acids biosynthesis, has been established as promising target of novel antimycobacterial drugs. The development of inhibitors active without previous activation by catalase/peroxidase system (e.g. isoniazid), seems to be rational approach. Catalase/peroxidase system is frequently responsible for resistance. We hereby present a review of direct mycobacterial enoyl-acyl carrier protein reductase inhibitors development in past decade. A special attention was paid to mechanism of inhibition, which shows relatively conserved interactions of inhibitors with Tyr 158 and cofactor. Hence, future developments of more effective antitubercular drugs should consider structural demands for potent direct mycobacterial enoyl reductase inhibitors.
Introduction
Mycobacterium tuberculosis (MT) infection is the cause of over two million deaths worldwide every year. According to WHO there will be about one billion more people newly infected with tuberculosis and approximately 36 million deaths in 2020, if the worldwide ravage of tuberculosis is left unchallenged. Moreover tuberculosis is leading opportunistic pathogen in patients with AIDS who are very susceptible. In these cases is MT usually cause of death. One third of the increase in the incidence of TB in the past five years can be linked to HIV co-infectionCitation1. The situation is exacerbated by occurrence of multiple drug resistant (MDR-TB) strains, extreme drug resistant (XDR-TB) strains and even total drug resistant (TDR-TB) strains that do not respond to some or most of the currently used antitubercular drugsCitation2. Growing incidence of drug resistance, severe side effects of current treatment and growing number of HIV patients makes the discovery of novel more potent antitubercular compounds an important issueCitation3.
The current front line chemotherapeutics consist of isoniazid (INH), pyrazinamide and rifampicine (). INH’s discovery was a cornerstone of modern antitubercular treatment. INH was the first antitubercular compound confirmed enoyl-ACP-reductase (InhA; EC 1.3.1.9) inhibitor. It disrupts the integrity of mycobacterial cell wall via inhibiting the biosynthesis of mycolic acids, a long chain fatty acids containing component of the cell envelope. InhA is the target of INH as well as ethionamide (ETH). INH and ETH are known to be prodrugs requiring activation by mycobacterial KatG (catalase/peroxidase enzyme) and EthA (flavin monooxidase), respectivelyCitation4,Citation5. After activation INH and ETH form adduct with nicotine adenine dinucleotide (NADH). The adducts are tight binding inhibitor of mycobacterial InhA. The mutations of KatG coding genes are often observed at INH and ETH resistant strains.
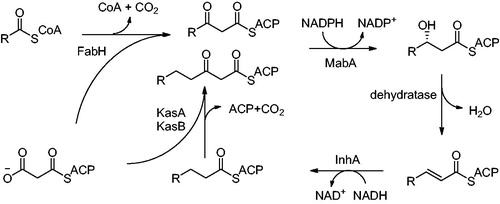
Mycolic acids are very long chain (C74–C90) α-alkyl β-hydroxy fatty acids covalently linked to arabino-galactan. They are the major components of the mycobacterial cell envelope providing protection from commonly used antibiotics. They are also responsible for a great part of Mycobacteria virulenceCitation6. The biosynthesis of mycolic acids is catalyzed by two enzyme systems: fatty acid synthase I (FAS I) producing shorter chain fatty acids and fatty acid synthase II (FAS II) involved in fatty acids chain elongation. InhA is responsible for trans-enoyl reduction, the final step of FAS II pathway (). C16 substrate, a product of FAS I, is utilized as a starting point for further elongation by FAS II cycle. Therefore InhA is specific for substrate with aliphatic chains C16 and longer. FAS II enzymes as depicted at have been evaluated as promising antimicrobial targets. InhA is the most prominent potential target among themCitation7.
Figure 1. Structures of currently used front line antituberculotics and direct InhA inhibitor triclosan.
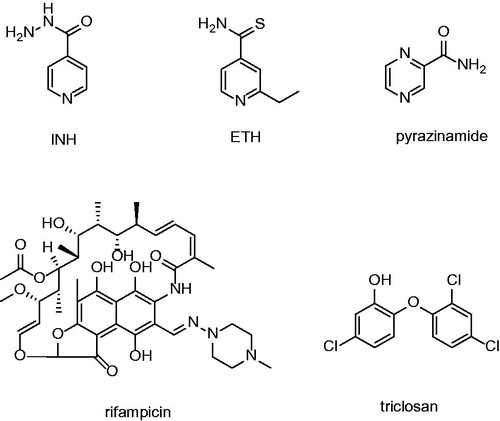
Figure 2. FAS II pathway; FabH: β-ketoacyl-ACP synthase; MabA: 3-ketoacyl-ACP reductase; InhA: Enoyl-ACP-reductase and KasA, KasB: β-ketoacyl-ACP synthase. KasA is specific for shorter chain substrates, KasB utitlizes longer chain substrates.
Enyol-reductases (FabI) could be found in large number of known organisms, the most notorious of which are Escherichia coli, Plasmodium falciparum, Staphylococcus aureus, Mycobacterium smegmatis and Mycobacterium tuberculosisCitation8. Mycobacterial InhA molecule is a homotetramer in aqueous solution, and its crystal structure in complex with NAD+ and a fatty acyl substrate reveals that the substrate binds in a general U-shaped conformationCitation9. The residues crucial for trans-enoyl reduction catalysis, Tyr 158 and Lys 165, are situated at the active site. Tyr 158 and Lys 165 residues promote deprotonation of the 2′-nicotinamide hydroxyl group. InhA facilitates reduction by a catalytic mechanism in which a hydride is transferred to the C3 carbon of the C2–C3 double bond. The C1 carbonyl oxygen accepts a proton from the Tyr 158 hydroxyl, which forms an enolate anionCitation10. The fatty acyl chain of the bound substrate is completely surrounded by hydrophobic residues, the majority of which are located on the substrate binding loop (Met 103, Phe 149, Tyr 158, Lys 165, Thr 196, Met 199, Leu 207 and Ile 215). The substrate binding loop of InhA is larger than that of other bacteria enoyl-ACP reductases (FabI) and creates a deeper substrate binding crevice. This fact is consistent with the ability of InhA to recognize longer chain fatty acyl substrates. Tyr 158 is conserved in the bacterial FabIs and plant enoyl-ACP reductases, interaction with Tyr 158 is likely to be a key feature of fatty acyl substrate binding common to other enoyl-ACP reductasesCitation10. The impaired action of InhA therefore leads to the loss cell integrity and consequently to the cell death. Since InhA plays a crucial role in the fatty acids elongation cycle it was validated as a promising target of novel antitubercular compoundsCitation11,Citation12.
Targeting mycobacterial enzymes with a small molecules and consequent disruption of cellular homeostasis remains a most promising approachCitation13–15. Other small molecules, which do not require the bioactivation by KatG and inhibit InhA directly are able to bypass mechanism of resistance to inhibitors such as INH and ETH. Such small molecules active without bioactivation are rational approach that may lead towards novel antitubercular compounds. This approach holds a promise of novel InhA inhibitors active against MDR-TB and XDR-TB strainsCitation16.
Medline, Scopus and Web of Knowledge were searched for “InhA inhibitors” and “enoyl-ACP inhibitors” concerned articles. In this work we present a review of notable direct InhA inhibitors groups with special attention to mechanism of inhibition. The in vitro results together with the inhbitors structures are summarized in .
Table 1. Structures and in vitro results of notable InhA inhibitors.
Diphenyl ethers
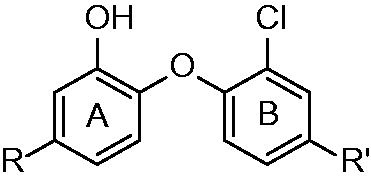
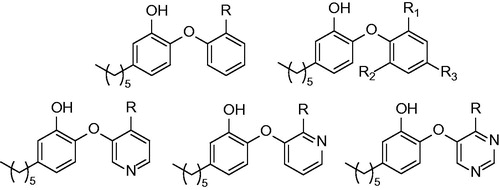
Triclosan (5-chloro-2-(2,4-dichloro-phenoxy)-phenol) can be exampled as such inhibitor active without prior activation. At first, it was thought that triclosan, being a small hydrophobic molecule, was absorbed by passive diffusion into the bacterial cell. The nonspecific damage to bacterial cell wall was believed to be the mechanism of antibacterial effect. Triclosan was later proven to target fatty acid synthesis via inhibition of enoyl-reductase in late 90’sCitation17,Citation18. However, its use is limited by its poor bioavailability. Triclosan was found to be a potent inhibitor of FabI of various organisms such as Brassica napus, Helicobacter pylori, Escherichia coli (Ki value of 38 pM) or Plasmodium falciparium (Ki value of 96 pM), however, its inhibitory potency against MT InhA was not as high (Ki value of 0.22 µM)Citation19. This can be explained by the mechanism of inhibition, triclosan was found to be rapid reversible inhibitor of InhA, however it acts as slow, tight binding inhibitor of E. coli FabICitation18. It has found its uses as an antimicrobial agent in locally acting medical preparations such as soaps, mouthwashes, shampoos, or toothpastesCitation20. Other triclosan modified structures with improved pharmacokinetics were synthesized over past decades. Importantly these compounds show sufficient MIC against INH-resistant strains. The mechanism of inhibition is well described: important interactions with InhA active site are π–π stacking of triclosan hydroxyl substituted A-ring and NAD+ pyridine. Moreover there is conserved hydrogen bonding with Tyr 158 in the catalytic site. The hydrogen bond network is further extended towards NAD+ ribose 2′-hydroxyl Hydrophobic contacts of B-ring chlorines oriented orthogonally to the A-ring are responsible for the orientation of the triclosan moleculeCitation21.
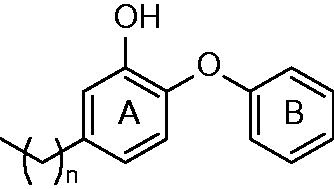
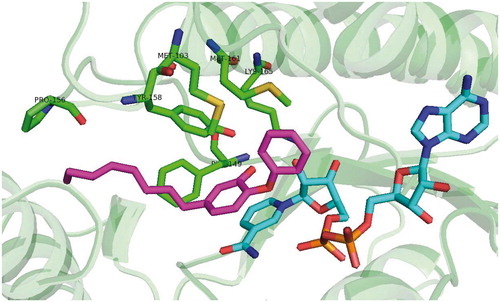
The first generation of alkyl substituted diphenyl ethers (general structure depicted at ) was prepared to improve affinity towards InhA. The structure based development has been used to design nanomolar range non-competitive InhA inhibitorsCitation22. The most potent compound, 5-octyl-2-phenoxyphenol (5) has a Ki value of 1 nM, IC50 value of 5 nM and MIC99 values of 2–3 μg/mL−1 (6–10 μM) for both drug-sensitive (H37RV) and drug-resistant strains of MT with differing drug resistance profiles. MIC99 values for growth inhibition are increased 9–12-fold upon overexpression of InhA in MT. This provides an evidence, that InhA is target and the mechanism of action is KatG independent. There is an apparent influence of the alkyl chain length. The InhA inhibitory potency is increasing from 5-ethyl-2-phenoxyphenol (1) to 5. Moreover, these inhibitors are able to circumvent the triclosan resistance mechanism, which is upregulation of a putative efflux pump and aromatic dioxygenaseCitation22,Citation23. Crystallographic study was performed for 5 and 5-pentyl-2-phenoxyphenol (3). The mode of binding was revealed to be similar to triclosan. π–π stacking of A-ring and hydrogen bonding network of phenolate group with Tyr 158 and 2′-hydroxyl group of NAD+ ribose could be observed (Scheme 1). Unlike the C16 substrate, which forms characteristic U-shape, the inhibitor alkyl chain takes up a linear conformation protruding into the acyl-binding pocket forming hydrophobic interactions with Phe 149, Met 155, Pro 156, Ile 215 and Leu 218. Conformational changes within substrate binding loop suggest, that these compounds are rapid and reversible inhibitors, rather than slow, tight bindingCitation24.
Figure 3. General structure of diphenyl ethers (1–6) published by Sullivan et al.; n = 1,3–5,7,13.
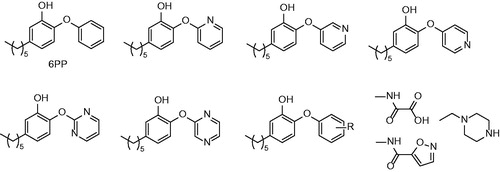
Another study was performed in order to determine in vivo efficacy and bioavailability. After the oral administration neither 5-hexyl-2-phenoxyphenol (4) nor 5 had the bioavailability comparable to that of INH. In contrast, the analogues could be detected in the serum 8 h after the administration, which is considerably longer compared with INH. Additionally, there were no adverse effects observed, showing the reduced toxicity of 4PP and 5PP. Transcriptional differences between 6PP, 8PP and triclosan suggest, that analogues have narrowed mode of action in contrast to triclosan, which seems to have more than one target in the cellCitation25. 4 and 5 were active against MT clinically isolated strains with different resistance profile. However, neither of novel inhibitors was effective in rapid mouse model. Limited bioavailability still remains severe drawback of these compoundsCitation26.
In order to improve pharmacokinetic properties of diphenyl ethers the series of B-ring modified analogues was prepared. Most of the previously prepared compounds have calculated log P (Clog P) value greater than five, which is considered to be a reason for their poor in vivo efficacyCitation26. Based on the relationship between log P and in vivo activity the series of less lipophilic diphenyl ethers were synthesizedCitation27. The structure of 4 compound was used as a starting point (IC50 = 11 nM; MIC90 = 2.1 µg/mL−1). Two series were designed. B ring was either replaced with aromatic nitrogen heterocyclus, or substituted with nitro, amino, piperazino or substituted carbamoyl group in ortho, meta and para position (). In general, none of the structure modification yielded inhibitors with better inhibitory properties. Most of the compounds showed significantly reduced both inhibitory properties and antimycobacterial activity. However, introduction of ortho or para amino or nitro group (12,13,14,15,17) were better tolerated. The most active compounds were 4-pyridinoyl isoster (8), meta nitro derivative (13), ortho and para amino derivative (15 and 17) with IC50 values ranging between 48 nM and 160 nM. MIC90 values of these most successful compounds were ranging between 12.5 µg/mL−1 and 3.13 µg/mL−1. Authors state, that three inhibitors are tight-binding, yet further evidence to support this mechanism of inhibition is neededCitation24. Values of Clog P of successful inhibitors were significantly lowered compared with parent compound, while possessing sufficient inhibitory and antitubercular properties. Such compounds are expected to possess more favorable pharmacokinetic profileCitation28.
Figure 4. Structures of InhA inhibitors developed by Ende et al. R: NO2, NH2 and -NHCOCH3.
A series of triclosan derivates with modified triclosan 5-position (replacing 5-chloro substitution with various aliphatic, aromatic or heteroaromatic substituents; ) was prepared with the aim to improve their InhA inhibitory activityCitation21. Similar compounds were previously evaluated by Freundlich et al. against P. falciparum FabICitation29. Inhibitors with hydrophobic substituent proved to be much more potent compared with those with hydrophilic substituent (compare 5-COOH substituted inhibitor with IC50 value higher than 1 µM and 5-phenylehtyl inhibitor (34) with IC50 value of 21 nM). None of the compounds with hydrophilic substitution was able to reach 1 µM IC50. These results are consistent with the hydrophobic nature of acyl binding pocket and the fact that 5-substituents protrude into that pocket. InhA inhibitory potency appeared to be improving with the growing alkyl chain from ethyl (24) IC50 = 120 nM to butyl (26) IC50 = 55 nM and isopentyl (28) IC50 = 63 nM. Longer alkyl chains are able to create more extensive hydrophobic interactions in the substrate binding pocket. However, branched chain do not seem to improve inhibitory properties. It is probably because of steric clashes with one or more binding loop residues. Crystalographic studies show that alkyl chain occupies the acyl binding pocketCitation21. Those findings are consistent with results presented by Sullivan et alCitation22. Triclosan analogues show better in vitro activity then their des-chloro analogues. B ring chlorines therefore seem to contribute to anchor the inhibitors within the active site via interactions with Phe 97 and Met 103. The most potent inhibitors were those with arylalkyl chains. Inhibitor 34 showed the IC50 value of 21 nM, 2-pyridylmethyl (30) IC50 was 29 nM against recombinant InhA. Crystals reveal close proximity of inhibitor with Leu 218, Ile 215, Phe 149, Pro 193 and Met 199 (pdb no. 3FNE, 3FNF, 3FNG and 3FNH; Scheme 2). However, it was not reported which type of inhibition is observed, whether fast, rapid, or slow and tight binding inhibition. Substrate binding loop in these crystallographic structures is ordered which might point towards slow, tight binding inhibitors. The most potent InhA inhibitors were tested against both INH-sensitive and INH-resistant strains whole cell mycobacteria. MIC values suggest the independence on KatG activation and therefore good activity against INH resistant strains. The third most potent inhibitor (phenylpropyl substituted; 35) with IC50 50 nM showed the best MIC H37Rv value of 13 µM, which represents tenfold improvement over triclosan. On the other hand the most potent in vitro inhibitor 34 showed the weakest antitubercular activity. That may possibly be because of non-favorable pharmacokinetic parametersCitation28.
Figure 5. Structures of triclosan analogues published by Freundlich et al.; R: aliphatic, aromatic or heteroaromatic substitution; R': Cl and CN.
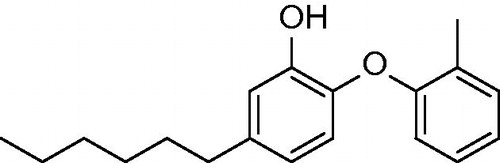
With respect to the findings highlighting the advantages of slow onset, tight binding inhibitors, the discovery of novel compounds with long residence time is of importance. It was shown on the example of FabI from Francisella tularensisCitation24,Citation30. For example INH-NAD adduct was demonstrated to be such inhibitor. Kisker et al. introduced 2-(o-tolyloxy)-5-hexylphenol (36; ) which is slow, tight binding inhibitor of mycobacterial InhA with Ki value of 22 pM. This is 430 fold improvement compared with the parent compound 4, which is fast reversible inhibitor. Methyl group in the B-ring ortho position causes reduced conformational flexibility and forces the substrate binding loop to order itself with respect to inhibitor. Moreover, methyl group forms a crucial van der Waals Interactions with NAD+ and Ala 198 and Gly 96 providing alongside with Met 199, Ile 202 and Val 203 a disordered substrate binding loop resulting in slow two steps (induced fit) tight-binding mechanism of inhibition (pdb no. 2X23; Scheme 3). Inhibitor 36 has residence time on InhA of 24 min, 14 000 times longer than parent compound 4Citation31.
Figure 6. Tight binding diphenyl ether InhA inhibitor PT70 (36).
Scheme 1. Alkyl substituted diphenylether inhibitor (5) binding mode (pdb no. 2B37).
Scheme 2. Triclosan binding mode, pdb no: 3FNF (24).
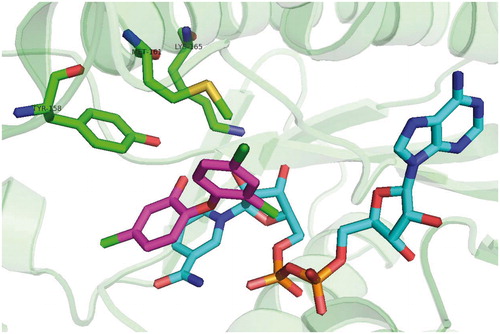
Scheme 3. PT70 (36) binding mode, pdb no. 2X23.
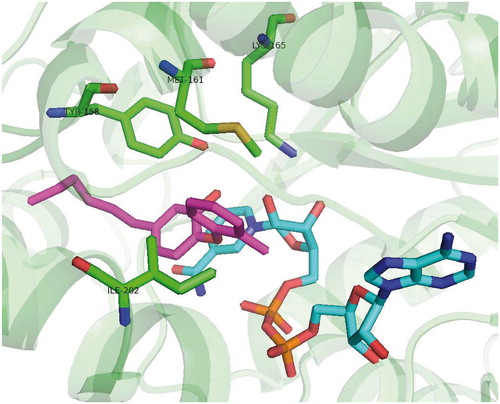
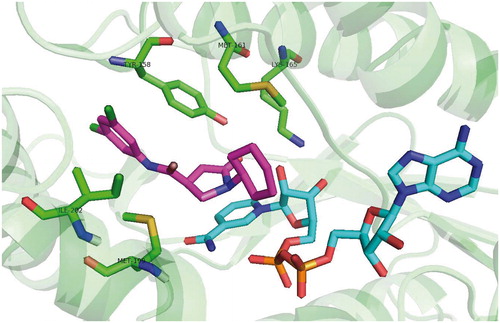
Scheme 4. Genz 10850 (45) binding mode, pdb no. 1P44.
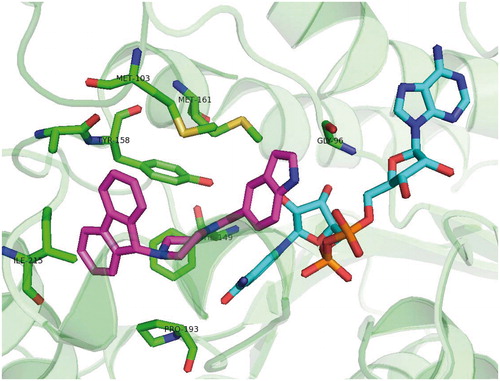
Based on the premise that, B-ring plays a crucial role in the tight-binding mechanism of inhibitionCitation32 a SAR study with series of B-ring modified diaryl ethers was performed (structures depicted at ). Results show that tight binding inhibition mechanism is indeed sensitive to B-ring substitutionCitation31,Citation33. Several of the ortho substituted derivatives of 36 (replacement of ortho methyl group) showed the residence time (tR) comparable or even higher than 36. Those were NO2 derivative (tR = 27 min), Cl derivative 38 (tR = 21 min) Br derivative 39 (tR = 30 min) and CN derivative 44 (tR = 80 min). However, only inhibitor 39 surpassed 36’s IC50 towards InhA and none of them showed lower Ki compared with 36. Crystal structures of 39 and 44 show the substrate binding loop is order in the similar way as other tight binding inhibitors-InhA complexes. Both crystal structures show the close interactions with B-ring. This loop ordering is in contrast to rapid-reversible inhibitors, where the loop is either disordered or forms alpha-helix in a much more open conformation. Compound 39 orders the substrate binding loop in very similarly to 36. The binding mode of compound 44 is somewhat different. It forms the van der Waals interactions with Ala 201 and Ile 202 instead of Ile 202 and Val 203. This different binding mode and loop ordering rationalizes the prolonged tR. However, decreased overall binding affinity of 44 indicates, that different loop ordering is also destabilized. Since the overall energy barrier between non-ordered and ordered conformation of the binding loop has increased for 44 it shows decreased overall binding affinity compared with 36. 44 appears to make weaker van der Waals interactions with binding loop residues. Moreover bulkier CN substituent approaches NAD+ backbone within a less favorable distance and these interactions provide plausible explanation for above mentioned destabilization.
Figure 7. General structures of compounds published by Pan and Tonge and colleagues in 2014; R: NO2, NH2, CH3, Cl, Br, CF3, I, F, OH and CN.
Unfortunately, none of these derivatives had improved activity compared with their parent compound. The docking studies showed that additional B-ring substitution leads to steric hindrance and unfavorable van der Waals interactions and therefore lowered binding affinity. The results of the binding studies are consistent with a previous proposal that B-ring substituents should generally be small in order to be accommodated in the InhA active site. SAR study revealed similar relationship to ortho B-ring substituted compounds. Large and hydrophilic substituents are not favorable. Halogen substituted B-rings are preferred. Although MIC values for the di-ortho analogues are 4–15-fold higher compared with 36. 36 and its chloro analogue 38 were also shown to decrease the bacterial levels in MT infected miceCitation33.
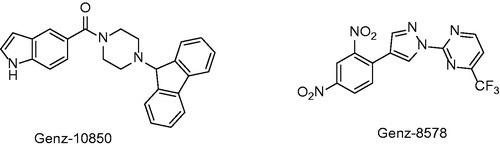
In 2003 Kuo and co-workersCitation34 presented results of high-throughput screening against InhA with two hits. Genz-8575 (46) and Genz-10850 (45) (pdb no. 1P44; ) showed promising activity against InhA. Moreover, they were active against both INH-sensitive and INH-resistant strains InhA. Reported IC50 values against recombinant InhA values were of 160 nM for 45 and 2400 nM for 45. These values are considerably higher compared with previously mentioned diphenyl-ethers. Ki values were determined suggesting non-competitive mechanism of inhibition. The crystallographic x-ray study was performed for 45 in complex with InhA. Despite the fact, that the structure of these compounds is very different from triclosan, the mechanism of inhibition is notably similar. Compound 45 carbonyl oxygen forms a hydrogen bond network with NAD+ 2′-ribose hydroxyl and catalytic site Tyr 158. The bulky fluorenyl group is located in the substrate binding pocket forming a hydrophobic interactions with substrate binding loop residues Gly 96, Met 103, Phe 149, Met 155, Pro 156, Ala 157, Met 161, Pro 193, Ala 198, Ile 215 and Leu 218 (Scheme 4). Superimposition with triclosan reveals that A and B rings are overlaid with piperazine and indol of 45. Triclosan phenolate group and 45 carbonyl form a similar hydrogen bond network with ribose and Tyr 158.
Figure 8. Structure of two inhibitors from Genzyme compound library.
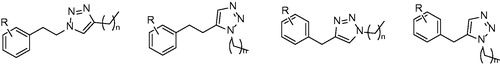
Triazole derivatives were synthesized in order to find novel group of inhibitors by replacing of diphenylether A-ring by triazole. The compounds with the general structure displayed at were assayed for their activity against mycobacterial InhA. Unfortunately, none of the compounds in this study showed any promising results. The best results were achieved with class b () compound bearing a nonyl substituent (50), however, it showed only 58% inhibition at 50 µM concentration. Good antimycobacterial activities compared in relation to poor InhA inhibition suggest presence of another target distinct from InhACitation35.
Figure 9. General structure of two triazole derivatives classes. R: H and Cl; R’: Me, OMe and NO2; R’’: phenyl, cyclohexyl and alkyl; n = 1–2.
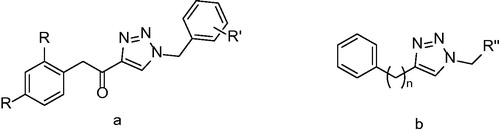
The series of class b derivatives () were consequently synthesized and evaluated for their effectivity against mycobacterial InhA. Some improvement in the inhibitory potency could be observed. More specifically, compounds with triazole core substituted with longer alkyl chains (C8–C12) showed better inhibition ranging between 58% and 75%. Docking studies suggest that binding mode of triazole inhibitors is similar to alkyl substituted diphenylethers, but the key interactions with Tyr 158 and ribose are considerably weaker. However, compared with IC50 values of diphenylethers the triazole IC50 values are insufficient and replacing phenyl core with triazole core does not seem to be effective approachCitation36.
Figure 10. General structure of triazole potential inhibitors derived from previously published class b compounds. R: halogen, Me and OMe and n: 5–11.
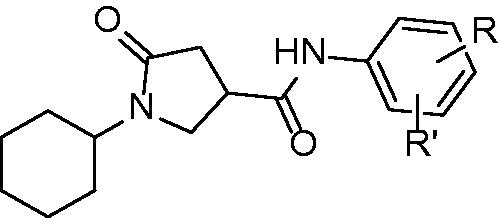
Pyrrolidine carboxamides (general structure displayed at ) are InhA inhibitors discovered by high-throughput screening of 30 000 compounds presented by He and co-workers in 2006Citation37. Compounds exhibiting at least 50% InhA inhibitory activity at 30 µM were labeled as hit compounds. Compounds identified in this study were active against mycobacterial InhA. Interestingly, none of the hits were active against E. coli FabI or P. falciparium FabI. The representative is 2,5-dimethyl derivative with IC50 of 10 µM yet other compounds are reported to be inhibitors at submicromolar range. The derivatization of anilide ring reveals that lipophilic, electron withdrawing substituent in the positions 3 and 5, preferably Cl, Br, or CF3, is the most beneficent. More specifically, compounds 52 3-Br, 53 2-Me, 54 3,5-Cl, and 55 3-Br, 5-CF3 have IC50 of 0.89 µM, 0.97 µM, 0.39 µM and 0.85 µM, respectively. The binding mode is conserved for most of the non-covalent InhA inhibitors. Hydrogen bond network between pyrrolidine carbonyl, Tyr 158 and NAD+ ribose is involved (pdb no. 2H7M; Scheme 5). The cyclohexyl ring forms a van der Waals interactions with Gly 96 and Phe 97. The substituents of anilide phenyl ring may contribute to inhibitory effect by forming the interactions with Tyr 158, Met 155 and Leu 218. On the other hand, superimposed poses of differently substituted phenyl rings are almost completely overlaid and this suggests, that these interactions are not crucialCitation37.
Figure 11. General structure of pyrrolidne carboxamide InhA inhibitors.
Scheme 5. Pyrrolidine carboxamide derivative binding mode, pdb no 2H7M.
Pyrrolidine diones (succinimides) are compounds reported by Matviiuk and colleagues in 2013Citation38. Based on the virtual screening of the succinimide compounds library 3-(9H-fluoren-9-yl)pyrrolidine-2,5-diones () were selected for further evaluation. These compounds are structurally very similar to 45 (). The most potent inhibitors within this family are those with lipophilic either aromatic or aliphatic substituent bound to the pyrrolidine. Two classes could be distinguished: acyl substituted pyrrolidinediones and alkyl/aryl substituted pyrrolidinediones. Within the group of acyl substituted compounds the best results were achieved with benzoyl and nonanoyl substituted inhibitors 56 and 57. Inhibition values at 50 µM inhibitor concentration were 71% and 73%, respectively. Nine carbon atoms residue proved to be beneficial for inhibitory properties and nonyl substituted inhibitor showed 86% inhibition. Indolylethyl substituted inhibitor showed activity of 79% at 50 µM. The dichlorophenyl substituted inhibitor 59 was the most promising compound evaluated at this study and showed inhibitory potency at 50 µM higher than 95%. Further evaluation showed 31% inhibition at 5 µM inhibitor concentration. The molecular docking study showed that 9H-fluoren attains similar pose as 45 as well as interaction with Tyr 158 and NAD+ ribose. Despite the relatively poor inhibitory properties pyrrolidinediones show sufficient antitubercular activities. The majority of compounds showed better MIC compared with 45 whose poor antitubercular properties (MIC value of >40 µM) are explained by poor membrane permeability. Replacing piperazine core of 45 with succinimide leads to compounds with improved antitubercular activities. Several potent InhA inhibitors such as benzoyl substituted were also potent inhibitors of mycobacterial cell growth (MIC value of 2 µM). The most potent 3,5-dichlorophenyl inhibitor (59) is only moderate inhibitor of mycobacterial growth. On the other hand, compounds with very poor inhibitory properties towards such as amide derivative bearing 3,5-dichlorophenyl moiety showed good potential to inhibit mycobacterial growth (MIC value of 4 µM). InhA inhibition and MIC not being in correlation suggest, that InhA inhibition may not be the only mechanism of actionCitation38.
Figure 12. General structure of pyrrolidine-2,5-diones. R: acyl group, akyl, aryl and β-amide.
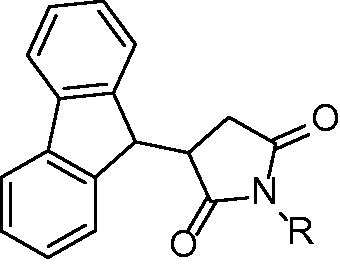
Consequently another series of 3-hetaryl pyrollidine-2,5-diones were synthesized but comparison of inhibitory properties towards InhA suggest, that novel pyrrolidinediones mechanism of action is other than InhA inhibitionCitation39.
Similar high-throughput experimental design published by He and co-workers in 2006Citation37 led to Arylamides a novel direct InhA inhibitors class discoveryCitation40. The largest subgroup of presented compounds are those containing piperazine () as a core amide fragment. Structure activity relationship study revealed, that the most favorable for InhA inhibition is small electron withdrawing substituent in the meta position (R2). The most successful compound in initial screening was structural type a R1: 4-CH3; R2: 3-Cl, compound 60 with IC50 value of 3 µM. This structure was consequently modified in order to yield more potent inhibitors. The electron-withdrawing nature of the group at the meta position of B-ring is an important determinant of activity, as many analogues with para or ortho substituents on B-ring were found to be inactive in the high-throughput screen. SAR study indeed revealed, that the most favorable for InhA inhibition is small electron withdrawing substituent in the B-ring meta position. Position of A-ring substituent seems to be also of importance. Transfer of the methyl substituent to other position of phenyl ring leads to the 3-fold decrease of inhibition potency. However, dimethyl derivatives (R1: 3,4-CH3; R2: 3-Cl and 3-CF3) compounds 61 and 62 were the compounds with the best inhibitory potency. Bulkier A-ring alkyls seemed to interfere with the binding within the active site and no further improvement could be observed. The insertion of one carbon atom had little effect on the activity and all the () structure derivatives exhibited similar potency with IC50 values ranging from 5 to 14 µM. Several 45 derivatives were also prepared. The best results were achieved with compound 64 which has indolyl cyclus reduced to phenyl. It showed 2-fold improvement compared with parent compound.
Figure 13. General structures of arylamides R1: small aliphatic or aromatic substituents and R2: Cl, CF3 and NO2.
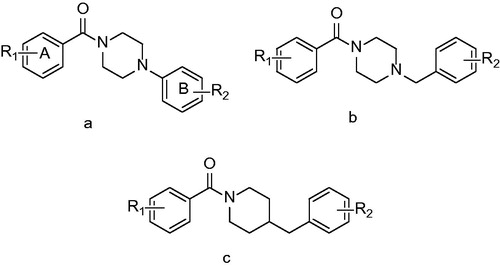
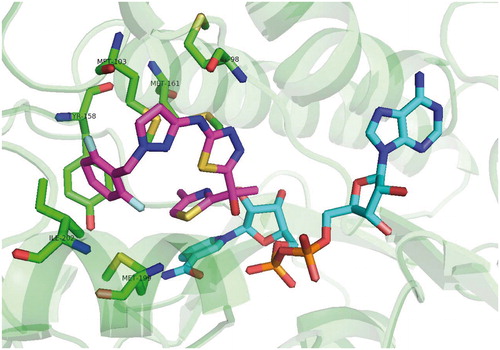
Crystallographic studies reveal the binding mode of arylamides very similar to all the previously mentioned inhibitor classes. Carbonyl oxygen forms a hydrogen bond network, which is extended to 2′-hydroxyl moiety of the NAD+ ribose. The B ring protrudes into the acyl binding pocket interacting with the hydrophobic residues of Phe 149, Pro 193, Leu 218 and Val 203. In agreement with in vitro results, the meta B-ring substituent strengthens these interactions being able to fill the void between inhibitor and acyl binding pocket residues. Van der Waals interactions of the methyl A-ring substituent further stabilize the inhibitor pose (Scheme 6).
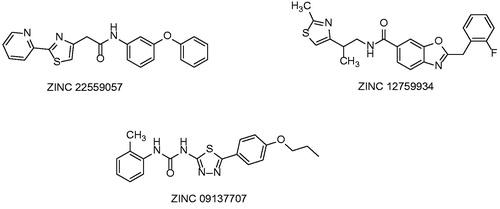
Two distinct computational modeling approaches were employed in the study of Pauli et al. in order to identify novel potent InhA inhibitorsCitation41. Structure based pharmacophore generation and docking using four established docking programs were used in this experimental setup. A database containing 999 853 entries was screened. First approach, the pharmacophore based search employed 36 known wild type or resistance based crystal InhA structures. The structural analysis of InhA in complex with the ligands bound to the substrate binding pocket indicated key amino acid residues that might be involved in the molecular recognition process. On that basis, a four-point 3-D pharmacophore model was developed. UNITY-3D flexible search was used to screen the library. High scored molecules, which matched the pharmacophore points were selected and subsequently docked into 1P44 derived InhA. The final selection of the molecules to be tested in vitro for its inhibitory activity against InhA was made according to in silico results. Structures of the in vitro evaluated compounds are displayed at .
Figure 14. Compounds selected from Pharmacophore modeling study.
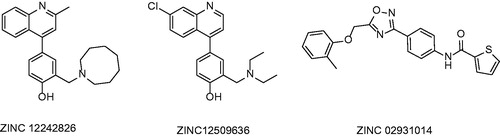
Second approach employed in order to identify novel potent InhA inhibitors was exhaustive molecular docking using various docking algorithms. Docking study was performed using widely used docking software: GOLD (CCDC, Cambridge, United Kingdom), Autodock (Scripps Research Institute, La Jolla, CA) and FlexX (BioSolveIT, Sankt Augustin, Germany). Protein receptor chosen for this study was 1P44 characterized by its wide open substrate binding loop conformation. Molecular dataset chosen was the same as the one used in first approach. First selection was performed by GOLD. 100 top scored molecules was then submitted to other programs using same parameters.
Molecules submitted to in vitro evaluation are summarized in . In vitro evaluation showed the poor inhibitory properties of these compounds. The best evaluated compound 67 showed IC50 value of 24 µM.
Figure 15. Compounds selected by exhaustive molecular docking published by Pauli et al.
Alternative approach for direct inhibition of InhA including allosteric modulators of InhA binding outside the active site was introduced by Shirude and colleagues together with AstraZeneca. Based on the previous research of potent direct InhA inhibitors with good antimycobacterial activitiesCitation42, methyl-thiazoles were introduced. The structures of novel methyl-thiazoles are displayed at . Methyl-thiazole series are characterized by to-date unknown mode of inhibition. The binding mode of methyl-thiazoles is completely distinct from other inhibitor groups. It includes a “Tyr 158 out” InhA conformation and neutrally charged molecule, which authors call “warhead”. Hydrophilic properties of methyl-thiazoles allow the favorable pharmacokinetic profile resulting in good antimycobacterial activityCitation43. Isothermal titration calorimetry, surface plasmon resonance and other experiments showed, that difluorophenyl lead structure (68) binds to InhA with both oxidized and reduced cofactor. However, it shows weak binding towards NAD+ bound form and tight binding (1000-fold increased affinity) to NADH bound InhA. This experiment also suggests, that lead methyl-thiazole structure acts as noncompetitive inhibitor. In vitro results show a good correlation between InhA inhibition and MIC. The crystal structures with InhA (pdb no. 4BQP) confirm the distinct mechanism of inhibition. The methyl-thiazole group engages with nicotinamide and ribose. Thiazole ring stacks itself via π–π interactions with nicotinamide ring. Thiazole nitrogen forms a hydrogen bond with ribose hydroxyl. Uniquely among the InhA inhibitors, the methyl group of methyl-thiazole forces Tyr 158 into “flipped-out” conformation preventing it to take “flipped-in” conformation observed with the most competitive InhA inhibitors. In this binding mode Tyr 158 adopts the conformation similar to apo form of the enzyme. Additionally hydrogen bond is formed between Met 98 backbone amide and thiadiazole nitrogen. This interaction allows difluorophenyl ring enter the hydrophobic pocket. Thiadiazole, pyrazole and difluorophenyl all come into close proximity of Met 103 forming various van der Waals interactions. In contrast to most of the competitive inhibitors with “Tyr 158 in” conformation the inhibitor binding in “Tyr 158 out” conformation allows to accept hydrogen bond from ribose hydroxyl, but remains uncharged (Scheme 7).
Figure 16. Methyl-thiazole lead sructure and its derivatives published by Shirude et al.
Scheme 6. Arylamides binding mode.
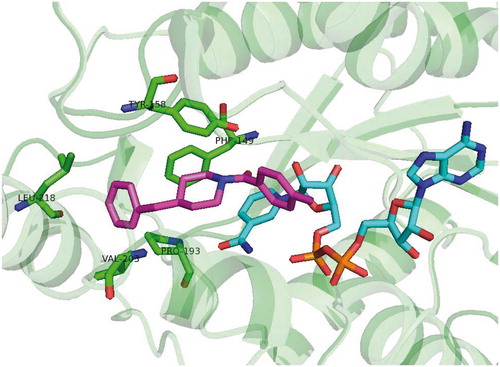
Scheme 7. Methyl-thiazole “Tyr158 out” binding mode.
Conclusion
INH resistance frequently observed at MDR-TB strains is usually associated with KatG alterations. From this point of view the development of novel direct inhibitors bypassing the KatG activation is a rational approach.
In this review we present the summary of direct InhA inhibitors introduced in the past 10 years. Some inhibitors exhibit better in vitro inhibitory potency compared with triclosan and INH. The most prominent of those are diphenyl ethers with IC50 values at nanomolar range and Ki values at subnanomolar range. Ortho substituted diphenyl ethers such as PT70 (introduced by Luckner and colleagues)Citation42 not only showed high inhibitory potency, but were also proven to have slow onset, tight binding mechanism of inhibition. Low Ki values (3500-fold improvement compared with INH, also a tight binding inhibitor) show very high affinity to InhA molecule. Such inhibitors present a good promise for future development. Drawback of novel diphenyl ethers lies in their less then optimal pharmacokinetics. High lipophilicity is usually reported.
Other inhibitor groups were not reported to possess inhibitory activity comparable to the most potent diphenyl ethers with the exception of methyl-thiazoles, which were reported to have IC50 values at nanomolar and some possibly at subnanomolar range.
Closer insight into mechanism of inhibition reveals that this mechanism is for the most of inhibitors very similar. Molecule with InhA inhibitory properties always contains some electronegative moiety, which creates the crucial hydrogen bond with Tyr 158 hydroxyl. This hydrogen bond network is also extended to 2′-hydroxyl group of NAD+ ribose. This electronegative group is usually in close proximity with cyclic non-saturated or aromatic system which interacts via π–π stacking with NAD+ pyridine. Exception is again presented by methyl thiazoles, whose crystallographic studies reveal novel atypical “Tyr 158 out” mechanism.
Currently known InhA inhibitors show sufficient activity against their target. However, inhibitory properties frequently are not in correlation with antimycobacterial and in vivo results. Further development should be focused on compounds with better pharmacokinetic profile and membrane penetration.
Declaration of interest
The authors report no declarations of interest. This work was supported by project no. CZ.1.07/2.3.00/20.0235. The Czech Ministry of Education, Youth and Sports project SVV 2014-260-062 is also acknowledged.
References
- World Health Organization (WHO). Global tuberculosis report 2013 [Online]. Geneva: WHO; 2013. Available at: http://apps.who.int/iris/bitstream/10665/91355/1/9789241564656_eng.pdf?ua=1
- Velasquez GE, Becerra MC, Gelmanova IY, et al. Improving outcomes for multidrug-resistant tuberculosis: aggressive regimens prevent treatment failure and death. Clin Infect Dis 2014;59:9–15
- Gandi NR, Moll A, Sturm AW, et al. Extensively drug-resistant tuberculosis as a cause of death in patients co-infected with tuberculosis and HIV in a rural area of South Africa. Lancet 2006;368:1575–80
- Rawat R, Whitty A, Tonge PJ. The isoniazid-NAD adduct is a slow, tight-binding inhibitor of InhA, the Mycobacterium tuberculosis enoyl reductase: adduct affinity and drug resistance. Proc Natl Acad Sci USA 2003;100:13881–6
- Vale N, Gomes P, Santos HA. Metabolism of the antituberculosis drug ethionamide. Curr Drug Metabol 2013;14:151–8
- Barry CE 3rd, Lee RE, Mdluli K, et al. Mycolic acids: structure, biosynthesis and physiological functions. Prog Lipid Res 1998;37:143–79
- Vilcheze C, Baughn AD, Tufariello J, et al. Novel inhibitors of InhA efficiently kill Mycobacterium tuberculosis under aerobic and anaerobic conditions. Antimicrob Agents Chemother 2011;55:3889–98
- Massengo-Tiassé RP, Cronan JE. Diversity in enoyl-acyl carrier protein reductases. Cell Mol Life Sci 2009;66:1507–17
- Rozwarski DA, Vilcheze C, Sugantino M, et al. Crystal structure of the Mycobacterium tuberculosis enoyl-ACP reductase, InhA, in complex with NAD+ and a C16 fatty acyl substrate. J Biol Chem 1999;274:15582–9
- Parikh S, Moynihan DP, Xiao G, Tonge PJ. Roles of tyrosine 158 and lysine 165 in the catalytic mechanism of InhA, the enoyl-ACP reductase from Mycobacterium tuberculosis. Biochem 1999;38:13623–34
- Lu XY, Huang K, You QD. Enoyl acyl carrier protein reductase inhibitors: a patent review (2006–2010). Expert Opin Ther Patents 2011;21:1007–22
- Tonge PJ, Kisker C, Slayden RA. Development of modern InhA inhibitors to combat drug resistant strains of Mycobacterium tuberculosis. Curr Top Med Chem 2007;7:489–98
- Maresca A, Vullo D, Scozzafava A, et al. Inhibition of the β-class carbonic anhydrases from Mycobacterium tuberculosis with carboxylic acids. J Enzyme Inhib Med Chem 2013;28:392–6
- Maresca A, Scozzafava A, Vullo D, Supuran CT. Dihalogenated sulfanilamides and benzolamides are effective inhibitors of the three β-class carbonic anhydrases from Mycobacterium tuberculosis. J Enzyme Inhib Med Chem 2013;28:384–7
- Maresca A, Carta F, Vullo D, Supuran CT. Dithiocarbamates strongly inhibit the β-class carbonic anhydrases from Mycobacterium tuberculosis. J Enzyme Inhib Med Chem 2013;28:407–11
- Pan P, Tonge PJ. Targeting InhA, the FASII enoyl-ACP reductase: SAR studies on novel inhibitor scaffolds. Curr Top Med Chem 2012;12:672–93
- McMurry LM, Oethinger M, Levy SB. Triclosan targets lipid synthesis. Nature 1998;394:531–2
- Ward WH, Holdgate GA, Rowsell S, et al. Kinetic and structural characteristics of the inhibition of enoyl (acyl carrier protein) reductase by triclosan. Biochem 1999;38:12514–25
- Pidugu LS, Kapoor M, Surolia N, et al. Structural basis for the variation in triclosan affinity to enoyl reductases. J Mol Biol. 2004;343:147–55
- Jones RD, Jampani HB, Newman JL, Lee AS. Triclosan: a review of effectiveness and safety in health care settings. Am J Infect Control 2000;28:184–96
- Freundlich JS, Wang F, Vilcheze C, et al. Triclosan derivatives: towards potent inhibitors of drug-sensitive and drug-resistant Mycobacterium tuberculosis. Chem Med Chem 2009;4:241–8
- Sullivan TJ, Truglio JJ, Boyne ME, et al. High affinity InhA inhibitors with activity against drug-resistant strains of Mycobacterium tuberculosis. ACS Chem Biol 2006;1:43–53
- Betts JC, McLaren A, Lennon MG, et al. Signature gene expression profiles discriminate between isoniazid-, thiolactomycin-, and triclosan-treated Mycobacterium tuberculosis. Antimicrob Agents Chemother 2003;47:2903–13
- Copeland RA, Pompliano DL, Meek TD. Drug-target residence time and its implications for lead optimization. Nat Rev Drug Dis 2006;5:730–9
- Boshoff HI, Myers TG, Copp BR, et al. The transcriptional responses of Mycobacterium tuberculosis to inhibitors of metabolism: novel insights into drug mechanisms of action. J Biol Chem 2004;279:40174–84
- Boyne ME, Sullivan TJ, am Ende CW, et al. Targeting fatty acid biosynthesis for the development of novel chemotherapeutics against Mycobacterium tuberculosis: evaluation of A-ring-modified diphenyl ethers as high-affinity InhA inhibitors. Antimicrob Agents Chemother 2007;51:3562–7
- am Ende CW, Knudson SE, Liu N, et al. Synthesis and in vitro antimycobacterial activity of B-ring modified diaryl ether InhA inhibitors. Bioorg Med Chem Lett 2008;18:3029–33
- Lipinski CA, Lombardo F, Dominy BW, Feeney PJ. Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Adv Drug Deliver Rev 2001;46:3–26
- Freundlich JS, Anderson JW, Sarantakis D, et al. Synthesis, biological activity, and X-ray crystal structural analysis of diaryl ether inhibitors of malarial enoyl acyl carrier protein reductase. Part 1: 4′-substituted triclosan derivatives. Bioorg Med Chem Lett 2005;15:5247–52
- Lu H, England K, am Ende C, et al. Slow-onset inhibition of the FabI enoyl reductase from francisella tularensis: residence time and in vivo activity. ACS Chem Biol 2009;4:221–31
- Luckner SR, Liu N, am Ende CW, et al. A slow, tight binding inhibitor of InhA, the enoyl-acyl carrier protein reductase from Mycobacterium tuberculosis. J Biol Chem 2010;285:14330–7
- Lu H, Tonge PJ. Drug-target residence time: critical information for lead optimization. Curr Opin Chem Biol 2010;14:467–74
- Pan P, Knudson SE, Bommineni GR, et al. Time-dependent diaryl ether inhibitors of InhA: structure-activity relationship studies of enzyme inhibition, antibacterial activity, and in vivo efficacy. Chem Med Chem 2014;9:776–91
- Kuo MR, Morbidoni HR, Alland D, et al. Targeting tuberculosis and malaria through inhibition of enoyl reductase. J Biol Chem 2003;278:20851–9
- Menendez C, Gau S, Lherbet C, et al. Synthesis and biological activities of triazole derivatives as inhibitors of InhA and antituberculosis agents. Eur J Med Chem 2011;46:5524–31
- Menendez C, Chollet A, Rodriguez F, et al. Chemical synthesis and biological evaluation of triazole derivatives as inhibitors of InhA and antituberculosis agents. Eur J Med Chem 2012;52:275–83
- He X, Alian A, Stroud R, Ortiz de Montellano PR. Pyrrolidine carboxamides as a novel class of inhibitors of enoyl acyl carrier protein reductase from Mycobacterium tuberculosis. J Med Chem 2006;49:6308–23
- Matviiuk T, Rodriguez F, Saffon N, et al. Design, chemical synthesis of 3-(9H-fluoren-9-yl)pyrrolidine-2,5-dione derivatives and biological activity against enoyl-ACP reductase (InhA) and Mycobacterium tuberculosis. Eur J Med Chem 2013;70:37–48
- Matviiuk T, Mori G, Lherbet C, et al. Synthesis of 3-heteryl substituted pyrrolidine-2,5-diones via catalytic Michael reaction and evaluation of their inhibitory activity against InhA and Mycobacterium tuberculosis. Eur J Med Chem 2014;71:46–52
- He X, Alian A, Ortiz de Montellano PR. Inhibition of the Mycobacterium tuberculosis enoyl acyl carrier protein reductase InhA by arylamides. Bioorg Med Chem 2007;15:6649–58
- Pauli I, dos Santos RN, Rostirolla DC, et al. Discovery of new inhibitors of Mycobacterium tuberculosis InhA enzyme using virtual screening and a 3D-pharmacophore-based approach. J Chem Inf Model 2013;53:2390–401
- Castro PJ, Fernandez MR, Fernandez VEP, et al. 3-Amino-pyrazole derivatives useful against tuberculosis. Patent no.: WO2012049161 A1. Google Patents; 2012. Available from: http://www.google.com/patents/WO2012049161A1?cl=fr
- Shirude PS, Madhavapeddi P, Naik M, et al. Methyl-thiazoles: a novel mode of inhibition with the potential to develop novel inhibitors targeting InhA in Mycobacterium tuberculosis. J Med Chem 2013;56:8533–42