
Abstract
Several new oxalamide and 2-butenediamide derivatives have been designed, synthesized and evaluated as the acetyl- and butyryl-cholinesterase inhibitors for Alzheimer’s disease. The enzyme inhibitory activity of the synthesized compounds was measured using Ellman’s colorimetric method. It was revealed that compound 1a (N,N′-bis-(4-chloro-benzyl)-N,N′-diphenyl-oxalamide) showed maximum activity against BuChE with a half maximal inhibitory concentration (IC50) = 1.86 µM and compound 2a (but-2-enedioic acid bis-[(4-chloro-benzyl)-phenyl-amide]) exhibited optimum AChE (IC50 = 1.51 µM) inhibition with a high-selectivity index. To better understand the enzyme–inhibitor interaction of the most active compounds towards cholinesterase, molecular modelling studies were carried out. Docking simulations revealed that inhibitors 1a and 2a targeted both the catalytic active site and the peripheral anionic site of 1ACJ and 1P0I.
Introduction
Dementia is a loss of brain function that occurs with certain diseases. Alzheimer’s disease (AD) is one form of dementia that gradually gets worse over time and it can cause symptoms such as the loss of memory, learning and language skillsCitation1–4. The progression and definite causes of AD are still mostly unknown, but the most common hypotheses to have been put forward are based on causative factors and include the cholinergic, amyloid, tau and metal hypothesesCitation5. Standard medical treatment for AD includes cholinesterase inhibitors (ChEIs) and partial N-methyl-d-aspartate (NMDA) antagonistsCitation6,Citation7. The cholinergic hypothesis is one of the oldest and most popular hypotheses outlining the pathogenesis of AD. According to the cholinergic hypothesis, the main approach of the current pharmacotherapy for AD is to increase the levels of acetylcholine (ACh) through the inhibition of cholinesterase enzymes (ChEs)Citation8–10. Acetylcholinesterase (AChE) and butyrylcholinesterase (BuChE), which are located in the central nervous system (CNS), are able to hydrolyse the neurotransmitter AChCitation11. Studies have shown that AD is defined by the rapid loss of AChE activity in the early stages of the disease, along with an increasing ratio of AChE as the disease progressesCitation12,Citation13. Furthermore, BuChE has been found to be capable of compensating for the missing AChE catalytic functions in the synaptic cleft, and its activity increases by 30–60% during ADCitation14–17. Due to the role of BuChE in the hydrolysis of ACh, the inhibition of both ChEs using a dual inhibitor should result in increased levels of ACh in the brain, which provide more successful clinical efficacy of ADCitation18,Citation19.
High levels of metals like mercury cause severe and fatal neurologic diseases, as well as learning and memory problems and movement disordersCitation20. However, some metals are essential for brain function. Imbalances in the level and distribution of these metals in the brain, especially zinc, copper and iron, may play a role in diseases like AD (this pertains to the so-called “metal hypothesis”)Citation21. The level of metal ions (Fe2+, Cu2+ and Zn2+) in AD patients is 3–7 times higher than it is in healthy individualsCitation22. For example, abnormally high levels of iron are often found in the brains of people who die of Alzheimer’s diseaseCitation23. In addition, copper, iron and zinc are all associated with abnormal clumps of brain proteins (plaques), which are a hallmark of Alzheimer’s diseaseCitation24–26.
The aim of this study was to synthesize and develop new oxalamide and 2-butenediamide derivatives as potential anticholinesterase and chelator agents.
Materials and methods
Experimental
The melting points were measured on an Electrothermal 9100 (Thomas Hoover, USA) melting-point apparatus. The 1H and 13C NMR spectra were recorded with a Bruker FT-400(100) MHz (Rheinstetten, Germany) spectrometer using deuterated chloroform (CDCl3) as the solvent. Mass spectra were recorded on an Agilent 1200 (Waldbronn, Germany) mass spectrometer at 10 eV. Benzaniline compounds were synthesized with a CEM 3100 (Matthews, NC) microwave oven. Reaction progress and product mixtures were routinely checked by thin-layer chromatography (TLC) on Merck SilicaGel F254 (Darmstadt, Germany) aluminium plates. The chemical reagents and solvents used in this study were purchased from Merck (Darmstadt, Germany) or Sigma Aldrich (Munich, Germany).
A general procedure for the synthesis of oxalamide compounds (1a–h)
(4-Chloro-benzyl)-(4-substituted-phenyl)-amine derivatives were prepared with microwave (150 W, 120 °C) irradiation under solvent-free phase transfer catalysis conditions. To synthesise the oxalamide derivatives, a mixture of (4-chloro-benzyl)-(4-substituted-phenyl)-amine (1.4 mmol), triethylamine (1.4 mmol) and oxalyl chloride (0.7 mmol) in tetrahydrofuran (10 ml) was stirred at room temperature for 12 h. The reaction mixture was quenched with 15 ml of distilled water, and the aqueous phase was extracted with two portions of CH2Cl2. The combined organic layers were dried over Na2SO4, filtered and concentrated. The obtained solid was recrystallized from ethanol. The synthesis pathway is shown in Scheme 1.
Scheme 1. Synthesis of the target compounds 1a–h and 2a–h.
N,N′-Bis-(4-chloro-benzyl)-N,N′-diphenyl-oxalamide (1a) was obtained from (4-chloro-benzyl)-phenyl-amine according to the general procedure as white solid. Yield 68%; m.p. 102–105 °C. The crude compound was recrystallized from ethanol. 1H-NMR (400 MHz, CDCl3) δ: 4.62 (s, 4H, 2× CH2-N), 6.66 (d, 4H, J = 8.40 Hz, Ar-H), 6.87 (d, 4H, J = 8.56 Hz, Ar-H), 7.06 (d, 4H, J = 8.40 Hz, Ar-H), 7.31–7.27 (m, 2H, Ar-H), 7.37–7.35 (m, 2H, Ar-H). 13C-NMR (100 MHz, CDCl3) δ: 51.37 (CH2–N), 128.50, 128.60, 129.14, 129, 17 129.54, 133.23, 134.81, 139.23, 164.34 (C=O). ESI-MS m/z [M+H] 489.4.
N,N′-Bis-(4-chloro-benzyl)-N,N′-di-p-tolyl-oxalamide (1b) was obtained from (4-chloro-benzyl)-p-tolyl-amine according to the general procedure as white solid. Yield 64%; m.p. 175–178 °C. The crude compound was recrystallized from ethanol. 1H-NMR (400 MHz, CDCl3) δ: 2.45 (s, 6H, 2× Ar-CH3), 4.58 (s, 4H, 2× CH2–N), 6.69 (d, 4H, J = 8.36 Hz, Ar-H), 6.75 (d, 4H, J = 8.28 Hz, Ar-H), 7.05 (d, 4H, J = 8.36 Hz, Ar-H), 7.07 (d, 4H, J = 8.28 Hz, Ar-H). 13C-NMR (100 MHz, CDCl3) δ: 21.19 (Ar-CH3), 51.41 (CH2–N), 128.37, 129.61, 129.71, 133.13, 135.03, 136.70, 138.65, 164.52 (C=O). ESI-MS m/z [M+H] 517.5.
N,N′-Bis-(4-chloro-benzyl)-N,N′-bis-(4-ethyl-phenyl)-oxalamide (1c) was obtained from (4-chloro-benzyl)-(4-ethyl-phenyl)-amine according to the general procedure as white solid. Yield 59%; m.p. 192–195 °C. The crude compound was recrystallized from ethanol. 1H-NMR (400 MHz, CDCl3) δ: 1.28–1.25 (t, 6H, 2× CH2–CH3), 2.72–2.66 (q, 4H, 2× CH2–CH3), 4.59 (s, 4H 2× CH2–N), 6.71 (d, 4H, J = 8.40 Hz, Ar-H), 6.77 (d, 4H, J = 8.32 Hz, Ar-H), 7.05 (d, 4H, J = 8.40 Hz, Ar-H), 7.09 (d, 4H, J = 8.30 Hz, Ar-H). 13C-NMR (100 MHz, CDCl3) δ: 15.18 (CH2–CH3), 28.41 (CH2–CH3), 51.46 (CH2–N), 128.36, 128.40, 129.67, 129.71, 133.12, 135.07, 136.85, 144.79, 164.55 (C=O). ESI-MS m/z [M+H] 545.7.
N,N′-Bis-(4-chloro-benzyl)-N,N′-bis-(4-methoxy-phenyl)-oxalamide (1d) was obtained from (4-chloro-benzyl)-(4-methoxy-phenyl)-amine according to the general procedure as white solid. Yield 44%; m.p. 187–190 °C. The crude compound was recrystallized from ethanol. 1H-NMR (400 MHz, CDCl3) δ: 3.83 (s, 6H, 2× OCH3), 4.58 (s, 4H, 2× CH2–N), 6.66 (d, 4H, J = 8.30 Hz, Ar-H), 6.77 (d, 4H, J = 9.30 Hz, Ar-H), 6.81 (d, 4H, J = 9.28 Hz, Ar-H), 7.06 (d, 4H, J = 8.30 Hz, Ar-H). 13C-NMR (100 MHz, CDCl3) δ: 51.40 (CH2-N), 51.42 (OCH3), 114.02, 128.43, 129.73, 130.08, 131.87, 133.18, 135.10, 159.59, 164.73 (C=O). ESI-MS m/z [M + H] 549.3.
N,N′-Bis-(4-chloro-benzyl)-N,N′-bis-(4-ethoxy-phenyl)-oxalamide (1e) was obtained from (4-chloro-benzyl)-(4-ethoxy-phenyl)-amine according to the general procedure as white solid. Yield 26%; m.p. 137–140 °C. The crude compound was recrystallized from ethanol. 1H-NMR (400 MHz, CDCl3) δ: 1.49–1.46 (t, 6H, 2× OCH2–CH3), 4.05–4.00 (q, 4H, 2× OCH2CH3), 4.57 (s, 4H, 2× CH2-N), 6.65 (d, 4H, J = 8.28 Hz, Ar-H), 6.75 (d, 4H, J = 9.24 Hz, Ar-H), 6.78 (d, 4H, J = 9.32 Hz, Ar-H), 7.06 (d, 4H, J = 8.32 Hz, Ar-H). 13C-NMR (100 MHz, CDCl3) δ: 14.81 (OCH2CH3), 51.40 (CH2–N), 63.74 (OCH2CH3), 114.41, 128.42, 129.69, 130.04, 131.68, 133.13, 135.14, 159.64, 164.82 (C=O). ESI-MS m/z [M+H] 577.5.
N,N′-Bis-(4-chloro-benzyl)-N,N′-bis-(4-fluoro-phenyl)-oxalamide (1f) was obtained from (4-chloro-benzyl)-(4-fluoro-phenyl)-amine according to the general procedure as white solid. Yield 52%; m.p. 147–150 °C. The crude compound was recrystallized from ethanol. 1H-NMR (400 MHz, CDCl3) δ: 4.59 (s, 4H, 2× CH2-N), 6.85 (d, 4H, J = 8.88 Hz, Ar-H), 6.86 (d, 4H, J = 8.88 Hz, Ar-H), 6.98 (d, 4H, J = 8.20 Hz, Ar-H), 7.12 (d, 4H, J = 8.36 Hz, Ar-H). 13C-NMR (100 MHz, CDCl3) δ: 51.37 (CH2–N), 115.98, 116.20, 128.66, 129.68, 130.59, 130.67, 133.67, 134.46 (C-F), 164.12 (C=O). ESI-MS m/z [M+H] 525.3.
N,N′-Bis-(4-chloro-benzyl)-N,N′-bis-(4-chloro-phenyl)-oxalamide (1g) was obtained from (4-chloro-benzyl)-(4-chloro-phenyl)-amine according to the general procedure as white solid. Yield 42%; m.p. 182–185 °C. The crude compound was recrystallized from ethanol. 1H-NMR (400 MHz, CDCl3) δ: 4.59 (s, 4H, 2× CH2–N), 6.69 (d, 4H, J = 8.40 Hz, Ar-H), 6.80 (d, 4H, J = 8.68 Hz, Ar-H), 7.14 (d, 4H, J = 8.40 Hz, Ar-H), 7.27 (d, 4H, J = 8.84 Hz, Ar-H). 13C-NMR (100 MHz, CDCl3) δ: 51.31 (CH2–N), 128.73, 129.40, 129.66, 129.95, 133.77, 134.33, 134.91, 137.53, 163.89 (C=O). ESI-MS m/z [M + H] 557.4.
N,N′-Bis-(4-bromo-phenyl)-N,N′-bis-(4-chloro-benzyl)-oxalamide (1h) was obtained from (4-bromo-phenyl)-(4-chloro-benzyl)-amine according to the general procedure as white solid. Yield 66%; m.p. 217–220 °C. The crude compound was recrystallized from ethanol. 1H-NMR (400 MHz, CDCl3) δ: 4.59 (s, 4H, 2× CH2–N), 6.69 (d, 4H, J = 8.40 Hz, Ar-H), 6.73 (d, 4H, J = 8.64 Hz, Ar-H), 7.16 (d, 4H, J = 8.40 Hz, Ar-H), 7.42 (d, 4H, J = 8.64 Hz, Ar-H). 13C-NMR (100 MHz, CDCl3) δ: 51.28 (CH2–N), 128.78, 128.82, 129.65, 132.42, 133.78, 134.32, 138.05, 163.80 (C=O). ESI-MS m/z [M + H] 645.7.
General procedure for the synthesis of 2-butenediamide derivatives (2a–h)
The mixture of (4-chloro-benzyl)-(4-substituted-phenyl)-amine (1.4 mmol), triethylamine (TEA) (1.4 mmol) and dry ethylacetate (5 ml) was cooled with an ice bath to 0–5 °C, and fumaryl chloride (0.65 mmol) in 5 ml dry ethylacetate was added dropwise by syringe over 30 min. The reaction mixture was then stirred at room temperature for 12 h. The reaction mixture was quenched with 50 ml of water, and the aqueous phase was extracted with two portions of CH2Cl2. The combined organic layers were dried over MgSO4, filtered and concentrated by rotary evaporation. The target compounds were recrystallized from ethanol. The synthesis procedure is described in our previously reported studyCitation27.
But-2-enedioic acid bis-[(4-chloro-benzyl)-phenyl-amide] (2a) was obtained from (4-chloro-benzyl)-phenyl-amine according to general procedure as bright yellow. Yield 52%; m.p. 203–206 °C. The crude compound was recrystallized from ethanol. 1H-NMR (400 MHz, CDCl3) δ: 4.85 (s, 4H, 2× CH2–N), 6.86 (s, 2H, fumaryl CH=CH), 6.96 (d, 4H, J = 7.88 Hz, Ar-H), 7.07 (d, 4H, J = 8.32 Hz, Ar-H), 7.19 (d, 4H, J = 8.32 Hz, Ar-H), 7.35 (d, 4H, J = 7.88 Hz, Ar-H). 13C-NMR (100 MHz, CDCl3) δ: 52.90 (CH2–N), 128.01, 128.43, 128.61, 129.87, 130.09, 132.06 (CH=CH), 133.40, 135.41, 140.97, 164.34 (C=O). ESI-MS m/z [M + H] 515.7.
But-2-enedioic acid bis-[(4-chloro-benzyl)-p-tolyl-amide] (2b) was obtained from (4-chloro-benzyl)-p-tolyl-amine according to general procedure as white solid. Yield 45%; m.p. 201–204 °C. The crude compound was recrystallized from ethanol. 1H-NMR (400 MHz, CDCl3) δ: 2.35 (s, 6H, 2× Ar-CH3), 4.83 (s, 4H, 2× CH2–N), 6.82 (d, 4H, J = 8.20 Hz, Ar-H), 6.86 (s, 2H, fumaryl CH=CH), 7.08 (d, 4H, J = 8.40 Hz, Ar-H), 7.13 (d, 4H, J = 8.08 Hz, Ar-H), 7.18 (d, 4H, J = 8.02 Hz, Ar-H). 13C-NMR (100 MHz, CDCl3) δ: 21.11 (Ar-CH3), 52.87 (CH2–N), 127.75, 128.56, 130.12, 130.47, 131.97 (CH=CH), 133.32, 135.54, 138.32, 138.41, 164.50 (C=O). ESI-MS m/z [M + H] 543.2.
But-2-enedioic acid bis-[(4-chloro-benzyl)-(4-ethyl-phenyl)-amide] (2c) was obtained from (4-chloro-benzyl)-(4-ethyl-phenyl)-amine according to general procedure as bright yellow. Yield 55%; m.p. 194–197 °C. The crude compound was recrystallized from ethanol. 1H-NMR (400 MHz, CDCl3) δ: 1.27–1.23 (t, 6H, 2× CH2–CH3), 2.68–2.62 (q, 4H, 2× CH2–CH3), 4.83 (s, 4H 2× CH2–N), 6.86 (d, 4H, J = 8.24 Hz, Ar-H), 6.89 (s, 2H, fumaryl CH=CH), 7.09 (d, 4H, J = 8.40 Hz, Ar-H), 7.16 (d, 4H, J = 8.24 Hz, Ar-H), 7.19 (d, 4H, J = 8.40 Hz, Ar-H). 13C-NMR (100 MHz, CDCl3) δ: 24.35 (CH2–CH3), 29.89 (CH2–CH3), 52.39 (CH2–N), 127.91, 128.36, 129.67, 129.71, 134.42 (CH=CH), 135.07, 136.45, 143.85, 163.35 (C=O). ESI-MS m/z [M + H] 571.4.
But-2-enedioic acid bis-[(4-chloro-benzyl)-(4-methoxy-phenyl)-amide] (2d) was obtained from (4-chloro-benzyl)-(4-methoxy-phenyl)-amine according to general procedure as bright yellow solid. Yield 52%; m.p. 160–163 °C. The crude compound was recrystallized from ethanol. 1H-NMR (400 MHz, CDCl3) δ: 3.80 (s, 6H, 2× OCH3), 4.81 (s, 4H, 2× CH2-N), 6.84 (s, 2H, fumaryl CH=CH), 6.85 (br s, 8H, Ar-H), 7.08 (d, 4H, J = 8.24 Hz, Ar-H), 7.19 (d, 4H, J = 8.28 Hz, Ar-H). 13C-NMR (100 MHz, CDCl3) δ: 52.95 (CH2–N), 55.44 (OCH3), 114.97, 128.58, 129.17, 130.24, 131.94 (CH=CH), 133.35, 133.52, 135.51, 159.29, 164.64 (C=O). ESI-MS m/z [M + H] 575.6.
But-2-enedioic acid bis-[(4-chloro-benzyl)-(4-ethoxy-phenyl)-amide] (2e) was obtained from (4-chloro-benzyl)-(4-ethoxy-phenyl)-amine according to general procedure as white solid. Yield 45%; m.p. 167–170 °C. The crude compound was recrystallized from ethanol. 1H-NMR (400 MHz, CDCl3) δ: 1.44–1.41 (t, 6H, 2× OCH2–CH3), 4.04–3.98 (q, 4H, 2× OCH2CH3), 4.80 (s, 4H, 2× CH2–N), 6.82 (s, 2H, fumaryl CH=CH), 7.07 (d, 4H, J = 6.56 Hz, Ar-H), 7.08 (d, 4H, J = 6.64 Hz, Ar-H), 7.19 (d, 4H, J = 6.52 Hz, Ar-H), 7.20 (d, 4H, J = 6.48 Hz, Ar-H). 13C-NMR (100 MHz, CDCl3) δ: 14.80 (OCH2CH3), 52.94 (CH2-N), 63.70 (OCH2CH3), 115.39, 128.56, 129.14, 130.25, 131.93 (CH=CH), 133.34, 135.54, 158.73, 165.21 (C=O). ESI-MS m/z [M + H] 603.5.
But-2-enedioic acid bis-[(4-chloro-benzyl)-(4-fluoro-phenyl)-amide] (2f) was obtained from (4-chloro-benzyl)-(4-fluoro-phenyl)-amine according to general procedure as white solid. Yield 27%; m.p. 194–197 °C. The crude compound was recrystallized from ethanol. 1H-NMR (400 MHz, CDCl3) δ: 4.82 (s, 4H, 2× CH2-N), 6.82 (s, 2H, fumaryl CH=CH), 6.92 (d, 4H, J = 8.84 Hz, Ar-H), 7.03 (d, 4H, J = 8.24 Hz, Ar-H), 7.06 (d, 4H, J = 8.36 Hz, Ar-H), 7.21 (d, 4H, J = 8.40 Hz, Ar-H). 13C-NMR (100 MHz, CDCl3) δ: 52.92 (CH2–N), 128.72, 129.92, 130.14, 130.17, 130.20, 131.99 (CH=CH), 133.63, 135.07 (C–F), 164.20 (C=O). ESI-MS m/z [M + H] 551.2.
But-2-enedioic acid bis-[(4-chloro-benzyl)-(4-chloro-phenyl)-amide] (2g) was obtained from (4-chloro-benzyl)-(4-chloro-phenyl)-amine according to general procedure as white solid. Yield 57%; m.p. 216–219 °C. The crude compound was recrystallized from ethanol. 1H-NMR (400 MHz, CDCl3) δ: 4.83 (s, 4H, 2× CH2–N), 6.82 (s, 2H, fumaryl CH=CH), 6.89 (d, 4H, J = 8.60 Hz, Ar-H), 7.07 (d, 4H, J = 8.31 Hz, Ar-H), 7.21 (d, 4H, J = 8.30 Hz, Ar-H), 7.33 (d, 4H, J = 8.60 Hz, Ar-H). 13C-NMR (100 MHz, CDCl3) δ: 52.83 (CH2-N), 128.76, 129.34, 130.15, 132,06 (CH=CH), 133.68, 134.96, 139.31, 164.02 (C=O). ESI-MS m/z [M + H] 583.4.
But-2-enedioic acid bis-[(4-bromo-phenyl)-(4-chloro-benzyl)-amide] (2h) was obtained from (4-bromo-phenyl)-(4-chloro-benzyl)-amine according to general procedure as bright yellow. Yield 27%; m.p. 213–216 °C. The crude compound was recrystallized from ethanol. 1H-NMR (400 MHz, CDCl3) δ: 4.83 (s, 4H, 2× CH2–N), 6.82 (d, 4H, J = 8.52 Hz, Ar-H), 6.83 (s, 2H, fumaryl CH=CH), 7.07 (d, 4H, J = 8.40 Hz, Ar-H), 7.21 (d, 4H, J = 8.36 Hz, Ar-H), 7.48 (d, 4H, J = 8.60 Hz, Ar-H). 13C-NMR (100 MHz, CDCl3) δ: 52.79 (CH2–N), 122.55, 128.77, 129.64, 130.13, 132.07 (CH=CH), 133.69, 134.94, 139.83, 163.95 (C=O). ESI-MS m/z [M + H] 670.8.
Inhibition studies on AChE and BuChE
AChE, BuChE, 5,5-dithiobis-(2-nitrobenzoic acid) DTNB, acetylthiocholine iodide (ATCI) and butyrylthiocholine iodide (BTCI) were purchased from Sigma Aldrich. The inhibitory activities of the AChE and BuChE test compounds were evaluated using Ellman’s colorimetric methodCitation28 with some modifications using commercially available neostigmine bromideCitation11 and ambenonium dichlorideCitation29 as the reference compounds. The test compounds were dissolved in dimethylsulfoxide and then diluted in a 50-mM Tris buffer (pH 8.0) to provide a final concentration range. In a 96-well plate, the assay medium in each well consisted of 50 µl of a Tris buffer, 125 µl of 3 mM DTNB (Ellman’s reagent), 25 µl of 0.2 U/ml enzyme (AChE or BuChE) and a 15-mM substrate (ATCI or BTCI). The assay mixture containing the enzyme, buffer, DTNB and 25 µl of the inhibitor compound was preincubated for 15 min at 37 °C before the substrate was added to begin the reaction. Neostigmine bromide, ambenonium dichloride and all test compounds were prepared at 11 different concentrations: 0.09, 0.195, 0.39, 0.78, 1.56, 3.13, 6.25, 12.5, 25, 50 and 100 µg/ml. The absorbance of the reaction mixture was then measured three times at 412 nm every 45 s using a microplate reader (Bio-Tek ELx800, USA). The results are presented as mean ± standard errors of the experiment. The IC50 values of the compounds showing percentage inhibition, the measurements and the calculations were evaluated by non-linear regression analysis using GraphPad Prism software.
Spectrophotometric measurement of chelation capacity
The ability of metal chelation was determined by measuring the formation of metal complexes in dimethylsulfoxide (DMSO) at room temperature using a UV–visible spectrophotometer (Thermo Electron Heλios) with a wavelength ranging from 190 to 380 nmCitation30,Citation31. The UV absorption of test compounds 1a and 2a, in the absence or presence of CuSO4, FeSO4 and ZnSO4, was recorded in a 1-cm quartz cuvette after 20 min at room temperature. The final volume of the reaction mixture was 3 ml, and the final concentrations of the test compounds and metals were 100 μM.
Docking procedure
The docking study was performed using Surflex-Dock in Sybyl-X 2.0 by Tripos Associates. 3D structures of compounds 1a and 2a were constructed using the Sybyl sketcher module. The structures were minimized using the steepest descent conjugated gradient method until the gradient was 0.001 kcal/mol and max iterations: 1000 with the Tripos force field with the Gasteiger–Huckel charge. The simulation system was built on the crystal structures of 1ACJ and 1P0I, which were obtained from the Protein Data Bank. At the commencement of docking, all the water and ligands were removed and the random hydrogen atoms were added. Docking calculations using Surflex-Dock for 1ACJ and 1P0I were performed through protomol generation by ligand. The parameters used were threshold 0.5 and bloat 0.
Result and discussion
Chemistry
Compounds 1a–h were synthesized by the reaction of oxalyl chloride with para-(4-chloro-benzyl)-(4-substituted-phenyl)-amines in tetrahydrofuran in the presence of triethylamine with moderate to good yields (26–68%). 2-Butenediamide derivatives (2a–h) were obtained in moderate yields (27–57%). The obtained spectroscopic data are in accordance with the predicted structures. The synthesis scheme of the compounds is presented in Scheme 1. In the proton nuclear magnetic resonance (1H-NMR) spectra, the resonance signals of ethylene bridge protons are registered as singlets in the range of 6.82 to 6.89 ppm for compounds 2a–f. The signals for aromatic protons were showed in the range of δ 6.65–7.42 and δ 6.82–7.48 ppm for compounds 1a–h and 2a–h, respectively. The N-CH2 protons appeared at δ 4.57–4.62 ppm for compounds 1a–h and δ 4.80–4.85 ppm for compounds 2a–h. The carbon nuclear magnetic resonance (13C-NMR) spectrum shows characteristic carbon resonance frequencies of carbonyl atoms in the range of 163.80–164.82 ppm for compounds 1a–h and 163.35–165.21 ppm for compounds 2a–h. The aromatic carbons appeared at δ 114.97–159.64 ppm and N–CH2 groups appeared at δ 51.28–52.90 ppm. In the positive electron impact (EI) mass spectra, the molecular ion peaks [M + H] show that the predicted compound has formed. Spectral data of the compounds are documented in Supplementary file.
In vitro cholinesterase enzymes inhibitory activity
The inhibitory activity of the compounds against AChE and BuChE was measured according to the colorimetric assay of EllmanCitation28. Neostigmine and ambenonium were used as the reference compounds. The IC50 values of all tested compounds and their selectivity index for AChE are summarized in . In general, butenediamide derivatives 2a–h showed moderate to good inhibitory activity on AChE with micromolar concentrations. The non-substituted compound among the butenediamide derivatives, 2a (IC50 = 1.51 µM), showed the most potent inhibitory activity against AChE, being 4.47- and 2.69-fold stronger than the reference compounds neostigmine bromide (IC50 = 6.76 µM) and ambenonium dichloride (IC50 = 4.07 µM), respectively. The other compound in the series, 2f, was found to be the second most powerful compound, with an IC50 value of 2.47 µM. In addition, the most potent compounds, 2a and 2f, were found to be more selective inhibitors on AChE than the other compounds with 62.11 and 40.48 selectivity index (SI) values, respectively. At the same time, compounds 2g and 2h showed similar activity to the reference compounds. Compounds 2b and 2c were found to have moderate inhibitory activity on AChE and BuChE, with an IC50 of 87.09 and 26.30 µM, respectively. A similar situation was seen on BuChE inhibition with IC50 values of 11.22 and 35.48 µM for the same compounds (2b and 2c). Compound 2g exhibited the strongest inhibition to BuChE, with an IC50 value of 7.07 µM. In addition, compound 2g exhibited 2.04-fold stronger inhibition against BuChE than the reference compound neostigmine bromide (IC50 = 14.45 µM).
Table 1. Anticholinesterase activity of IC50 (μM) and selectivity of compounds 1a–h and 2a–h on AChE and BuChE.
On the other hand, 1a exhibited the strongest inhibition to BuChE, with an IC50 value of 1.86 µM, which was 7.76- and 3.23-fold more potent than those of neostigmine (IC50 = 14.45 µM) and ambenonium (IC50 = 6.02 µM). In addition, compound 1a had a high level of BuChE inhibitor selectivity. Compound 1b was found to have moderate inhibitory activity on BuChE, with an IC50 of 72.02 µM. The other oxalamide derivatives did not exhibit inhibitory activity against both ChEs (). These compounds in the series were considered to be inactive since their IC50 values were more than 100 µM.
From the IC50 values, the butenediamide derivatives (except for 2d and 2e) seemed to be more effective than the oxalamide derivatives on AChE inhibition due to the ethylene bridge for the linker between the two carbonyl groups. This finding suggests that acetylcholinesterase’s inhibitory activity is strongly dependent on the presence of an ethylene moiety in the inhibitors. In our previously reported study, the results of anticholinesterase activity showed that the α,β-unsaturated moiety contributed to the interaction with cholinesterase enzymesCitation27.
The target compounds (1a–h and 2a–h) were synthesized using different para-substituted anilines with varying electronic properties. The target compounds 2f–h were found to be effective inhibitors against AChE, which have various electron-drawing halogen groups (F, Cl and Br) at the para-position of the phenyl ring. To support this approach, as shown in , 2b–d possessing an electron-donating group (–CH3, –C2H5, –OCH3, –OC2H5) at the para-position of the aniline ring moiety showed a decreased AChE inhibitory activity. These results imply that an electron-drawing group at the para-position of the aniline ring is beneficial to AChE inhibitory activity.
Docking simulation
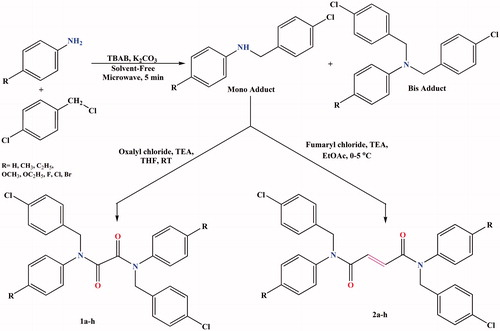
To explore the possible interaction mode of the most potent compounds with AChE and BuChE, molecular modelling simulations were performed between the most active compounds, 2a with AChE and 1a with BuChE, using SYBYL X 2.0 Surflex-Dock software. The enzyme structures were obtained from the Protein Data Bank. The docking results showed that compound 2a displayed multiple binding patterns with Torpedo californica (TcAChE-1ACJ), as shown in . In the 1ACJ-2a complex, the oxygen atom from the carbonyl group created two hydrogen bonds with an OH group of Ser200 (2.54 Å) and an NH group of Gly118 (2.16 Å) CAS. The chloro group in the para-positions on benzyl rings created an H-bond, such as the OH group of Tyr334 (2.55 Å) into the PAS. The benzyl moiety of compound 2a was located on the residues of Trp84 and Phe330 in a “sandwich” form. The p-chlorobenzyl moiety of 2a was observed to bind to CAS via parallel π–π stacking interactions between Trp84 (3.75 Å) and Phe330 (3.23 Å) in a “sandwich” form.
Figure 1. Docking model of compound 2a and TcAChE complex.
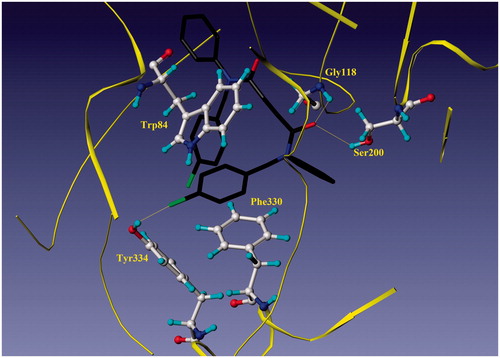
The most potent BuChE inhibitor, 1a, showed interactions with Gly116, Gly117, Ala199 and Trp82 residues of human butyrylcholinesterase enzyme (HuBuChE-1P0I) (). Hydrogen bond interactions occurred between the carbonyl group of compound 1a and the NH groups of Gly116 (1.92 Å), Gly117 (2.06 Å) and Ala199 (2.51 Å) in an oxyanion hole of the CAS of HuBuChE. In addition, one of the phenyl rings entered the PAS of BuChE and π–π stacking interactions occurred with the indole moiety of Trp82 (3.30–5.25 Å).
Figure 2. Docking model of compound 1a and HuBuChE complex.
Metal chelating effect
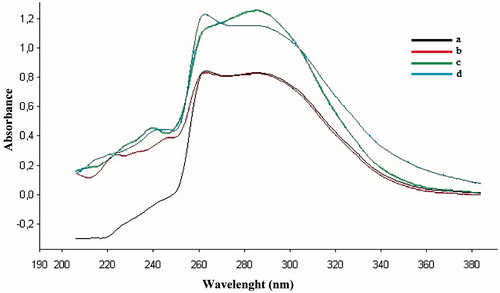
The complexation abilities of the most potent compounds, 1a and 2a, for metals such as Cu2+, Fe2+ and Zn2+ in DMSO were studied using a UV–visible spectrophotometer. The UV absorption of the compounds in DMSO changed with the titration of Cu2+, Fe2+ and Zn2+, and the results are shown in and . The absorption peaks of compounds 1a and 2a in DMSO increased after the addition of Cu2+ and Fe2+. Only compound 1a showed an increased absorption peak after adding ZnSO4. A change in the absorption intensity was observed after adding CuSO4, FeSO4 and ZnSO4, indicating the formation of all biometal complexes for compound 1a (Cu2+, Fe2+ and Zn2+).
Figure 3. (a) UV spectrum of compound 1a (100 μM). (b) Spectrum of a mixture of 1a (100 μM) and ZnSO4 (100 μM). (c) Spectrum of a mixture of 1a (100 μM) and CuSO4. (d) Spectrum of a mixture of 1a (100 μM) and FeSO4 (100 μM).
Figure 4. (a) UV spectrum of compound 2a (100 μM). (b) Spectrum of a mixture of 2a (100 μM) and ZnSO4 (100 μM). (c) Spectrum of a mixture of 2a (100 μM) and CuSO4. (d) Spectrum of a mixture of 2a (100 μM) and FeSO4 (100 μM).
Conclusion
In conclusion, our study involved the synthesis of new oxalamide and 2-butenediamide compounds. These compounds were evaluated as potential inhibitors of ChE and as chelators of biometals. Among the synthesized 2-butenediamide derivatives, compound 2a exhibited the greatest inhibitory potency towards AChE (IC50 value: 1.51 µM) and good Fe2+ and Cu2+ chelating ability. In addition, the most potent oxalamide derivative, compound 1a, showed inhibitory activity towards BuChE (IC50 value: 1.86 µM). In addition, compound 1a exhibited high-metal chelation ability with all of the biometals. Docking simulations showed that compounds 1a and 2a were bound mainly with the catalytic active sites of AChE and BuChE, respectively. Modelled derivatives created lots of hydrogen bonding interactions with the CAS and π–π stacking interactions with the PAS of both ChEs, which confirms their high-inhibitory potency. As a result, further investigations of AD candidates based on these results are in progress.
Supplementary material available online
Supplemental Material.pdf
Download PDF (4.9 MB)Declaration of interest
This research work was supported by Ataturk University Research Fund (Project No: 2013/271), Turkey. The authors have declared no conflicts of interest with the presented data from this article.
References
- Lambert J, Heath S, Even G, et al. Genome-wide association study identifies variants at CLU and CR1 associated with Alzheimer’s disease. Nat Genet 2009;41:1094–9
- Naj AC, Jun G, Beecham GW, et al. Common variants at MS4A4/MS4A6E, CD2AP, CD33 and EPHA1 are associated with late-onset Alzheimer’s disease. Nat Genet 2011;43:436–41
- Piazzi L, Rampa A, Bisi A, et al. Extensive SAR and computational studies of 3-{4-[(benzylmethylamino)methyl]phenyl}-6,7-dimethoxy-2H-2-chromenone (AP2238) derivatives. J Med Chem 2003;46:2279–82
- Seshadri S, Fitzpatrick AL, Ikram MA, et al. Genome-wide analysis of genetic loci associated with Alzheimer disease. J Am Med Assoc 2010;303:1832–40
- Singh M, Kaur M, Kukreja H, et al. Acetylcholinesterase inhibitors as Alzheimer therapy: from nerve toxins to neuroprotection. Eur J Med Chem 2013;70:165–88
- Massoud F, Léger G. Pharmacological treatment of Alzheimer disease. Can J Psychiatry 2011;56:579–88
- Winslow B, Onysko M, Stob C, Hazlewood K. Treatment of Alzheimer disease. Am Fam Physician 2011;83:1403–12
- Akasofu S, Kimura M, Kosasa I, et al. Study of neuroprotection of donepezil, a therapy for Alzheimer’s disease. Chem Biol Interact 2008;175:222–6
- Dumas JA, Newhouse PA. The cholinergic hypothesis of cognitive aging revisited again: cholinergic functional compensation. Pharmacol Biochem Behav 2011;99:254–61
- Tai HC, Serrano-Pozo A, Hashimoto T, et al. The synaptic accumulation of hyperphosphorylated tauoligomers in Alzheimer disease is associated with dysfunction of the ubiquitin-proteasome system. Am J Pathol 2012;181:1426–35
- Skrzypek A, Matysiak J, Niewiadomy A, et al. Synthesis and biological evaluation of 1,3,4-thiadiazole analogues as novel AChE and BuChE inhibitors. Eur J Med Chem 2013;62:311–19
- Darvesh S, Hopkins DA, Geula C. Neurobiology of butyrylcholinesterase. Nat Rev Neurosci 2003;4:131–8
- Greig NH, Utsuki T, Ingram DK, et al. Selective butyrylcholinesterase inhibition elevates brain acetylcholine, augments learning and lowers Alzheimer beta-amyloid peptide in rodent. Proc Natl Acad Sci USA 2005;102:17213–18
- Bullock R, Lane R. Executive dyscontrol in dementia, with emphasis on subcortical pathology and the role of butyrylcholinesterase. Curr Alzheimer Res 2007;4:277–93
- Jhee SS, Shiovitz T, Hartman RD, et al. Centrally acting antiemetics mitigate nausea and vomiting in patients with Alzheimer’s disease who receive rivastigmine. Clin Neuropharmacol 2002;25:122–3
- Kamal MA, Klein P, Luo W, et al. Kinetics of human serum butyrylcholinesterase inhibition by a novel experimental Alzheimer therapeutic, dihydrobenzodioxepine cymserine. Neurochem Res 2008;33:745–53
- Mesulam MM, Guillozet A, Show P, et al. Acetylcholinesterase knockouts establish central cholinergic pathways and can use butyrylcholinesterase to hydrolyze acetylcholine. Neuroscience 2002;110:627–39
- Basiri A, Murugaiyaha V, Osman H, et al. An expedient, ionic liquid mediated multi-component synthesis of novel piperidone grafted cholinesterase enzymes inhibitors and their molecular modeling study. Eur J Med Chem 2013;67:221–9
- Khoobi M, Alipour M, Moradi A, et al. Design, synthesis, docking study and biological evaluation of some novel tetrahydrochromeno [3′,4′:5,6]pyrano[2,3-b]quinolin-6(7H)-one derivatives against acetyl- and butyrylcholinesterase. Eur J Med Chem 2013;68:291–300
- Budimir A. Metal ions, Alzheimer’s disease and chelation therapy. Acta Pharm 2011;61:1–14
- Liu G, Huang W, Moir RD, et al. Metal exposure and Alzheimer’s pathogenesis. J Struct Biol 2006;155:45–51
- Zatta P, Drago D, Bolognin S, Sensi SL. Alzheimer’s disease, metal ions andmetal homeostatic therapy. Trends Pharmacol Sci 2009;30:346–55
- Loef M, Walach H. Copper and iron in Alzheimer’s disease: a review and its dietary implications. Br J Nutr 2012;107:7–19
- Dong J, Atwood CS, Anderson VE, et al. Metal binding and oxidation of amyloid-beta within isolated senile plaque cores: Raman microscopic evidence. Biochemistry 2003;42:2768–73
- Opazo C, Huang X, Cherny RA, et al. Cu-dependent catalytic conversion of dopamine, cholesterol, and biological reducing agents to neurotoxic H(2)O(2). J Biol Chem 2002;277:40302–8
- Yan H, Yao PF, Chen SB, et al. Synthesis and evaluation of 7,8-dehydrorutaecarpine derivatives as potential multifunctional agents for the treatment of Alzheimer’s disease. Eur J Med Chem 2013;63:299–312
- Ellman GL, Courtney D, Andies V, Featherstone RM. A new and rapid colorimetric determination of acetylcholinesterase activity. Biochem Pharmacol 1961;7:88–95
- Musileka K, Komloova M, Holas O, et al. Preparation and in vitro screening of symmetrical bis-isoquinolinium cholinesterase inhibitors bearing various connecting linkage – implications for early Myasthenia gravis treatment. Eur J Med Chem 2011;46:811–18
- Huang W, Lv D, Yu H, et al. Dual-target-directed 1,3-diphenylurea derivatives: BACE 1 inhibitor and metal chelator against Alzheimer’s disease. Bioorg Med Chem 2010;18:5610–15
- Joseph R, Ramanujam B, Acharya A, et al. Experimental and computational studies of selective recognition of Hg2+ by amide linked lower rim 1,3-dibenzimidazole derivative of calix[4]arene: species characterization in solution and that in the isolated complex, including the delineation of the nanostructures. J Org Chem 2008;73:5745–58
- Yerdelen KO, Gul HI. Synthesis and anticholinesterase activity of fumaramide derivatives. Med Chem Res 2013;22:4920–9