
Abstract
Novel 1-(1,3-disubstituted-imidazolidyn-2-ylidene)-3-ethoxycarbonylmethylurea derivatives (3a–3j) were obtained from appropriate 1-aryl-3-arylsulfonyl-1H-imidazolidine-2-imines (1a–1j) and ethyl isocyanatoacetate (2), which were subjected to condensation. Seven compounds were tested for their antiviral activity against HSV-1 and CVB3 viruses. Among the tested compounds, 3c was found to be active against HSV-1, proving that 4-methoxy substituent as R and 4-methyl substituent as R1 are most beneficial for activity against this virus. Furthermore, 3e and 3g were active against CVB3, which demonstrated that both 4-methyl and 4-chloro substitution is tolerated as R1, whereas 4-chloro and 2-methoxy substituents are best as R. It was also shown that the active compounds are characterized by relatively big surface area, small ovality, and greatest HOMO and LUMO energies in comparison to the rest of the compounds.
Introduction
Viral infections are a permanent health problem of mankind. The increasing danger of viral infections generates the need to search for new antiviral drugs, which are non-toxic for human beings. Designing safe and effective antiviral drugs is difficult, because viruses use the host’s cells to replicate. Moreover, the major difficulty in developing vaccines and antiviral drugs is due to viral variation. Nowadays, searching for new potential antiviral agents follows two main strategies. One trend deals with the synthesis of new derivatives of already existing antiviral drugs; the chemical synthesis of such compounds is oriented towards creating a new, more effective drug. The other trend of working on new antiviral drugs is to look for natural compounds, mainly of plant origin, aimed to obtain potential chemotherapeuticsCitation1. In order to examine the antiviral activity of new substances, their influence on the propagation of various experimental models of RNA viruses simultaneously with the cytotoxicity of these preparations is defined.
Human herpesvirus 1 (HHV-1), also known as Herpes simplex virus 1 (HSV-1), is a neurotropic virus that establishes latent, life-long infection in dorsal root ganglia. This key feature of HSV-1 can be well summarized by a quote by the British scientist James Lovelock: “An inefficient virus kills its host. A clever virus stays with it”. Most primary infections occur during the first two decades of life and in most cases are asymptomaticCitation2. Reactivation of HSV leads to symptomatic disease. Besides lesions on mucocutaneous surfaces, HSV-1 reactivation may lead to encephalitis, corneal blindness and several peripheral nervous system disordersCitation2,Citation3. There are three classes of drugs licensed for the treatment of HSV-1 infections: guanosine analogs, including acyclovir, valacyclovir, famcilovir and ganciclovir; acyclic nucleotide analog – cidofovir and pyrophosphate analog – foscarnet. All these drugs target the viral DNA replicationCitation4. Among immunocompetent patients, HSV-1 reactivations are usually sparser and have self-limited course, whereas, among patients undergoing immunosuppression reactivations, they are very common and lead to high morbidity. Frequent and long-lasting antiviral treatment of patients with an impaired immune system may lead to the selection and emergence of drug-resistant mutants complicating the treatment. Different resistance phenotypes of HSV-1 show mutations of thymidine kinase (TK, UL23) and DNA polymerase (pol, UL30) genes. HSV-1 isolates cross-resistant to acyclovir, penciclovir and brivudin exhibit mutations in TK gene. HSV-1 resistant to acyclovir, penciclovir, foscarnet and brivudin-sensitive have substitutions in DNA polCitation5. Due to the ongoing emergence of drug-resistant strains of HSV-1, it is crucial to seek for new compound possessing antiviral activity.
Coxsackievirus B3 (CVB3) is a single-stranded RNA enterovirus belonging to the Picornavirdae family, associated with a significant percentage of patients diagnosed with myocarditis and dilated cardiomyopathy (DCM)Citation6. Unfortunately, there is no clinically proven specific treatment for viral myocarditis and DCM, and most patients with DCM eventually require heart transplantation. Possible options for the management of -induced myocarditis involve supportive therapies (hemodynamic support, diuretics, digoxin, angiotensin-converting enzyme inhibitors, angiotensin-II receptor blockers and beta-adrenergic blockers) in combination with non-specific antiviral agents to decrease the viral load (type I interferon or nucleotide analogs such as ribavirin)Citation7,Citation8. Recently, pleconaril was shown to interact with picornavirus anti-receptor and block CVB3 entry into the host, resulting in a reduced viral load in the heart. However, due to its high toxicity, pleconaril was not approved by the American FDA and is used only in a compassionate mannerCitation8. Because of the lack of specific antiviral therapy for viral myocarditis it is crucial to look for new compounds with activity against CVB3.
The aim of the present work was to search for novel antiviral compounds among substituted imidazoline derivatives. In recent years, a number of simple and fused derivatives of substituted imidazoline have been synthesized. Substituted imidazoline are an important class of molecules with a large spectrum of biological properties. Arylsulfonylimidazolinones were reported as analogs possessing broad spectrum of potent activity against various human cancersCitation9–13 as well as hypoglycemic activityCitation14,Citation15; however, to our best knowledge, they were not tested for antiviral activity. In particular, 1-(1-benzoylindoline-5-sulfonyl)-4-phenylimidazolidinonesCitation11 were found to have cytotoxic activity and it was justified to check if similar compounds will have antiviral activity. The presented series of compounds is a part of our large-scale project on compounds against HSV and CVB3 viruses and subsequent reports will be published soon. This constitutes the rationale of this work. Furthermore, the set of the substituents was selected in order to enable to study the effect of substituents on antiviral activity.
Methods
Chemistry
All the commercial reagents and solvents were purchased from Sigma-Aldrich (St. Louis, MO) and used without purification. Reactions were routinely monitored by thin-layer chromatography (TLC) in silica gel (60 F254 Merck plates, Darmstadt, Germany) and the products were visualized with ultraviolet light of 254 nm wavelength. All NMR spectra were acquired on Bruker AVANCE III 300 MHz spectrometer (Billerica, MA) equipped with BBO Z-gradient probe. Spectra were recorded at 25 °C using DMSO as a solvent with a non-spinning sample in 5 mm NMR-tubes. MS spectra were recorded on Bruker microTOF-Q II (Billerica, MA) and processed using Compass Data Analysis software. The elementary analysis was performed with the application of Perkin-Elmer analyzer (Waltham, MA). Melting points were determined with Boetius apparatus (Jena, Germany).
General procedure for the synthesis of compounds 3a–3j
A total of 1.29 g (0.01 mol) of ethyl isocyanatoacetate (2) was dissolved in 25 mL of dichloromethane under the atmosphere of dry nitrogen and added to the solution free base (0.01 mol) of 1-aryl-3-arylsulfonyl-1H-imidazolidine-2-imines (1a–1j) dissolved in 50 mL of dichloromethane. The mixture was shaken for 24 h at room temperature. Solvent was removed by distillation and the rubber-like residue was treated with warm propan-2-ol. The solid product 1-(1-aryl-3-arylsulfonylimidazolidin-2-ylidene)-3-ethoxycarbonylmethylurea (3a–3j) was filtrated out and purified by crystallization from propan-2-ol.
1-[1-phenyl-3-(4-methylphenylsulfonylimidazolidyn-2-ylidene)]-3-ethoxycarbonylmethylurea (3a)
From a general procedure with 3.15 g (0.01 mol) of 1a and 1.29 g (0.01 mol) of 2 obtaining 1.42 g of 3a (32% yield), white crystalline solid, m.p. 260–262 °C; 1H NMR (DMSO-d6, 300 MHz): δ (ppm): 8.15 (s, 1H, NH); 7.06–7.90 (m, 9H, H-Ar); 4.02 (dd, 2H, CH2, J = 9.1, J′ = 7.6 Hz); 4.20 (dd, 2H, CH2, J = 9.0, J′ = 7.4 Hz); 3.84–3.97 (m, 2H, CH2); 3.30 (s, 2H, CH2); 2.56 (s, 3H, CH3); 1.15–1.20 (t, 3H, CH3, J = 7.3 Hz); 13C NMR (DMSO-d6, 75 MHz): δ (ppm): 14.6 (CH3), 21.7 (CH3), 42.1 (CH2), 43.7 (CH2), 44.1 (imidazolidine C-4), 48.7 (imidazolidine C-5), 123.1, 123.3, 125.8, 125.9, 128.5, 128.8 (C-Ar), 159.0 (imidazolidine-C-2), 170.9 (C=O), 171.5 (C=O); EIMS m/z 445.1 [M + H]+. HREIMS (m/z): 444.1620 [M+] (calcd for C21H24N4O5S 444.5210); Anal. Calcd for: C21H24N4O5S: C, 56.74; H, 5.44; N, 12.61; S, 7.21. Found: C, 56.99; H, 5.36; N, 12.89; S, 7.28.
1-[1-(2-mehoxylphenyl)-3-(4-methylphenylsulfonylimidazolidyn-2-ylidene)]-3-ethoxycarbonylmethylurea (3b)
From a general procedure with 3.45 g (0.01 mol) of 1b and 1.29 g (0.01 mol) of 2 obtaining 1.61 g of 3b (34 % yield), white crystalline solid, m.p. 240–241 °C; 1H NMR (DMSO-d6, 300 MHz): δ (ppm): 8.53 (s, 1H, NH); 6.83–7.89 (m, 8H, H-Ar); 4.05 (dd, 2H, CH2, J = 8.9, J′ = 7.4 Hz); 4.15 (dd, 2H, CH2, J = 9.0, J′ = 7.6 Hz); 3.68–3.82 (m, 2H, CH2); 3.71 (s, 2H, CH2); 3.29 (s, 3H, OCH3); 2.44 (s, 3H, CH3); 1.15–1.22 (t, 3H, CH3, J = 7.2 Hz); 13C NMR (DMSO-d6, 75 MHz,): δ (ppm): 13.9 (CH3), 14.5 (CH3), 21.0 (OCH3), 41.3 (CH2), 42.8 (CH2), 44.1 (imidazolidine C-4), 46.1 (imidazolidine C-5), 115.4, 116.5, 116.9, 118.1. 124.1, 124.9, 128.5, 128.8, 130.1, 132.9 (C-Ar), 164.6 (imidazolidine-C-2), 159.1 (imidazolidine-C-2), 170.5 (C=O), 171.8 (C=O); EIMS m/z 475.1 [M + H]+. HREIMS (m/z): 474.5330 [M+] (calcd for C22H26N4O6S 474.5480); Anal. Calcd for: C22H26N4O6S: C, 55.68; H, 5.52; N, 11.81; S, 6.76. Found: C, 55.49; H, 5.46; N, 11.89; S, 6.83.
1-[1-(4-methoxyphenyl)-3-(4-methylphenylsulfonylimidazolidyn-2-ylidene)]-3-ethoxycarbonylmethylurea (3c)
From a general procedure with 3.45 g (0.01 mol) of 1c and 1.29 g (0.01 mol) of 2 obtaining 1.61 g of 3c (36% yield), white crystalline solid, m.p. 220–221 °C; 1H NMR (DMSO-d6, 300 MHz): δ (ppm): 8.35 (s, 1H, NH); 6.89–7.59 (m, 8H, H-Ar); 4.04 (dd, 2H, CH2, J = 8.8, J′ = 7.4 Hz); 4.14 (dd, 2H, CH2, J = 9.1, J′ = 7.6 Hz); 3.81–3.92 (m, 2H, CH2); 3.79 (s, 2H, CH2); 3.19 (s, 3H, OCH3); 2.30 (s, 3H, CH3); 1.25–1.29 (t, 3H, CH3, J = 7.3 Hz); 13C NMR (DMSO-d6, 75 MHz): δ (ppm): 14.2 (CH3), 14.8 (CH3), 21.6 (OCH3), 41.9 (CH2), 42.7 (CH2), 44.2 (imidazolidine- C-4), 45.3 (imidazolidine C-5), 114.1, 114.5, 115.6, 118.1. 125.8, 125.9, 128.5, 128.8, 130.1, 132.9 (C-Ar), 164.6 (imidazolidine-C-2), 170.9 (C=O), 171.5 (C=O); EIMS m/z 475.1 [M + H]+. HREIMS (m/z): 474.4671 [M+] (calcd for C22H26N4O6S 474.5480); Anal. Calcd for: C22H26N4O6S: C, 55.68; H, 5.52; N, 11.81; S, 6.76. Found: C, 55.89; H, 5.31; N, 11.59; S, 6.65.
1-[1-(2,3-dimethylphenyl)-3-4-(methylphenylsulfonylimidazolidyn-2-ylidene)]-3-ethoxycarbonylmethylurea (3d)
From a general procedure with 3.15 g (0.01 mol) of 1d and 1.29 g (0.01 mol) of 2 obtaining 1.86 g of 3d (42% yield), white crystalline solid, m.p. 231–232 °C; 1H NMR (DMSO-d6, 300 MHz): δ (ppm): 8.26 (s, 1H, NH); 7.09–7.80 (m, 7H, H-Ar); 4.06 (dd, 2H, CH2, J = 9.0, J′ = 7.5 Hz); 4.19 (dd, 2H, CH2, J = 9.1, J′ = 7.6 Hz); 3.73–3.99 (m, 2H, CH2); 3.69 (s, 2H, CH2), 2.44 (s, 3H, CH3); 2.34 (s, 3H, CH3); 2.05 (s, 3H, CH3); 1.14–1.19 (t, 3H, CH3, J = 7.1 Hz); 13C NMR (DMSO-d6, 75 MHz): δ (ppm): 12.1 (CH3), 12.9 (CH3), 14.9 (CH3), 21.66 (CH3), 41.9 (CH2), 42.7 (CH2), 44.3 (imidazolidine C-4), 45.8 (imidazolidine C-5), 112.4, 120.4, 121.9, 128.6, 128.8, 129.1, 129.9 (C-Ar), 164.8 (imidazolidine C-2), 170.7 (C=O), 171.9 (C=O); EIMS m/z 473.1 [M + H]+. HREIMS (m/z): 472.1630 [M+] (calcd for C23H28N4O3S 472.5750); Anal. Calcd for: C23H28N4O5S: C, 58.45; H, 5.97; N, 11.85; S, 6.78. Found: C, 58.59; H, 5.86; N, 11.71; S, 6.83.
1-[1-(4-chlorophenyl)-3-(4-methylphenylsulfonylimidazolidyn-2-ylidene)]-3-ethoxycarbonylmethylurea (3e)
From a general procedure with 3.49 g (0.01 mol) of 1e and 1.29 g (0.01 mol) of 2 obtaining 1.86 g of 3e (39% yield), white crystalline solid, m.p. 253–255 °C; 1H NMR (DMSO-d6, 300 MHz): δ (ppm): 8.11 (s, 1H, NH); 7.06–7.89 (m, 8H, H-Ar); 4.01 (dd, 2H, CH2, J = 9.1, J′ = 7.6 Hz); 4.17 (dd, 2H, CH2, J = 9.0, J′ = 7.5 Hz); 3.89–3.98 (m, 2H, CH2); 3.40 (s, 2H, CH2); 2.45 (s, 3H, CH3); 1.17–1.22 (t, 3H, CH3, J = 7.1 Hz); 13C NMR (DMSO-d6, 75 MHz): δ (ppm): 14.5 (CH3), 15.1 (CH3), 41.1 (CH2), 43.9 (CH2), 44.1 (imidazolidine C-4), 45.8 (imidazolidine C-5), 119.1, 120.4, 121.8, 122.3, 123.2, 126.6,128.9, 130.1 (C-Ar), 164.8 (imidazolidine C-2), 170.9 (C=O), 171.6 (C=O); EIMS m/z 479.3 [M + H]+. HREIMS (m/z): 478.5316 [M+] (calcd for C21H23N4ClO5S 478.9700); Anal. Calcd for: C21H23N4ClO5S: C, 52.66; H, 4.84; N, 11.69; Cl, 7.40; S, 6.69. Found: C, 52.59; H, 4.96; N, 11.54; Cl, 7.35; S, 6.81.
1-[1-phenyl-3-(4-chlorophenylsulfonylimidazolidyn-2-ylidene)]-3-ethoxycarbonylmethylurea (3f)
From a general procedure with 3.35 g (0.01 mol) of 1f and 1.29 g (0.01 mol) of 2 obtaining 2.27 g of 3f (49% yield), white crystalline solid, m.p. 242–243 °C; 1H NMR (DMSO-d6, 300 MHz): δ (ppm): 8.39 (s, 1H, NH); 6.93–7.64 (m, 9H, H-Ar); 4.03 (dd, 2H, CH2, J = 9.0, J′ = 7.5 Hz); 4.16 (dd, 2H, CH2, J = 8.9, J′ = 7.6 Hz); 3.77–3.89 (m, 2H, CH2); 3.71 (s, 2H, CH2), 1.16–1.20 (t, 3H, CH3, J = 7.1 Hz); 13C NMR (DMSO-d6, 75 MHz): δ (ppm): 14.3 (CH3), 41.1 (CH2), 42.9 (CH2), 44.1 (imidazolidine C-4), 45.8 (imidazolidine C-5), 120.1, 120.5, 121.7, 122.9, 123.2, 125.6, 127.3, 129.1, 129.6 (C-Ar), 165.6 (imidazolidine C-2), 170.4 (C=O), 171.5 (C=O); EIMS m/z 465.3 [M + H]+. HREIMS (m/z): 464.1655 [M+] (calcd for C20H21N4ClO5S 464.9430); Anal. Calcd for: C20H21N4ClO5S: C, 51.66; H, 4.55; N, 12.05; Cl, 7.62; S, 6.89. Found: C, 51.69; H, 4.36; N, 12.09; Cl, 7.81; S, 6.77.
1-[1-(2-methoxyphenyl)-3-(4-chlorophenylsulfonylimidazolidyn-2-ylidene)]-3-ethoxycarbonylmethylurea (3g)
From a general procedure with 3.65 g (0.01 mol) of 1g and 1.29 g (0.01 mol) of 2 obtaining 1.86 g of 3 g (45% yield), white crystalline solid, m.p. 283–284 °C; 1H NMR (DMSO-d6, 300 MHz): δ (ppm): 8.50 (s, 1H, NH); 6.81–7.63 (m, 8H, H-Ar); 4.07 (dd, 2H, CH2, J = 8.9, J′ = 7.4 Hz); 4.19 (dd, 2H, CH2, J = 9.0, J′ = 7.6 Hz); 3.83–4.01 (m, 2H, CH2); 3.56 (s, 2H, CH2); 3.36 (s, 3H, OCH3); 1.19–1.22 (t, 3H, CH3, J = 7.2 Hz); 13C NMR (DMSO-d6, 75 MHz): δ (ppm): 14.5 (CH3), 21.4 (OCH3), 41.3 (CH2), 42.9 (CH2), 44.1 (imidazolidine C-4), 46.1 (imidazolidine C-5), 114.4, 115.5, 118.9, 119.1.119.5, 124.1, 124.9, 128.5, 128.3, 130.5, 132.9 (C-Ar), 159.1 (imidazolidine C-2), 170.5 (C=O), 171.8 (C=O); EIMS m/z 495.1 [M + H]+. HREIMS (m/z): 494.1430 [M+] (calcd for C21H23N4ClO6S 494.9700); Anal. Calcd for: C21H23N4ClO6S: C, 50.95; H, 4.68; N, 11.33; Cl, 7.16; S, 6.47. Found: C, 50.99; H, 4.76; N, 11.39; Cl, 7.24; S, 6.54.
1-[1-(4-methoxyphenyl)-3-(4-chlorophenylsulfonylimidazolidyn-2-ylidene)]-3-ethoxycarbonylmethylurea (3h)
From a general procedure with 3.65 g (0.01 mol) of 1h and 1.29 g (0.01 mol) of 2 obtaining 3.21 g of 3h (65% yield), white crystalline solid, m.p. 216–219 °C; 1H NMR (DMSO-d6, 300 MHz): δ (ppm): 8.31 (s, 1H, NH); 7.11–7.64 (m, 8H, H-Ar); 4.06 (dd, 2H, CH2, J = 8.8, J′ = 7.4 Hz); 4.21 (dd, 2H, CH2, J = 9.1, J′ = 7.6 Hz); 3.64–3.82 (m, 2H, CH2); 3.75 (s, 2H, CH2); 3.49 (s, 3H, OCH3); 1.21–1.29 (t, 3H, CH3, J = 7.3 Hz); 13C NMR (DMSO-d6, 75 MHz): δ (ppm): 14.7 (CH3), 21.6 (OCH3), 41.9 (CH2), 42.9 (CH2), 44.3 (imidazolidine C-4), 46.3 (imidazolidine C-5), 115.1, 115.5, 118.6, 118.8. 122.8, 122.9, 127.5, 128.8, 130.1, 132.9 (C-Ar), 164.6 C-2, 158.2 (imidazolidine C-2), 170.1 (C=O), 171.5 (C=O); EIMS m/z 495.6 [M + H]+. HREIMS (m/z): 494.3369 [M+] (calcd for C21H23N4ClO6S 494.9700); Anal. Calcd for: C21H23N4ClO6S: C, 50.95; H, 4.68; N, 11.33; Cl, 7.16; S, 6.47. Found: C, 50.83; H, 4.54; N, 11.43; Cl, 7.11; S, 6.39.
1-[1-(2,3-dimethoxyphenyl)-3-(4-chlorophenylsulfonylimidazolidyn-2-ylidene)]-3-ethoxycarbonylmethylurea (3i)
From a general procedure with 3.63 g (0.01 mol) of 1i and 1.29 g (0.01 mol) of 2 obtaining 3.0 g of 3i (61% yield), white crystalline solid, m.p. 206–208 °C; 1H NMR (DMSO-d6, 300 MHz): δ (ppm): 8.56 (s, 1H, NH); 7.05–7.69 (m, 7H, H-Ar); 4.07 (dd, 2H, CH2, J = 9.0, J′ = 7.5 Hz); 4.16 (dd, 2H, CH2, J = 9.1, J′ = 7.4 Hz); 3.77–3.90 (m, 2H, CH2); 3.44 (s, 2H, CH2); 2.34 (s, 3H, CH3); 2.15 (s, 3H, CH3); 1.12–1.17 (t, 3H, CH3, J = 7.1 Hz); 13C NMR (DMSO-d6, 75 MHz): δ (ppm): 14.1 (CH3), 14.9 (CH3), 21.66 (CH3), 41.6 (CH2), 42.7 (CH2), 44.3 (imidazolidine C-4), 45.8 (imidazolidine C-5), 111.4, 112.4, 112.9, 120.6, 120.8, 125.1, 129.9 (C-Ar), 164.8 (imidazolidine C-2), 170.7 (C=O), 171.9 (C=O); EIMS m/z 493.4 [M + H]+. HREIMS (m/z): 492.1455 [M+] (calcd for C22H25N4ClO5S 492.9970); Anal. Calcd for: C22H25N4ClO5S: C, 53.59; H, 5.11; N, 11.36; Cl, 7.19; S, 6.50. Found: C, 53.43; H, 5.14; N, 11.31; Cl, 7.09; S, 6.69.
1-[1-(4-chlorophenyl)-3-(4-chlorophenylsulfonylimidazolidyn-2-ylidene)]-3-ethoxycarbonylmethylurea (3j)
From a general procedure with 3.70 g (0.01 mol) of 1j and 1.29 g (0.01 mol) of 2 obtaining 2.39 g of 3j (48% yield), white crystalline solid, m.p. 217–218 °C; 1H NMR (DMSO-d6, 300 MHz): δ (ppm): 8.19 (s, 1H, NH); 6.83–7.74 (m, 8H, H-Ar); 4.05 (dd, 2H, CH2, J = 9.0, J′ = 7.6 Hz); 4.25 (dd, 2H, CH2, J = 9.0, J′ = 7.5 Hz); 3.61–3.82 (m, 2H, CH2);3.85 (s, 2H, CH2), 1.21–1.30 (t, 3H, CH3, J = 7.1 Hz); 13C NMR (DMSO-d6, 75 MHz,): δ (ppm): 14.9 (CH3), 41.0 (CH2), 43.7 (CH2), 44.3 (imidazolidine C-4), 45.8 (imidazolidine C-5), 122.3, 122.5, 121.9, 122.5, 123.1, 125.6 (C-Ar), 165.6 (imidazolidine C-2), 170.4 (C=O), 171.8 (C=O); EIMS m/z 500.4 [M + H]+. HREIMS (m/z): 499.1843 [M+] (calcd for C20H20N4Cl2O5S 499.3920); Anal. Calcd for: C20H20N4Cl2O5S: C, 48.10; H, 4.03; N, 11.33; Cl, 17.70; S, 6.42. Found: C, 48.19; H, 4.09; N, 11.31; Cl, 17.79; S, 6.63.
Molecular modeling
The studied compounds were modeled applying the LigPrep protocol from the Schrödinger SuiteCitation16. In order to sample different protonation states of ligands in physiological pH, the Epik module was usedCitation17. The energy and geometry of the compounds were further optimized using the Hartree-Fock approach and 6-31g(d,p) basis set of Spartan 10Citation18. Parameters to evaluate drug-likeness were calculated using VegaZZ v. 3.0.1 (Milano, Italy)Citation19 (number of atoms), Discovery Studio v. 3.1 (Accellys, San Diego, CA)Citation20 (molar mass, number of rings, lipophilicity, number of rotatable bonds), ACD Labs (Ontario, Canada) (molar refractivity, number of hydrogen bond donors and acceptors), and the Schrödinger Suite (Cambridge, MA) (a number of rigid bonds) as described previouslyCitation21–23. Drug-likeness was also evaluated with Osiris Property Explorer (Allschwil, Switzerland)Citation24. This approach is based on a list of about 5300 distinct substructure fragments with associated drug-likeness scores. The drug-likeness is calculated by summing up score values of those fragments that are present in the molecule under investigation. ADMET parameters were calculated with Discovery Studio 3.1 (solubility, blood-brain permeation) or Osiris Property ExplorerCitation24 (toxicity risks). The prediction of toxicity by this tool relies on a precomputed set of structural fragment that gives rise to toxicity alerts in case they are encountered in the investigated structure. In order to perform structure–activity relationship studies, HOMO and LUMO energies, lipophilicity and polarizability were calculated with Discovery Studio 3.1Citation20. HOMO and LUMO orbitals as well as a map of the electrostatic potential (ESP) onto a surface of the electron density were visualized with ArgusLab (Seattle, WA)Citation25. Molecular surface area, polar surface area, molecular volume and ovality were calculated with VegaZZCitation19.
Antiviral activity
Antiviral activity assay
Antiviral activity assays were similar for HSV-1 and CVB3. After 24 h of incubation, the cell culture was infected with appropriate virus in the dose of 100 TCID50/mL. After 1 h incubation at 37 °C, the suspension of the virus was removed and the media with 2% of serum together with the tested compounds in the maximum non-toxic concentration were added to the cell cultures. The virus diluted in the culture media without tested compounds was used as a control. Acyclovir and ribavirin were used as a reference compounds. After 48 h of incubation at 37 °C, the cells were frozen and after thawing the virus was titrated in the Vero cell culture (ECACC No. 84113001 – established from the kidney of a normal adult African Green monkey). The cytopathic effect (CPE) of the virus was examined by a light microscope and the titer of virus was estimated according to the Reed-Muench methodCitation26. Viral titers were determined by tissue culture infection dose (TCID50) assays.
Cell cultures and viruses
The Vero cell culture (ECACC No. 84113001 - established from the kidney of a normal adult African Green monkey) was used in the experiment. The media in the culture (Dulbecco's Modified Eagle Medium – DMEM, Cytogen, Sinn, Germany) were supplemented with 10% fetal bovine serum (FBS, Sigma), 100 U/mL of penicillin and 0.1 mg/mL of streptomycin (Polfa-Tarchomin, Warsaw, Poland). The cell culture was incubated at 37 °C in the 5% CO2 atmosphere. For antiviral activity of examined compounds the HSV-1 (ATCC No. VR-260) and Coxsackievirus B3 – CVB3 (ATCC No. VR-30) from the American Type Culture Collection were used. The viruses were propagated in the Vero cell culture. Viruses stock was stored at −70 °C until used.
Cytotoxicicty assay
Compounds were dissolved in dimethyl sulfoxide (DMSO – POCH, Poland) in the concentration of 50 mg/mL and further diluted with a complete test medium. 100 µL of the Vero cell suspension was plated into 96-well plastic plates (NUNC) at a cell density 1.5 × 104 cells per well. After 24 h of incubation at 37 °C, the media were removed and the cells were treated with examined substances diluted in the media with 2% of serum. The cells were submitted to a series of compound concentrations, from 1000 µg/mL to 1.9 µg/mL. Two-fold serial dilutions of compounds were added to the cells in triplicates. The culture cells were incubated for 72 h at 37 °C in the 5% CO2 atmosphere.
Cytotoxicity of tested compounds was estimated with the use of the MTT method, described by Takenouchi and MunekataCitation27. The MTT method is a quantitative colorimetric toxicity test, based on the transformation of yellow, soluble tetrazolium salts (3-[4,5-dimethylthiazol-2-yl]-2,5-diphenyltetrazolium bromide) to purple-blue insoluble formasane. This process occurs naturally in mitochondria of living cells. After 72 h of incubation with compounds cell, cultures were supplemented with 10 µL per well of 5 mg/mL MTT (Sigma) stock in PBS (BIOMED, Poland), and the incubation was continued for 4 h at 37 °C. Then, 100 µL per well of aqueous solution containing 50% dimethylformamide (POCH, Poland) and 20% SDS (Sigma) to solubilize the insoluble formasane precipitates produced by MTT was added. After the all-night incubation the absorbance was measured using plate reader (Epoch, BioTek, Winooski, VT) at two wavelengths – 540 and 620 nm. On the basis of the results, the cytotoxic concentration (CC50), which is the amount of tested substance that is required to reduce the number of viable cells by 50% compared to the control culture, was determined and was calculated by using the Gen 5 software (version 2.01.14) BioTek. The investigation was carried out in triplicates.
Results and discussion
Chemistry
The synthetic route employed for the preparation of title 1-(1,3-disubstitutedimidazolidyn-2-ylidene)-3-ethoxycarbonymethylurea derivatives is shown in . As it can be seen, a simple synthesis was carried out, from appropriate 1-aryl-3-arylsulfonyl-1H-imidazolidine-2-imines (1a–1j)Citation28–30 and ethyl isocyanatoacetate (2) which were subjected to condensation. At the end of the reaction, the isolated 1-(1,3-disubstytuted-imidazolidyn-2-ylidene)-3-ethoxycarbonylmethylurea derivatives (3a–3j) were obtained as colorless solids.
Scheme 1. The scheme of synthesis of the investigated compounds.
Estimation of drug-likeness
The descriptors applied for the estimation of drug-likeness are presented in . Drug-likeness was assessed using Lipinski’s rule as well as the placement of the investigated compounds in the chemical space determined by the databases of the pharmacologically active compounds (CMC, Comprehensive Medicinal Chemistry Database, containing about 7000 compounds and MDDR, MACCS-II Drug Data Report, containing about 100 000 compounds) according to the methodology of PREADMETCitation31 service as described previouslyCitation21–23. Concerning Lipinski’s rule, all the compounds possess the molar mass below 500, the number of hydrogen bond donors below 5, the number of hydrogen bond acceptors below 10, and the lipophilicity below 5.
Table 1. Parameters for drug-likeness estimation.
Regarding subsequent criteria of drug-likeness, most compounds collected in the CMC database has lipophilicity from −0.4 to 5.6, molar refractivity in the range of 40–130, the number of atoms from 20 to 70 and molar mass from 160 to 480Citation21–23. All the investigated compounds fulfill the first three criteria, whereas compounds 3g, 3h, 3i and 3j have slightly to high molecular mass.
Concerning the compounds in MDDR database, the drug-like substances have the number of rings equal to or greater than 3, the number of rigid bonds equal to or greater than 18, and the number of rotatable bonds equal to or greater than 6Citation21–23. All the compounds fulfill these criteria.
Finally, molecule drug-likeness score (fragment-based score) was calculated using Osiris Property ExplorerCitation24. According to this score, compounds 3f, 3g, 3i and 3j are more drug-like than the rest of the compounds. In summary, the investigated compounds may be termed drug-like.
Prediction of ADMET properties
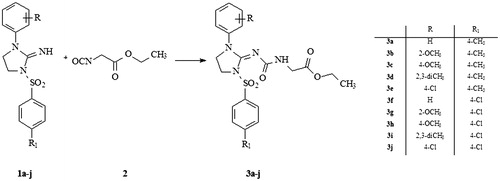
In order to facilitate the selection of compounds for antiviral activity assessment, some ADMET parameters were calculated (). The plot presented in confirms that most of the tested compounds possess reasonably favorable ADMET properties. Comparing the plot in with lipophilicity values from and polar surface areas from , it can be concluded that compounds from 3d, 3i and 3j have less favorable blood-brain permeation properties. All compounds are well absorbed (); however, most compounds, in particular, derivatives 3d, 3e, 3i and 3j are not enough soluble in water as they have values of log S below -4Citation24. Moreover, compounds 3d and 3i have lower overall drug score which combines drug-likeness, cLogP, logS, molecular weight and toxicity risks in one convenient value than may be used to judge the compound's overall potential to qualify as a drugCitation24. Importantly, most compounds are predicted to be non-toxic (all scores equal to 1.00 in ) whereas compounds 3d and 3i have middle risk (score 0.6) irritant properties.
Figure 1. Evaluation of ADMET properties of the studied compounds.
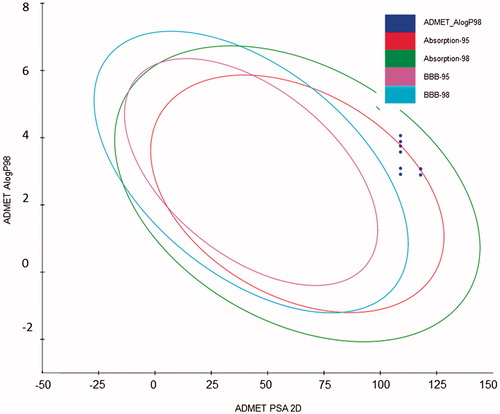
Figure 2. HOMO (A, C) and LUMO (B, D) orbitals for 3c (A, B) and 3g (C, D).
Table 2. ADMET parameters of the studied compounds.
Table 6. Molecular descriptors for structure–activity determination.
Antivral activity
Seven compounds were tested for their antiviral activity. On the basis of calculation of ADMET parameters and our earlier experience on the effect of substituents on the activity, we decided to exclude compounds 3h, 3i and 3j from antiviral activity determination.
The aim of the study was to evaluate the biological activity of 3a–3g against the HSV-1 and CVB3 viruses. CC50 values of compounds 3a–3g ranged from 107.97 to 759.1 µg/mL (). Among tested compounds, compound 3c in the concentration of 250 µg/mL influenced the HSV-1 replication by reducing the virus replication level by 1.5 log, which resulted in reducing the titer by 23.1% (). Compounds 3e and 3g exhibited antiviral activity against Coxsackievirus B3. The compound 3e in the concentration of 125 µg/mL decreased the titer of CVB3 by 0.69 log (9.6%). In the case of the compound 3g, tested in the concentrations of 62 i 125 µg/mL, the titer of CVB3 was decreased by 1.01 log (14%) and 1.34 log (18.5%), respectively ().
Table 3. Cytotoxicity of compounds 3a–3g.
Table 4. Antiviral activity of the compounds against HSV-1.
Table 5. Antiviral activity of the compounds against CVB3.
Structure–activity relationship
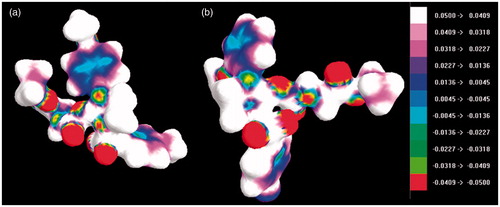
The most favorable pattern of substituents for the activity against HSV-1 was found for 3c with 4-methoxy substituent as R and 4-methyl substituent as R1. The most active compounds against CVB3 are 3e and 3g, proving that both 4-methyl and 4-chloro substituents are tolerated as R1. Regarding R substituent, the best are 4-chloro (3e) and 2-methoxy (3g). Molecular descriptors for structure–activity relationship are presented in . Analysis of data in makes it possible to conclude that most active compounds 3c, 3e and 3g are characterized by relatively big surface area, small ovality and greatest HOMO and LUMO energies. No trend was found for other calculated descriptors. depicts HOMO and LUMO orbitals for most active compounds 3c and 3g, whereas presents maps of electrostatic potential for these derivatives.
Figure 3. The map of the electrostatic potential (ESP) onto a surface of the electron density for 3c (a) and 3g (b).
Conclusions
In this work, we synthesized 10 novel compounds and tested seven of them for the antiviral activity against the HSV-1 and CVB3 viruses. The selection of compounds for antiviral activity determination was facilitated by in silico drug-likeness and ADMET properties calculation. In general, the compounds were found drug-like, although most of them have slightly too low solubility. Moreover, the compounds were predicted to be non-toxic, but two of them may exhibit moderate irritant properties. Among the tested compounds, one, bearing 4-methoxy substituent as R and 4-methyl substituent as R1 was found to be active against HSV-1. Two other compounds were active against CVB3 showing that both 4-methyl and 4-chloro substitution is tolerated as R1, whereas 4-chloro and 2-methoxy substituents are best as R. It was also demonstrated that the active compounds exhibit relatively big surface area, small ovality and greatest HOMO and LUMO energies in comparison to the rest of the compounds.
Declaration of interest
The paper was developed using the equipment purchased within the project “The equipment of innovative laboratories doing research on new medicines used in the therapy of civilization and neoplastic diseases” with the Operational Program Development of Eastern Poland 2007–2013, Priority Axis I modern Economy, operations I.3 Innovation promotion. The research was partially performed during the postdoctoral fellowship of Agnieszka A. Kaczor at University of Eastern Finland, Kuopio, Finland under Marie Curie fellowship. Part of the calculations was performed under a computational grant by Interdisciplinary Center for Mathematical and Computational Modelling (ICM), Warsaw, Poland, grant number G30-18 and under resources and licenses by CSC, Finland.
References
- Krawczyk E, Łuczak M. Antiviral activity of natural compounds and their derivatives. Post Mikrobiol 2005;44:239–52
- Steiner I, Benninger F. Update on Herpes virus infections of the nervous system. Curr Neurol Neurosci Rep 2013;13:414–20
- Hill GM, Ku ES, Dwarakanathan S. Herpes simplex keratitis. Dis Mon 2014;60:239–46
- Wilson SS, Fakioglu E, Herold BC. Novel approaches in fighting herpes simplex virus infections. Expert Rev Anti Infect Ther 2009;7:559–68
- Schubert A, Gentner E, Bohn K, et al. Single nucleotide polymorphisms of thymidine kinase and DNA polymerase genes in clinical herpes simplex virus type 1 isolates associated with different resistance phenotypes. Antiviral Res 2014;107:16–22
- Yajima T, Knowlton KU. Viral myocarditis: from the perspective of the virus. Circulation 2009;19:2615–24
- Andréoletti L, Lévêque N, Boulagnon C, et al. Viral causes of human myocarditis. Arch Cardiovasc Dis 2009;102:559–68
- Yang D, Zhang HM, Ye X, et al. New trends in the development of treatments of viral myocarditis. In: Milei J, ed. Diagnosis and treatment of myocarditis. InTech 2013; DOI: 10.5772/54103. Available online: http://www.intechopen.com/books/diagnosis-and-treatment-of-myocarditis/new-trends-in-the-development-of-treatments-of-viral-myocarditis
- Hui-Soon L, Kyung-Lae P, Sang-Un C, et al. Effect of substituted on benzenesulfonyl motif of 4-phenyl-1-arylsulfonylimidazolidinones for their cytotoxity. Arch Pharm Res 2000;23:579–84
- Il-Whan K, Sang-Hun J. Recognition of the importance of imidazolidinone motif for cytotoxity of 4-phenyl-1-arylsulfonylimidazolidinones using thiazolidine-1,1-dioxide analogs. Arch Pharm Res 2002;25:421–7
- Jung SH, Lee HS, Kim NS, et al. Synthesis and cytotoxic activity of 1-(1-benzoylindoline-5-sulfonyl)-4-phenylimidazolidinones. Arch Pharm Res 2004;27:478–84
- Son-Hyok K, Seong-Cheol B. Evaluation of anticancer activity of 4-vinyl-1-arylsulfonylimidazolidinones. Arch Pharm Res 2006;29:721–7
- El-Deeb IM, Bayoumi SM, El-Sherbeny MA, Abdel-Aziz AA. Synthesis and antitumor evaluation of novel cyclic arylsulfonylureas: ADME-T and pharmacophore prediction. Eur J Med Chem 2010;45:2516–30
- Iqbal Z, Akhtar T, Hendssbee AH, et al. Synthesis, characterization, and hypoglycemic activity of 3-(arylsulfonyl)spiroimidazolidine-2,4-diones. Monatsh Chem 2012;143:497–504
- Hussain A, Kashif MK, Naseer MM, et al. Synthesis and in vivo hypoglycemic activity of new imidazolidine-2,4-dione derivatives. Res Chem Intermed 2014. [Epub ahead of print]. doi: 10.1007/s11164-014-1814-3
- LigPrep, Version 2.4. New York: Schrödinger, LLC; 2010
- Epik, Version 2.1. New York: Schrödinger, LLC; 2010
- Spartan 10. Available from: www.wavefun.com
- Pedretti A, Villa L, Vistoli G. VEGA – an open platform to develop chemo-bio-informatic applications, using plug-in architecture and script programming. J Comput Aided Mol Des 2004;18:167–73
- Discovery Studio 3.1, Accelrys. San Diego, CA; 2009
- Rządkowska M, Szacoń E, Kaczor AA, et al. Synthesis, pharmacological activity and molecular modeling of 1-aryl-7-hydroxy-2,3-dihydroimidazo[1,2-a]pyrimidine-5(1H)-ones and their 6-substituted derivatives. Med Chem 2014;10:460–75
- Rządkowska M, Szacoń E, Kaczor AA, et al. Synthesis, central nervous system activity, and structure–activity relationship of 1-aryl-6-benzyl-7-hydroxy-2,3-dihydroimidazo[1,2-a]pyrimidine-5(1H)-ones. Med Chem Res 2014;23:4221–37
- Szacoń E, Rządkowska M, Kaczor AA, et al. Synthesis, central nervous system activity and structure-activity relationship of N-substituted derivatives of 1-arylimidazolidyn-2-ylideneurea and products of their cyclization. J Enzyme Inhib Med Chem 2015. [Epub ahead of print]. doi:10.3109/14756366.2014.965699
- Osiris Property Explorer. Available from: http://www.organic-chemistry.org/prog/peo/logS.html [last accessed 1 Dec 2014]
- http://www.arguslab.com/arguslab.com/ArgusLab.html [last accessed 1 Dec 2014]
- Reed LJ, Muench MA. A simple method of estimating fifty percent end points. Am J Hyg 1938;27:493–7
- Takenouchi T, Munekata E. Amyloid beta-peptide-induced inhibition of MTT reduction in PC12h and C1300 neuroblastoma cells: effect of nitroprusside. Peptides 1998;19:365–72
- Tkaczyński T, Rządkowska M. Polish patent PL 169902; 1996
- Tkaczyński T, Rządkowska M. Polish patent PL 170445; 1996
- Rządkowska M, Tkaczyński T. Synthesis of new 1-aryl-2-imino-3-arylsulfonylimidazolidines and their pharmacological activity. Pharmazie 1997;50:822
- Lee SK, Chang GS, Lee IH, et al. The PreADME:pc-based program for batch prediction of ADME properties. Istanbul, Turkey: EuroQSAR 2004, 9.5-10