
Abstract
A new series of sulfonamide derivatives of pyrazolo[4,3-e][1,2,4]triazine with chiral amino group has been synthesized and characterized. The compounds were tested for their relaxant effects in the rat aorta. Evaluation of prepared derivatives demonstrated that compound (8a) is probably a non-selective phosphodiesterase (PDE) inhibitor, as it induced aortic relaxation through endothelium-independent mechanism.
Introduction
Cyclic nucleotide phosphodiesterases (PDEs) are key enzymes that control cellular concentrations of the second messengers cyclic adenosine 3′,5′-monophosphate (cAMP) and cyclic guanosine 3′,5′-monophosphate (cGMP)Citation1. To date, 11 families of PDEs have been classified according to the sequence homology and biochemical propertiesCitation2,Citation3. PDE molecules contain a variable regulatory domain and a conserved catalytic domain. However, each PDE family shows characteristic substrate specificity and inhibitor selectivity. Phosphodiesterase 5 (PDE5) is widely distributed in vascular and visceral smooth muscle cells, platelets, kidney, lung and corpus cavernosum, and plays a key role in the regulation of the cellular level of cGMP. The main therapeutic indications for PDE5 inhibitors are the treatment of erectile dysfunction [sildenafil (Viagra), vardenafil (Levitra), and tadalafil (Cialis), udenafil (Zydena)] and idiopathic pulmonary hypertension [sildenafil (Revatio)], although several other potentials have also been identified, such as memory improvement, anticancer therapy and treatment of heart diseasesCitation4. PDE5 is a therapeutic target of considerable research interest as can be seen by numerous publications regarding the design, synthesis and optimization of new PDE5 inhibitorsCitation5,Citation6.
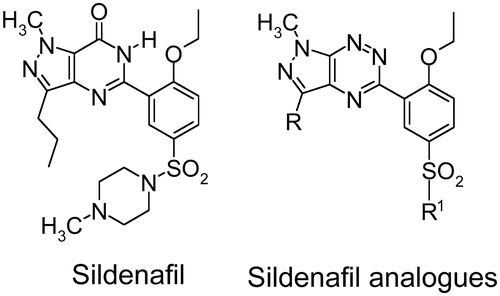
To develop novel PDE5 inhibitors with improved therapeutic efficacy based on the structure of sildenafil, we have prepared a series of sildenafil analogues in which the iminocarbonyl group was replaced with the nitrogen atoms of the triazine ring. Moreover, the methylpiperazine moiety was replaced with various amines (). Because sildenafil is being used not only for treatment of male erectile dysfunction but also for pulmonary hypertension, we have evaluated the relaxant effects of the newly synthesized compounds in the rat aorta.
Figure 1. Structure of sildenafil and its new analogues. The data are the means of four experiments ± S.E.M.
Experimental
Chemistry
General
Melting points were determined on a Mel-Temp apparatus and are uncorrected. 1H and 13C NMR spectra were recorded on a Varian spectrometer (400 MHz for 1H and 100 MHz for Citation13C). The chemical shift values are expressed in ppm (part per million) with tetramethylsilane (TMS) as internal reference. The relative integrals of peak areas agreed with those expected for the assigned structures. Molecular weight of final compounds was assessed by electrospray ionization mass spectrometry (ESI/MS) on a Agilent Technologies 6538 UHD Accurate Mass Q-TOF LC/MS (Lublin, Poland). Elemental compositions are within ±0.4% of the calculated values. For preparation and spectroscopic data of compounds 2a–5a and 8a–c see literatureCitation7,Citation8.
Synthesis of 1-(3-methylsulfanyl-1,2,4-triazin-5-yl)butan-1-one oxime (2b). To a stirred suspension of powdered potassium hydroxide (8.0 g) in dry dimethyl sulfoxide (20 mL) a solution of the 1,2,4-triazine (1) (2.0 g, 15.74 mmol) and nitrobutane (3 mL, 23.88 mmol) in dimethyl sulfoxide (1–2 mL) was added at once at room temperature. The mixture was stirred at room temperature for 2 h, then was poured into ice/water (200 mL) and acidified with acetic acid (pH 6.9–7.0). The product (2b) was precipitated and was collected by filtration and washed with water. The crude product was purified by chromatography (silica gel; CH2Cl2/EtOH 50:1) to afford (2) as a yellowish powder. Yield 86%; m.p. 126–127 °C; 1H NMR (400 MHz, CDCl3) δ: 0.97 (t, J = 7.2 Hz, 3H), 1.57–1.66 (m, 2H), 2.67 (s, 3H), 2.86 (t, 2H, J = 7.2 Hz), 9.45 (s, 1H), 9.96 (bs, 1H). Citation13C NMR (100 MHz, CDCl3) δ: 13.7, 14.2, 19.4, 24.9, 141.7, 153.0, 157.3, 173.7. HRMS (ESI, m/z) Calcd for C8H13N4OS [M + H] 213.0810. Found [M + H] 213.0805. Anal. Calcd for C8H12N4OS: C, 45.27; H, 5.70; N, 26.39. Found: C, 45.30; H, 5.76; N, 26.30.
Synthesis of 1-(3-methylsulfanyl-1,2,4-triazin-5-yl)butan-1-one (3b). The oxime (2b) (2.12 g, 10 mmol) was dissolved in dioxane (40 mL) and treated with a solution of hydrosulfite Na2S2O4 (4.6 g, 26.4 mmol) in water (40 mL). The resulting mixture was stirred at room temperature for 24 h. Dioxane was then removed in vacuo and the residue was treated with 10% HCl (pH ∼3) and bubbled by air for 10 min. Then the mixture was made neutral by addition of solid NaHCO3 and extracted with CH2Cl2. Evaporation of the organic solvent gave a crude product, which was purified by chromatography (silica gel; CH2Cl2/hexane 1:2) to afford ketone (3b) as a yellow oil. Yield 55%. 1H NMR (400 MHz, CDCl3) δ: 1.00 (t, 3H, J = 7.6 Hz), 1.72–1.78 (m, 2H), 2.72 (s, 3H), 3.12 (t, 2H, J = 7.6 Hz), 9.37 (s, 1H). Citation13C NMR (100 MHz, CDCl3) δ: 13.6, 14.0, 16.8, 39.4, 140.9, 147.3, 174.7, 200.9. HRMS (ESI, m/z) Calcd for C8H12N3OS [M + H] 198.0701. Found [M + H] 198.0696. Anal. Calcd for C8H11N3OS: C, 48.71; H, 5.62; N, 21.30. Found: C, 48.80; H, 5.75; N, 21.10.
Synthesis of methylhydrazone of 5-butanoyl-3-methylsulfanyl-1,2,4-triazine (4b). To a solution of (3b) (2.2 g, 11.16 mmol) and methylhydrazine (0.77 g, 16.73 mmol) in 10 mL of ethanol p-toluenosulfonic acid monohydrate (200 mg) was added and the resulting reaction mixture was stirred for 15 min at room temperature (rt). After that time the precipitated solid was filtered off. The crude product was recrystallized from ethanol–water mixture to obtain the pure product (4b) as a yellow solid. Yield 50%; m.p. 129–131 °C. 1H NMR (400 MHz, CDCl3) δ: 0.96 (t, 3H, J = 7.6 Hz), 1.46–1.56 (m, 2H), 2.60–2.64 (m, 5H), 3.28 (s, 3H), 9.40 (s, 1H). Citation13C NMR (100 MHz, CDCl3) δ: 13.7, 14.3, 18.0, 23.8, 38.3, 138.7, 141.6, 153.9, 172.1. HRMS (ESI, m/z) Calcd for C9H16N5S [M + H] 226.1126. Found [M + H] 226.1121. Anal. Calcd for C9H15N5S: C, 47.98; H, 6.71; N, 31.08. Found: C, 48.13; H, 6.90; N, 31.00.
Synthesis of 1-methyl-5-methylsulfanyl-3-propyl-1H-pyrazolo[4,3-e][1,2,4]triazine (5b). Method A: A solution of (4b) (155 mg, 0.78 mmol) and 10% hydrochloric acid (0.1 mL) in ethanol (5 mL) was refluxed for 1 h. After that time the solvent was evaporated in vacuo and the crude product was purified by column chromatography (silica gel, chloroform) to afford the final product (5b) as a yellow oil (130 mg, 0.58 mmol, 58%).
Method B: A mixture of (4b) (1 mmol) and p-toluenosufonic acid (2 mmol) was mixed and heated at 140 °C for 1 min under solvent free reaction condition. After cooling, the solid residue was treated with chloroform and then the major product was isolated by chromatography on silica gel with chloroform to give the corresponding pyrazolotriazine (5b) in 61% yield. 1H NMR (400 MHz, CDCl3) δ: 1.00 (t, 3H, J = 7.6 Hz), 1.83–1.90 (m, 2H), 2.70 (s, 3H), 2.98 (t, 2H, J = 7.6 Hz), 4.23 (s, 3H). Citation13C NMR (100 MHz, CDCl3) δ: 13.9, 14.2, 21.6, 27.9, 34.6, 134.5, 144.3, 147.1, 166.8. HRMS (ESI, m/z) Calcd for C9H14N5S [M + H] 224.0969. Found [M + H] 224.0964. Anal. Calcd for C9H13N5S: C, 48.41; H, 5.87; N, 31.36. Found: C, 48.33; H, 5.97; N, 31.20.
General method synthesis of derivatives (6ab). To a mixture of (5a) or (5b) (3.14 mmol, 1.0 equiv), CuMeSal (1.68 g, 7.85 mmol, 2.5 equiv), 2-ethoxyphenylboronic acid (1.3 g, 7.85 mmol, 2.5 eqiuv) in dry tetrahydrofuran (THF) (25 mL) under argon Pd(PPh3)4 (0.36 g, 0.31 mmol, 0.1 equiv) were added. The reaction mixture was stirred overnight at reflux. The reaction was quenched with a Na2CO3 saturated solution and extracted with dichloromethane. The combined organic phases were dried over MgSO4 and concentrated in vacuo. After purification by column chromatography on silica gel, (hexane:CH2Cl2, 5:1), the desired product (6) was obtained.
3-(2-Ethoxyphenyl)-1,3-dimethyl-1H-pyrazolo[4,3-e][1,2,4]-triazine (6a). Yellow powder. Yield 75%; m.p. 85–86 °C. 1H NMR (400 MHz, CDCl3): δ 1.29–1.33 (t, 3H, J = 14 Hz), 2.70 (s, 3H), 4.10–4.15 (q, 2H, J = 14 Hz), 4.32 (s, 3H); 7.06–7.12 (m, 2H), 7.42–7.46 (m, 2H), 7.74–7.75 (d–d, 1H, J1 = 2.4 Hz, J2 = 8.8 Hz). Citation13C NMR (CDCl3): δ 11.05, 14.69, 34.68, 64.61, 113.47, 120.79, 126.88, 131.09, 131.92, 134.48, 141.97, 146.96, 157.31, 160.10. HRMS (ESI, m/z) Calcd for C14H15N5O [M+] 269.1276. Found [M+] 269.1284. Anal. Calcd for C14H15N5O: C, 62.44; H, 5.61; N, 26.01. Found: C, 62.30; H, 5.70; N, 25.93.
3-(2-Ethoxyphenyl)-1-methyl-3-propyl-1H-pyrazolo[4,3-e][1,2,4]-triazine (6b). Yellow oil. Yield 80%. 1H NMR (400 MHz, CDCl3) δ: 1.03 (t, 3H, J = 7.6 Hz), 1.31 (t, 3H, J = 7.2 Hz), 1.88–1.98 (m, 2H), 3.08 (t, 2H, J = 7.6 Hz), 4.12 (q, 2H, J = 7.2 Hz), 4.32 (s, 3H), 7.06–7.12 (m, 2H), 7.42–7.46 (m, 1H), 7.78 (dd, 1H, J1 = 7.6 Hz, J2 = 1.6 Hz). Citation13C NMR (100 MHz, CDCl3) δ: 14.0, 14.8, 22.0, 28.2, 34.8, 64.7, 113.5, 120.9, 127.0, 131.2, 132.1, 134.5, 146.2, 147.1, 157.4, 160.2. HRMS (ESI, m/z) Calcd for C16H20N5O [M + H] 298.1667. Found [M + H] 298.1662. Anal. Calcd for C16H19N5O: C, 64.63; H, 6.44; N, 23.55. Found: C, 64.55; H, 6.56; N, 23.42.
General method synthesis of derivatives (7ab). Compound (6a) or (6b) (1.88 mmol) was added portion wise to stirred and cooled chlorosulfonic acid (2 mL) in an ice bath under argon atmosphere; the reaction mixture was then warmed to room temperature gradually for 2 h after the addition. The reaction solution was cautiously added to ice-water (15 mL), and the aqueous mixture was extracted with dichloromethane. The combined extracts were dried over anhydrous Na2SO4 and evaporated under vacuum to give the required sulfonyl chloride (7a) or (7b), respectively.
4-Ethoxy-3-(1,3-dimethyl-1H-pyrazolo[4,3-e][1,2,4]-triazin-5-yl)benzene-1-sulfonyl chloride (7a). Yellow crystals. Yield 85%; m.p. 119–120 °C. 1H NMR (400 MHz, CDCl3) δ: 1.37–1.40 (t, 3H, J = 14 Hz), 2.72 (s, 3H), 3.64 (s, 3H), 4.23–4.28 (q, 2H, J = 14 Hz), 7.20–7.22 (d, 1H, J = 9.2 Hz), 3.45–8.46 (d, 1H, J = 2.4 Hz), 8.12–8.15 (dd, 1H, J1 = 2.8 Hz, J2 = 8.8 Hz); Citation13C NMR (CDCl3) δ: 11.21, 14.48, 34.99, 65.57, 113.15, 127.92, 130.75, 131.71, 134.64, 136.12, 142.45, 147.13, 157.87, 162.69. HRMS (ESI, m/z) Calcd for C14H14N5O3ClS [M+] 367.0505. Found [M] 367.0514. Anal. Calcd for C14H14N5O3ClS: C, 45.72; H, 3.84; N, 19.04. Found: C, 45.60; H, 3.90; N, 18.90.
4-Ethoxy-3-(1-methyl-3-propyl-1H-pyrazolo[4,3-e][1,2,4]-triazin-5-yl)benzene-1-sulfonyl chloride (7b). Yellow solid. Yield 95%; m.p. 65–66 °C. 1H NMR (400 MHz, CDCl3) δ: 1.01 (t, 3H, J = 7.2 Hz), 1.36 (t, 3H, J = 7.2 Hz), 1.87–1.94 (m, 2H), 3.07 (t, 2H, J = 7.2 Hz), 4.23 (q, 2H, J = 7.2 Hz), 7.21 (d, 1H, J = 8.8 Hz), 8.14 (dd, 1H, J1 = 8.8 Hz, J2 = 2.4 Hz), 8.47 (d, 1H, J = 2.4 Hz). Citation13C NMR (100 MHz, CDCl3) δ: 13.8, 14.2, 21.7, 27.9, 34.7, 65.3, 112.9, 127.5, 130.4, 131.4, 134.2, 135.7, 146.1, 146.8, 157.4, 162.4. HRMS (ESI, m/z) Calcd for C16H19ClN5O3S [M + H] 396.0897. Found [M + H] 396.0892.
Synthesis of sulfonamides (8d–g). A mixture of chlorosulfonyl chloride (7a) (100 mg, 0.29 mmol) and amine (100 mg, 1 mmol) in anhydrous acetonitrile (5 mL) was stirred overnight at room temperature, and then the reaction mixture was concentrated in vacuo to afford the crude sulfonamide, as a yellow solid. The residue was purified on silica gel using a mixture of CH2Cl2:EtOH (25:1) as eluent to give the titled compounds as a yellow solid.
(S)-3-(1,3-dimethyl-1H-pyrazolo[4,3-e][1,2,4]triazin-5-yl)-4-ethoxy-N-(1-hydroxy-propan-2-yl)benzenesulfonamide (8d). Yield 92%; m.p. 131–136 °C. 1H NMR (CDCl3) δ: 1.09 (d, 3H, J = 6.4 Hz), 1.35 (t, 3H, J = 6.8 Hz), 2.63 (bs, 1H, OH, exchanged with D20), 2.70 (s, 3H), 3.39–3.46 (m, 2H), 3.56 (d, 1H, J = 7.6 Hz), 4.19 (q, 2H, J = 6.4 Hz), 4.31 (s, 3H), 5.11 (d, 1H, J = 6.0 Hz, NH, exchanged with D2O), 7.14 (d, 1H, J = 9.2 Hz), 7.98 (dd, 1H, J1 = 8.4 Hz, J2 = 2.4 Hz), 8.28 (d, 1H, J = 2.4 Hz).Citation13C NMR (CDCl3) δ: 11.05, 14.43, 17.93, 34.82, 51.55, 64.94, 65.99, 112.79, 126.97, 130.48, 131.39, 132.15, 134.56, 142.21, 146.78, 158.42, 160.46. HRMS (ESI, m/z) Calcd for C17H22N6O4S [M+] 406.1423. Found [M+] 406.1427. Anal. Calcd for C17H22N6O4S: C, 50.23; H, 5.46; N, 20.68. Found: C, 50.00; H, 5.49; N, 20.50.
(R)-3-(1,3-dimethyl-1H-pyrazolo[4,3-e][1,2,4]triazin-5-yl)-4-ethoxy-N-(1-hydroxy-propan-2-yl)benzenesulfonamide (8e). Yield 89%; m.p. 119–122 °C. 1H NMR (CDCl3) δ: 1.09 (d, 3H, J = 6.4 Hz), 1.34 (t, 3H, J = 6.8 Hz), 2.69 (s, 3H), 2.84 (bs, 1H, OH, exchanged with D20), 3.38–3.45 (m, 2H), 3.55 (d, 1H, J = 7.6 Hz), 4.18 (q, 2H, J = 6.4 Hz), 4.32 (s, 3H), 5.24 (d, 1H, J = 6.4 Hz, NH, exchanged with D2O), 7.13 (d, 1H, J = 8.8 Hz), 7.98 (dd, 1H, J1 = 8.8 Hz, J2 = 2.4 Hz), 8.27 (d, 1H, J = 2.4 Hz). Citation13C NMR (CDCl3) δ: 11.04, 14.42, 17.89, 34.80, 51.55, 64.92, 65.96, 112.78, 126.91, 130.47, 131.35, 132.18, 134.57, 142.19, 146.74, 158.40, 160.42. HRMS (ESI, m/z) Calcd for C17H22N6O4S [M+] 406.1423. Found [M+] 406.1426. Anal. Calcd for C17H22N6O4S: C, 50.23; H, 5.46; N, 20.68. Found: C, 49.89; H, 5.49; N, 20.55.
(S)-3-(1,3-dimethyl-1H-pyrazolo[4,3-e][1,2,4]triazin-5-yl)-4-ethoxy-N-(1-hydroxy-3-methylbutan-2-yl)benzenesulfonamide (8f). Yield 91%; m.p. 115–122 °C. 1H NMR (CDCl3) δ: 0.83 (dd, 6H, J1 = 6.8 Hz, J2 = 2.8 Hz), 1.34 (t, 3H, J = 6.8 Hz), 1.82 (sec, 1H, J = 6.8 Hz), 2.69 (s, 3H), 3.07–3.11 (m, 1H), 3.52–3.55 (m, 2H), 4.17 (q, 2H, J = 6.8 Hz), 4.32 (s, 3H), 5.23 (d, 1H, J = 8.4 Hz, NH, exchanged with D2O), 7.11 (d, 1H, J = 8.8 Hz), 7.96 (dd, 1H, J1 = 8.8 Hz, J2 = 2.8 Hz), 8.27 (d, 1H, J = 2.8 Hz). Citation13C NMR (CDCl3) δ: 11.03, 14.40, 18.56, 19.12, 29.62, 34.80, 60.98, 62.50, 64.91, 112.65, 126.79, 130.45, 131.42, 132.49, 134.57, 142.18, 146.69, 158.40, 160.33. HRMS (ESI, m/z) Calcd for C19H26N6O4S [M+] 434.1736. Found [M+] 434.1741. Anal. Calcd for C19H26N6O4S: C, 52.51; H, 6.03; N, 19.35. Found: C, 52.18; H, 6.06; N, 19.22.
(R)-3-(1,3-dimethyl-1H-pyrazolo[4,3-e][1,2,4]triazin-5-yl)-4-ethoxy-N-(1-hydroxy-3-methylbutan-2-yl)benzenesulfonamide (8g). Yield 72%; m.p. 115–120 °C. 1H NMR (CDCl3) δ: 0.84 (dd, 6H, J1 = 6.8 Hz, J2 = 3.6 Hz), 1.35 (t, 3H, J = 6.8 Hz), 1.82 (sec, 1H, J = 6.8 Hz), 2.70 (s, 3H), 3.08–3.11 (m, 1H), 3.50–3.58 (m, 2H), 4.18 (q, 2H, J = 6.8 Hz), 4.32 (s, 3H), 5.16 (d, 1H, J = 8.8 Hz, NH, exchanged with D2O), 7.11 (d, 1H, J = 8.8 Hz), 7.96 (dd, 1H, J1 = 8.8 Hz, J2 = 2.4 Hz), 8.28 (d, 1H, J = 2.4 Hz). Citation13C NMR (CDCl3) δ: 11.04, 14.41, 18.56, 19.13, 29.65, 34.81, 60.98, 62.53, 64.92, 112.66, 126.81, 130.46, 131.44, 132.47, 134.57, 142.20, 146.72, 158.40, 160.36. HRMS (ESI, m/z) Calcd for C19H26N6O4S [M+] 434.1736. Found [M+] 434.1741. Anal. Calcd for C19H26N6O4S: C, 52.51; H, 6.03; N, 19.35. Found: C, 52.18; H, 6.10; N, 19.15.
Pharmacology
Materials and methods
Animals
The experiments were carried out using male Wistar rats (Krf:(WI) WU), 200–250 g. The animals were housed in constant temperature facilities exposed to 12:12 h light/dark cycles and were maintained on a standard pellet diet with tap water given ad libitum. All procedures were conducted according to the Animal Care and Use Committee Guidelines and approved by the Local Ethics Committee of the Jagiellonian University in Kraków.
Drugs and chemicals
The materials used included 3-isobutyl-1-methylxanthine (IBMX), acetylcholine hydrochloride, glibenclamide, isoprenaline hydrochloride, Nω-nitro-L-arginine methyl ester hydrochloride (l-NAME), 1H-[1,2,4]oxadiazolo[4,3-a]quinoxalin-1-one (ODQ), phentolamine hydrochloride, sildenafil citrate (Sigma-Aldrich, Germany), thiopental sodium (Biochemie GmbH, Austria). Other chemicals used were obtained from POCH (Polish Chemical Reagents, Poland). The drugs were dissolved in dimethyl sulfoxide for the in vitro studies or suspended in 0.5% methylcelulose for the in vivo studies immediately before use.
Influence on isolated rat thoracic aorta contracted by KCl
Rats were anesthetized with thiopental sodium (60 mg/kg i.p.) and the thoracic aorta was dissected and placed in a Krebs–Henseleit solution (NaCl 119 mM, KCl 4.7 mM, CaCl2 1.9 mM, MgSO4 1.2 mM, KH2PO4 1.2 mM, NaHCO3 25 mM, glucose 11 mM, EDTA 0.05 mM) and cleaned of surrounding fat tissues and cut into approximately 4 mm long rings. Special care was taken to avoid any damage to the endothelium. In some aortic rings, the endothelial layer was mechanically removed by gently rubbing the luminal surface of the aortic ring back and forth several times with plastic tubing. The aorta rings were incubated in 30 mL chambers filled with a Krebs–Henseleit solution at 37 °C and pH 7.4 with constant oxygenation (O2/CO2, 19:1). Two stainless steel pins were inserted through the lumen of each arterial segment: one pin was attached to the bottom of the chamber and the other to an isometric FDT10-A force displacement transducer (BIOPAC Systems, Inc., COMMAT Ltd., Turkey). The aortic rings were stretched and maintained at optimal tension of 2 g and allowed to equilibrate for 2 h. The aortic rings were contracted to submaximal tension with KCl (60 mM). The depolarising solution KCl (60 mM) was obtained by equimolar substitution of NaCl for KCl. Once the plateau was attained, concentration–relaxation curves were obtained by the addition of cumulative doses of tested compounds to the precontracted preparations.
Influence on isolated rat thoracic aorta contracted by phenylephrine
Rats were anesthetized with thiopental sodium (60 mg/kg i.p.) and the aorta was isolated, cut, mounted and incubated as described in a method above. Special care was taken to avoid any damage to the endothelium. In some aortic rings, the endothelial layer was mechanically removed by gently rubbing the luminal surface of the aortic ring back and forth several times with plastic tubing. The aorta rings were stretched and maintained at optimal tension of 2 g and allowed to equilibrate for 2 h. Endothelial integrity or functional removal was verified by the presence or absence of the relaxant response to acetylcholine (1 μM) on the phenylephrine (1 μM) contracted vessels.
In the first series of experiments, the effects of compound (8a) on vascular tension were defined. Aortic rings were contracted with phenylephrine (10 μM) to obtain maximal response. Once the maximal response to phenylephrine had been obtained, the aortic rings were exposed to cumulative doses of compound (8a) and the responses were recorded. Additionally, to observe whether the vasorelaxant effects of tested compounds are affected by IBMX, a nonselective PDE inhibitor, the action of compound (8a) in the presence of IBMX (1 µM) was investigated.
In another experiment, the effect of compound (8a) on the vasorelaxant responses to isoprenaline, a β-adrenoceptor-mediated cAMP-dependent vasodilator was investigated by incubating the endothelium denuded aortic rings with 30 µM of compound (8a) for 30 min before either isoprenaline (0.01–10 µM) was added.
To define the mechanisms by which compound (8a) relaxes vascular smooth muscle, another series of experiments were done in aortic rings. The rings were exposed to various modulating agents such as l-NAME (10 μM), ODQ (1 μM), glibenclamide (1 μM) for 20 min, and then vascular relaxation was carried out by cumulative additions of compound (8a) at the plateau of the phenylephrine-induced contractions.
Measurement of blood pressure and heart rate
Rats were anesthetized with thiopental (60 mg/kg i.p.). The right carotid artery was cannulated with polyethylene tube filled with heparin in saline to facilitate pressure measurements using a Datamax apparatus (Columbus Instruments). Compound (8a) was administered at a dose of 10 mg/kg i.p. after a 15 min stabilization period at a volume equivalent to the 1 mL/kg.
Data analysis
The results are presented as the means ± S.E.M. Statistically significant differences between groups were calculated using one-way analysis of variance (ANOVA) and the post-hoc Dunnett's multiple comparison test. The criterion for significance was set at p < 0.05. Concentration–relaxation curves were analysed using GraphPad Prism 4.0 software (GraphPad Software Inc., San Diego, CA). Relaxations are expressed as a percentage of inhibition of the maximal tension obtained with the contractile agent (Emax = 100%). Data are the means ± S.E.M. of four separate experiments. The concentrations needed to produce 50% of the maximum relaxation (IC50) were calculated.
Results
Chemistry
The synthesis of target aza-analogues of sildenafil (8a–m) was achieved by a convenient multiple procedure starting from 3-methylsulfanyl-1,2,4-triazine (1) as shown in Scheme 1. First, using our previous established procedureCitation9, the reaction of (1) with nitrobutane or nitroethane and KOH in DMSO at room temperature gave appropriate oximes (2ab) as main products. In the next step, the readily available oximes (2ab) were transformed into ketones (3ab) in good yieldCitation10, which reacted with methylhydrazine in the presence of acidic media according to standard procedure to give suitable hydrazones (4ab) as key intermediates for the preparation of 1H-pyrazolo[4,3-e][1,2,4]triazine derivatives (5ab). The hydrazones (4ab) could be converted into derivatives (5) under conventional heating (10% HCl, EtOH, reflux, 1 h)Citation11 or under solvent free reaction conditions according to our previous published procedureCitation12. Using Guillaumet and co-workers methodCitation13 for the palladium-catalyzed cross-coupling reaction of 3-methylsulfanyl-1,2,4-triazine with boronic acid derivatives we have reacted 5-methylsulfanyl-1H-pyrazolo[4,3-e][1,2,4]triazines (5ab) with 2-ethoxyphenylboronic acid in the presence of copper (I) 3-methylsalicylate to obtain derivatives (6ab) in excellent yield. Chlorosulfonylation reaction of compounds (6ab) in neat chlorosulfonic acid at 0 °C proceeded smoothly and selectively at the 5′-position of the phenyl ring to give the desired products (7ab) in excellent yield. The chlorosulfonyl derivatives (7ab) were readily coupled with appropriate amines in acetonitrile at room temperature to produce the final sulfonamides (8a–m) as new chiral analogues of sildenafil in high yield.
Scheme 1. Synthetic pathway to the sildenafil analogues (8a–m). Reagents and conditions: (a) CH3CH2CH2CH2NO2 (for R = C3H7) or CH3CH2NO2 (for R = CH3), KOH, DMSO, 2 h, 80–86%; (b) Na2S2O4, H2O/dioxane, rt, 12 h, 55–65%; (c) CH3NH–NH2, PTSA, EtOH, rt, 1 h, 50–55%; (d) method A: 10% HCl, EtOH, reflux, 1 h, 58–61%; method B: PTSA, 140 °C, 1 min, 61%; (e) ethoxyphenylboronic acid, Pd(PPh3)4, CuMeSal, THF, Ar, reflux, overnight, 75–80%; (f); ClSO3H, 0 °C to rt, 2 h, 75–95%; (g) appropriate amine, anhydrous MeCN, rt, overnight, 72–93%.
Pharmacology
The aim of this work was to assess vasorelaxant properties of seven new structural analogues of sildenafil (marked with the numbers: 8a, 8b, 8c, 8d, 8e, 8f and 8g) and to explain their mechanism of action.
Influence on isolated rat thoracic aorta contracted by KCl
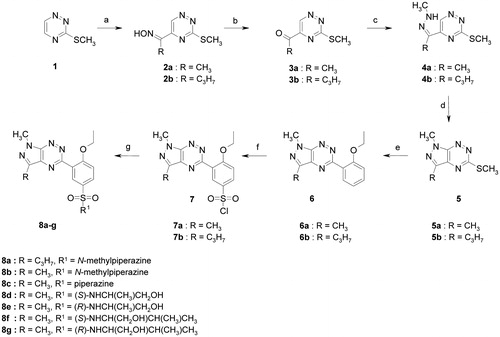
The vasorelaxant effects of selected sildenafil analogues, sildenafil (a selective PDE5 inhibitor) and IBMX (non-selective PDE inhibitor) in isolated aortic rings precontracted by KCl with and without endothelium were investigated. Compounds 8a, 8b, 8c, 8f and 8g (1–300 µM) relaxed KCl (60 mM)-precontracted endothelium intact aortic rings in a dose-dependent manner and they were able to inhibit the contractile response completely (). The IC50 values were at the range of 48.9–178.1 µM (). In endothelium denuded aorta cumulative concentrations of compounds 8a, 8b, 8c, 8f and 8g (1–300 µM) produced concentration-dependent vasorelaxations with IC50 values of 39.7–143.8 µM, indicating that the relaxant responses of these agents are likely to be endothelium independent. Compounds (8d) and (8e) relaxed the KCl depolarising solution precontracted vessels only by 52.4–65.6% in endothelium intact aorta and by 37–48% in endothelium denuded aortic rings (, ). Sildenafil inhibited contraction induced by KCl depolarising solution by 85–90% with IC50 values of 7.0 and 14.3 µM in endothelium intact and endothelium denuded aorta, respectively, suggesting that its vasorelaxant effect is endothelium dependent. For comparison, IBMX, relaxed the endothelium intact and denuded aorta with IC50 values of 39.3 and 26.4 µM, respectively, suggesting that its vasorelaxant effect is endothelium independent (). Since compound (8a) turned out to be the most active, with vasorelaxant properties similar to those of IBMX, it was selected for further pharmacological research aiming to explain its mode of action.
Figure 2. Effects of tested compounds on vascular tension in the endothelium-intact, KCl (60 mM) precontracted aortic rings. The data are the means of four experiments ± S.E.M.
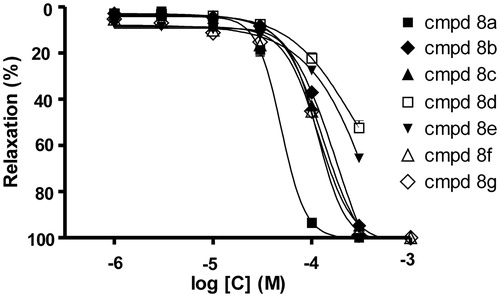
Table 1. Vasorelaxant potencies of tested compounds on sustained contraction of aortic rings induced by KCl depolarising solution (60 mM).
Influence on isolated rat thoracic aorta contracted by phenylephrine
Compound (8a) (0.1–300 µM) produced a concentration dependent inhibition of the contractile response induced by another stimulatory agent, phenylephrine. In phenylephrine-induced contractions the relaxation was again endothelium independent, with the IC50 values of 27.8 µM for endothelium intact and 25.3 µM for endothelium denuded aorta.
In the contractions induced by both stimulatory agents (KCl or phenylephrine), with intact or denuded endothelium-ring, the relaxant effect of compound (8a) was higher in phenylephrine than in KCl-induced contraction ().
Figure 3. Vasorelaxant effects of compound (8a) on KCl and phenylephrine-induced contractions in isolated endothelium-intact E(+) and denuded E(−) rat aorta. (♦) KCl E(+), (▪) KCl E(−), (▴) phenylephrine E(+), (□) phenylephrine E(−). The data are the means of 4–8 experiments ± S.E.M.
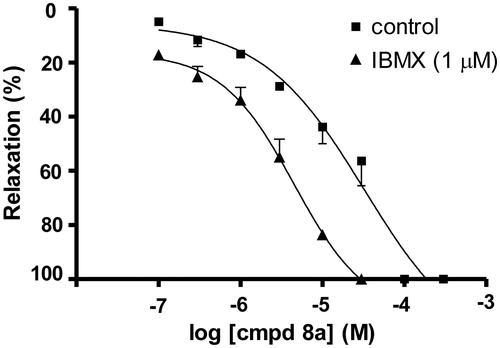
The effects of compound (8a) were also investigated after pretreatment with PDE nonselective inhibitor IBMX. Vasorelaxation induced by compound (8a) had an additive effect in the presence of IBMX ().
Figure 4. Additive effects of compound (8a) and IBMX (1 µM) on rat aortic rings precontracted by phenylephrine (10 µM). The data are the means of four experiments ± S.E.M.
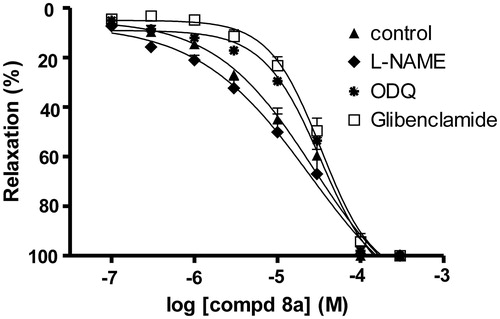
In endothelium intact rat aorta, the vasorelaxant effect of compound (8a) was reduced by pretreatment with the soluble guanylyl cyclase (sGC) inhibitor ODQ and the KATP channel blocker glibenclamide. Nevertheless, the vasorelaxation of compound (8a) was not affected by the nitric oxide synthase (NOS) inhibitor l-NAME ().
Figure 5. The vasorelaxant effect of compound (8a) on endothelium intact rat aorta precontracted by phenylephrine (10 μM) in the absence and presence of l-NAME (10 μM), ODQ (1 μM), glibenclamide (1 μM). The data are the means of four experiments ± S.E.M.
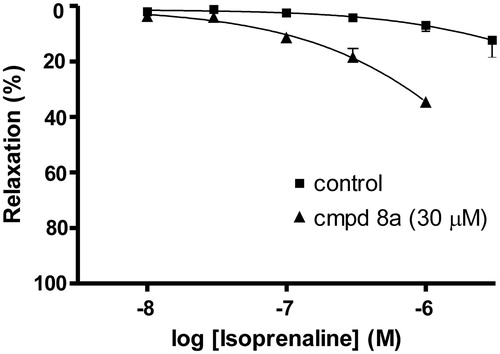
Denuded rat aorta was pretreated with compound (8a) and its response to isoprenaline, a cAMP-dependent vasodilator was studied. Compound (8a) (30 µM) significantly increased the potency of isoprenaline ().
Figure 6. Effect of compound (8a) (30 µM) on isoprenaline-induced vasorelaxation in endothelium denuded rat aorta precontracted by phenylephrine (10 µM).
Influence on the blood pressure and heart rate
The effects on the blood pressure and heart rate of compound (8a) and sildenafil were determined after i.p. administration at the dose of 10 mg/kg. Saline was used as a control without drug treatment.
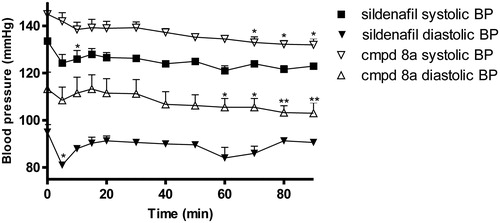
Compound (8a) reduced blood pressure slightly but significantly by 8% (systolic blood pressure) and 7% (diastolic blood pressure) in 60–70 min after its administration (). For comparison, sildenafil initially reduced systolic and diastolic pressure by 6% and 15%, respectively, but the pressure returned to the baseline value within next couple of minutes after administration (). Neither sildenafil nor compound (8a) influenced the heart rate, however, compound (8a) increased myocardial contractility index (LV (dP/dt) max/LVP) in 80 min after administration.
Figure 7. The effect of compound (8a) and sildenafil on blood pressure in anesthetized rats at a dose of 10 mg/kg i.p. The data are the means of 5–6 experiments ± S.E.M. Statistical analyses were performed using a one-way ANOVA test, post hoc Dunnet test; *p < 0.05; **p < 0.02.
Discussion
In the cardiovascular system, modulation of cyclic nucleotides is involved in regulation of vascular smooth muscles function and agents that inhibit PDE and increase cAMP and cGMP levels produce vasodilatationCitation14.
Compounds 8a, 8b, 8c, 8f and 8g produced concentration dependent aortic relaxation against the contraction induced by KCl depolarising solution in endothelium intact and denuded aortic rings. PDE inhibitors that increase cyclic nucleotides (cAMP i cGMP) produce vasodilatation by activating the appropriate protein kinases and decreasing intracellular calciumCitation14. Our results showed that the tested compounds (except for derivative (8a), which acts as potent PDE inhibitor) were less active than reference compounds, sildenafil and IBMX. Compound (8a) was the most active, with IC50 values comparable with that one of IBMX. It may be related to its chemical structure and similarity to sildenafilCitation15. Other compounds and different sildenafil chemical modifications turned to be less active regarding their vasorelaxant properties.
PDE1 and PDE3 play the important functional role in rat aorta smooth muscle, whereas in vascular endothelium PDE5 and PDE4 isoforms are relevantCitation14,Citation16. It has been proven that the activity of the tested compounds in the contractions induced by KCl is endothelium independent. On the basis of our experiment we may state that their vasorelaxant effect is not only due to PDE5 inhibition. If they had acted as selective PDE5 inhibitors, their vasorelaxant activity would have been endothelium dependent, what was observed in a case of sildenafil. In endothelium-devoid arteries the relaxant response to sildenafil was partially inhibited. Sildenafil vasorelaxant properties result from PDE5 inhibition and depend on endothelium and nitric oxide (NO)—soluble guanylyl cyclase (sGC)—cyclic guanosine monophosphate pathway (cGMP). NO activation of the sGC results in an increase in intracellular cGMP levels, which elicits cGMP-dependent protein kinase (PKG) signalling. PKG inhibits Ca2+ influx, augments Ca2+ sequestration and decrease the sensitivity of contractile elements to Ca2+. Selective PDE5 inhibition increase cGMP breakdown reduction and potentiate the vasorelaxant effectCitation17.
Since compound (8a) showed the highest activity in the model of contraction induced by KCl, the screening assay, it was selected for further pharmacological studies.
In the model of vasoconstriction induced by phenylephrine the vasorelaxant properties of compound (8a) were endothelium independent, similar to results obtained from the model of contraction induced by KCl. Comparing the effects of compound (8a) in both model of vasoconstrictions, the IC50 values in the contraction induced by phenylephrine were lower than the IC50 values in the contraction induced by KCl. This may be due to the fact that high K+ solution induces a cell membrane depolarization and calcium influx from the outside, so in order to achieve vasorelaxation higher concentrations of PDE inhibitor are required. In contrast, the model of vasoconstriction induced by phenylephrine is mainly dependent on intracellular stores of calcium ions, so the lower concentration of PDE inhibitor is sufficientCitation14.
We have demonstrated that the combination of compound (8a) and IBMX, a non-selective PDE inhibitor, had an additive effect on vasorelaxation what suggest that vasodilator effect of compound (8a) may be mediated by PDE inhibition.
In order to assess the involvement of endothelium-dependent signalling in compound (8a) induced relaxation, the effect of l-NAME, a non-selective NOS inhibitor, was tested. Our results showed that compound (8a) induced vasorelaxation was not inhibited by l-NAME, what confirms the effects observed in preliminary test and indicates that the endothelium-dependent NO–sGC–cGMP pathway may not be implicated in compound (8a) vasorelaxant properties. However, ODQ, a selective inhibitor of sGC markedly reduced the relaxation induced by compound (8a). This indicates that vasorelaxant effect of compound (8a) depends on sGC–cGMP signalling, at least partially. sGC and cGMP are located not only in endothelium but also in vascular smooth muscle cells and also PDE isoforms are differentially distributed both in endothelial and vascular smooth muscle cellsCitation16,Citation18,Citation19. In vascular smooth muscle, PDE1 (hydrolyzes cGMP and cAMP) and PDE3 (hydrolyzes cAMP preferentially but not exclusively) isoenzymes play an important role as modulators of rat aorta contractility. PDE4 and PDE5 isoforms have been also found but in the absence of endothelium their functional role is not relevantCitation14. On the basis of our results we can assume that compound (8a) may be a degradating cGMP PDE isoforms inhibitor, particularly PDE1 and PDE3.
In this work, we have also demonstrated that a KATP channel (an ATP-sensitive potassium channel) blocker glibenclamide significantly inhibited the vasorelaxant effect of compound (8a). K+ channels play a major role in the regulation of the resting membrane potential and modulate vascular smooth muscle tone. Endothelium-derived hyperpolarizing factor (EDHF) activates the K+ channels, and the subsequent K+ efflux causes hyperpolarization closing voltage-operated Ca2+ channels and decreasing Ca2+ entrance that promotes muscle relaxation. Endothelial and smooth muscle cells express ATP-sensitive and Ca2+ activated K+ channels. It is suggested that activation of KATP channels may also release endothelium-derived NO or EDHFCitation20. Therefore, we can state that compound (8a) induced vasorelaxation might partially occur via activation of KATP channels.
In order to check whether the vasodilatory activity of compound (8a) depends on cAMP, we determined its modulating effect on a cAMP-dependent vasodilator. Compound (8a) enhanced the vasorelaxant effect of a cAMP-dependent vasodilator, isoprenaline. This may suggest that compound (8a) is a cAMP degrading PDE inhibitor, presumably PDE3, which dominate in vascular smooth muscle cells.
Summarizing the biofunctional studies, in the case of compound (8a) we may suspect that its vasorelaxant response depends on cyclic nucleotides pathway since the pretreatment with the sGC inhibitor ODQ attenuated the vasorelaxant response and also, compound (8a) enhanced the vasorelaxant response to cAMP-dependent vasodilator isoprenaline. However, compound (8a) activates not only the sGC/cGMP and adenylyl cyclase (AC)/cAMP pathways, but also may activate K+ channels. The vasorelaxant mechanism of compound (8a), sildenafil analogue, through sGC/cGMP and AC/cAMP pathways is probably a result of its non selective PDE inhibitory activity. Compound (8a) induced vasorelaxation is endothelium independent and is not only due to PDE4 and PDE5 inhibitory activity, since these subtypes of PDE have been found mainly in endotheliumCitation14.
In in vivo study, compound (8a) moderately decreased the systolic and diastolic blood pressure and increased heart contractility. In rat heart, PDE2, PDE3 and PDE4 existCitation16,Citation18 and their inhibition by compound (8a) may increase cyclic nucleotides concentration and as a result compound (8a) produces vasodilatation and increases heart contractility.
In conclusion, the studies of seven new sildenafil analogues showed their moderate and endothelium-independent vasorelaxant properties. The strongest, comparable with IBMX effect was shown for compound (8a), being the closest structural sildenafil analogue. Compound (8a) may be a non-selective PDE inhibitor, and may inhibit isozymes degrading both cAMP and cGMP. Its vasorelaxant activity may also involve K+ channel activation.
Conclusions
We have described facile and efficient method for the preparation of new 1H-pyrazolo[4,3-e][1,2,4]triazine sulfonamides from simple available starting materials. Our biological study with sildenafil analogues indicated one compound (8a), which exhibited micromolar and endothelium-independent vasorelaxant properties in the rat aorta. The compound is undergoing further modification of the structure to increase the relaxation potency.
Declaration of interest
The authors declare no conflicts of interest. The authors alone are responsible for the content and writing of this article.
This research was partially funded by the National Science Centre, Poland (grant NN405 092340). The authors gratefully acknowledge use of the mass spectrometry services and facilities of the Center for Interdisciplinary Research of The John Paul II Catholic University of Lublin, Lublin, Poland, funded by POPW.01.03.00-06-003/09-00.
References
- Omori K, Kotera J. Overview of PDEs and their regulation. Circ Res 2007;100:309–27
- Soderling SH, Beavo JA. Regulation of cAMP and cGMP signaling: new phosphodiesterases and new functions. Curr Opin Cell Biol 2000;12:174–9
- Cote RH. Characteristics of photoreceptor PDE (PDE6): similarities and differences to PDE5. Int J Impotence Res 2004;16:S28–33
- Bella AJ, Deyoung LX, Al-Numi M, Brock GB. Daily administration of phosphodiesterase type 5 inhibitors for urological and nonurological indications. Eur Urol 2007;52:990–1005
- Yoo J, Thai K-M, Kim D-K, et al. 3D-QSAR studies on sildenafil analogues, selective phosphodiesterase 5 inhibitors. Bioorg Med Chem Lett 2007;17:4271–4
- Xia G, Li J, Peng A, et al. Synthesis and phosphodiesterase 5 inhibitory activity of novel pyrido[1,2-e]purin-4(3H)-one derivatives. Bioorg Med Chem Lett 2005;15:2790–4
- Mojzych M, Rykowski A. One-step synthesis and regioselective alkylation of substituted 1H-pyrazolo[4,3-e][l,2,4]triazine. Polish J Chem 2003;77:1797–804
- Mojzych M, Karczmarzyk Z, Wysocki W, et al. New approaches to the synthesis of sildenafil analogues and their enzyme inhibitory activity. Bioorg Med Chem 2015. [Epub ahead of print]. doi:10.1016/j.bmc.2015.02.026
- Rykowski A, Makosza M. Reaction of 1,2,4-triazines with nitronate anions, direct nucleophilic acylation of 1,2,4-triazines. Tetrahedron Lett 1984;25:4795–7
- Rykowski A, Lipinska A. A concise route to a key intermediate in the total synthesis of Sempervirens. Synth Commun 1996;26:4409–14
- Rykowski A, Mojzych M, Karczmarzyk Z. A new synthesis of pyrazolo[4,3-e][1,2,4]triazines via acid promoted ring closure of the phenylhydrazones of 5-acyl-1,2,4-triazines. Heterocycles 2000;53:2175–85
- Mojzych M, Rykowski A. Transformations of phenylhydrazones of 5-acyl-1,2,4-triazines to pyrazolo[4,3-e][1,2,4]triazines or 4-cyanopyrazole. J Heterocycl Chem 2007;44:1003–7
- Alphonse F-A, Suzenet F, Keromnes A, et al. Palladium-catalyzed 3-thiomethyltriazine-boronic acid cross coupling: easy access to 3-substituted-1,2,4-triazines. Synlett 2002;3:447–50
- Noguera MA, Ivorra MD, Lugnier C, D'Ocon P. Role of cyclic nucleotide phosphodiesterase isoenzymes in contractile responses of denuded rat aorta related to various Ca2+ sources. Naunyn Schmiedebergs Arch Pharmacol 2001;363:612–19
- Mojzych M, Dolashki A, Voelter W. Synthesis of pyrazolo[4,3-e][1,2,4]triazine sulfonamides, novel Sildenafil analogues with tyrosinase inhibitory activity. Bioorg Med Chem 2014;22:6616–24
- Lugnier C. Cyclic nucleotide phosphodiesterase (PDE) superfamily: a new target for the development of specific therapeutic agents. Pharmacol Therap 2006;109:366–98
- Salom JB, Burguete MC, Pérez-Asensio FJ, et al. Relaxant effect of sildenafil in the rabbit basilar artery. Vascul Pharmacol 2006;44:10–16
- Bender AT, Beavo JA. Cyclic nucleotide phosphodiesterases: molecular regulation to clinical use. Pharmacol Rev 2006;58:488–520
- Lo YC, Tsou HH, Lin RJ, et al. Endothelium-dependent and -independent vasorelaxation by a theophylline derivative MCPT: roles of cyclic nucleotides, potassium channel opening and phosphodiesterase inhibition. Life Sci 2005;76:931–44
- Wu BN, Chen IC, Lin RJ, et al. Aortic smooth muscle relaxants KMUP-3 and KMUP-4, two nitrophenylpiperazine derivatives of xanthine, display cGMP-enhancing activity: roles of endothelium, phosphodiesterase, and K+ channel. J Cardiovasc Pharmacol 2005;46:600–8