
Abstract
Because of the complexity of Alzheimer's disease (AD), the multi-target-directed ligand (MTDL) strategy is expected to provide superior effects for the treatment of AD, instead of the classic one-drug-one-target strategy. In this context, we focused on the design, synthesis and evaluation of homoisoflavonoid derivatives as dual acetyl cholinesterase (AChE) and monoamine oxidase (MAO-B) inhibitors. Among all the synthesized compounds, compound 10 provided a desired balance of AChE and hMAO-B inhibition activities, with IC50 value of 3.94 and 3.44 μM, respectively. Further studies revealed that compound 10 was a mixed-type inhibitor of AChE and an irreversible inhibitor of hMAO-B, which was also confirmed by molecular modeling studies. Taken together, the data indicated that 10 was a promising dual functional agent for the treatment of AD.
Introduction
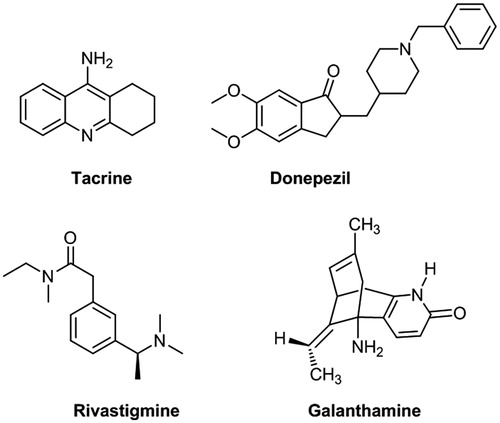
Alzheimer's disease (AD), characterized by memory loss and other cognitive impairments, is currently one of the most difficult and baffling diseases to treatCitation1–3. In the past decades, various pathogenesis hypothesis of AD have been proposed, such as cholinergic hypothesisCitation4,Citation5, amyloid cascade hypothesisCitation6–8, oxidative stress hypothesisCitation9–12, tau protein hypothesisCitation13,Citation14 and so on. Among them, cholinergic hypothesis was a widely accepted theory, which suggests that the low level of ACh in specific regions of the brain is the major cause results in learning and memory dysfunctions. Based on the cholinergic hypothesis, one possible approach to treat AD is to restore the level of acetylcholine, such as using reversible inhibitors to inhibit acetyl cholinesterase (AChE). Up to now, AChE inhibitors, including tacrine (Cognex®, ), donepezil (Aricept®, ), rivastigmine (Exelon®, ) and galanthamine (Reminyl®, ) are approved by Food and Drug Administration to use in clinic, which could effectively improve memory in some AD patientsCitation15,Citation16. The crystal structures of AChE with inhibitors revealed that there are two binding sites, a peripheral anionic site (PAS) and a catalytic active site (CAS)Citation17–19. The PAS region of AChE, located at the rim of the gorge, was involved in the interaction with Aβ peptide, which may induce the deposition of neurotoxic Aβ fibrilsCitation20,Citation21. Recent studies showed that AChE inhibitors, blocking both the catalytic and peripheral sites simultaneously, might not only alleviate the cognitive defect of AD patients by elevating ACh levels, but also act as disease-modifying agents delaying amyloid plaque formationCitation22,Citation23. For example, donepezil, a mixed-type inhibitor, has a small effect on decreasing Aβ aggregation induced by AChE (about 22%), which can be attributed to its higher affinity for the active site than for the PASCitation24.
Figure 1. Structure of tacrine, donepezil, rivastigmine and galanthamine.
Monoamine oxidases A and B (MAO-A and MAO-B), which could catalyze the oxidative deamination of neuroactive and vasoactive amines, are thought to play an important role in the metabolism of biogenic amines in the central nervous system and in peripheral tissuesCitation25,Citation26. Recent studies have revealed that MAOs are associated with psychiatric and neurological disorders, including depression, Parkinson’s disease (PD) and ADCitation27,Citation28. MAO-A inhibitors are used as antidepressants and anti-anxiety agents in the clinic, while MAO-B inhibitors are used as therapeutics for PDCitation29. As oxidative stress has been implicated in central nervous system degenerative disorders, MAO-B inhibitors are also potential candidates as anti-AD drugs due to their regulation of neurotransmitters and capacity to inhibit oxidative damageCitation30.
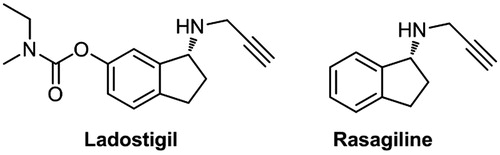
Because AD is a complex neurodegenerative disorder resulting from multiple factors, molecules that modulate the activity of a single protein target might not be able to significantly alter the progression of the disease. In recent years, a multi-target-directed ligand (MTDL) strategy has been developed to approach this multifaceted disease. Based on the MTDL drug design strategy, Sterling et al. incorporated the pharmacophores of rivastigmine (an AChE inhibitor, ) and rasagiline (an MAO inhibitor, ) to a single molecular entity to give a novel dual AChE and MAO-B inhibitors, ladostigil (), the Phase II clinical trials of which is in progressCitation31.
Figure 2. Chemical structures of ladostigil and rasagiline.
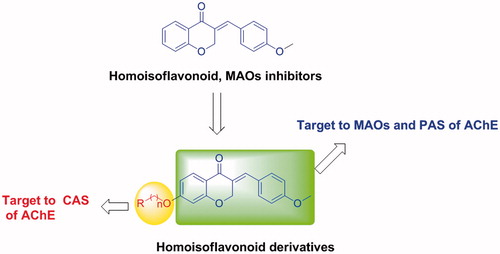
Homoisoflavonoids, which have been found to possess various biological properties, such as antifungalCitation32, antiviralCitation33,Citation34, anti-proliferativeCitation32, antioxidantCitation35 and anti-inflammatoryCitation36 activities, were also proven to be potent and selective MAO-B inhibitorsCitation37. Inspired by the outcomes of ladostigil, we have developed a series of tacrine-contained hybrids targeted to both AChE and MAO-B in earlier studiesCitation38,Citation39. Herein, we described the synthesis and evaluation of a series of homoisoflavonoid derivatives as dual AChE and MAO-B inhibitors (), which were expected to bind to MAO-B, PAS and CAS of AChE.
Figure 3. Design strategy for homoisoflavonoid derivatives.
Materials and methods
Materials
1H NMR and 13C NMR spectra were recorded on a Bruker BioSpin GmbH spectrometer (Östliche Rheinbrückenstr, Germany) at 400.132 and 100.614 MHz, respectively, using tetramethylsilane as the internal standard. Coupling constants are given in Hz. MS spectra were recorded on a Agilent LC–MS 6120 instrument (Santa Clara, CA) with an ESI mass selective detector. Flash column chromatography was performed with silica gel (200–300 mesh) purchased from Qingdao Haiyang Chemical Co. Ltd. (Qingdao, China). The chemicals used in the chemistry section were procured from Sinopharm Chemical Reagent Co., Ltd. (Shanghai, China) and all of the chemicals are analytical reagents. The purities of the synthesized compounds were confirmed to be higher than 95% by analytical HPLC (Agilent technologies 1200 series system) with a TC-C8 column (4.6 × 150 mm, 5 μm) and the compounds were eluted with CH3OH/water (0.1% TFA, w/v) in ratios of 70:30–40:60 at a flow rate of 1.0 mL/min.
General procedures for the preparation of 5–8
To a stirred suspension of compounds 4 (10 mmol) and K2CO3 (15 mmol) in butanone (50 mL), KI (1 mmol) and α,ω-dibromo alkane (12 mmol) were added. The mixture was stirred at room temperature for 1–4 h, filtered, and then evaporated under vacuum. The crude product was chromatographed on a silica gel column, eluted with EtOAc/petroleum ether as eluent to afford the proposed compound.
(E)-7-(2-bromoethoxy)-3-(4-methoxybenzylidene)chroman-4-one (5)
Compound 4 was treated with 1,2-dibromoethane according to the general procedure to give the desired product 5 as yellow solid, yield 74%; 1H NMR (400 MHz, CDCl3) δ: 7.77 (d, J = 8.4 Hz, 1H), 7.65 (s, 1H), 7.30 (d, J = 8.6 Hz, 2H), 7.04 (d, J = 8.6 Hz, 2H), 6.34 (d, J = 8.4 Hz, 1H), 6.31 (s, 1H), 5.30 (s, 2H), 4.51 (t, J = 6.0 Hz, 2H), 3.89 (t, J = 6.0 Hz, 2H), 3.81 (s, 3H).
(E)-7-(3-bromopropoxy)-3-(4-methoxybenzylidene)chroman-4-one (6)
Compound 4 was treated with 1,3-dibromopropane according to the general procedure to give the desired product 6 as yellow solid, yield 65%; 1H NMR (400 MHz, CDCl3) δ: 7.80 (d, J = 8.8 Hz, 1H), 7.68 (s, 1H), 7.34 (d, J = 8.4 Hz, 2H), 7.09 (d, J = 8.4 Hz, 2H), 6.38 (d, J = 8.8 Hz, 1H), 6.34 (s, 1H), 5.35 (s, 2H), 4.37 (t, J = 6.4 Hz, 2H), 3.83 (s, 3H), 3.70 (t, J = 6.4 Hz, 2H), 2.46–2.51(m, 2H).
(E)-7-(4-bromobutoxy)-3-(4-methoxybenzylidene)chroman-4-one (7)
Compound 4 was treated with 1,4-dibromobutane according to the general procedure to give the desired product 7 as yellow solid, yield 59%; 1H NMR (400 MHz, CDCl3) δ: 7.76–7.81 (m, 1H), 7.62 (s, 1H), 7.29 (d, J = 8.6 Hz, 2H), 7.04 (d, J = 8.6 Hz, 2H), 6.31 (d, J = 8.0 Hz, 1H), 6.29 (s, 1H), 5.32 (s, 2H), 4.26 (t, J = 6.4 Hz, 2H), 3.80 (s, 3H), 3.52 (t, J = 6.4 Hz, 2H), 2.11–2.28 (m, 4H).
(E)-7-(5-bromopentyloxy)-3-(4-methoxybenzylidene)chroman-4-one (8)
Compound 4 was treated with 1,5-dibromopentane according to the general procedure to give the desired product 8 as yellow solid, yield 51%; 1H NMR (400 MHz, CDCl3) δ: 7.81 (d, J = 8.6 Hz, 1H), 7.64 (s, 1H), 7.33 (d, J = 8.6 Hz, 2H), 7.07 (d, J = 8.6 Hz, 2H), 6.29–6.31 (m, 1H), 6.30 (s, 1H), 5.34 (s, 2H), 4.22 (t, J = 6.4 Hz, 2H), 3.80 (s, 3H), 3.47 (t, J = 6.8 Hz, 2H), 1.96–2.05 (m, 4H), 1.57–1.82 (m, 2H).
General procedure for the synthesis of 9–18
The substituent amine (2.4 mmol) was added to a magnetically stirred solution of 5, 6, 7 or 8 (2 mmol) in acetonitrile (30 mL). The mixture was refluxed for 12–24 h, and monitored by TLC. The mixture evaporated under vacuum. The residue was purified by column chromatography (petroleum ether/ethyl acetate, 2:1–10:1, plus 10 mL triethylamine per 1000 mL).
(E)-3-(4-methoxybenzylidene)-7-(2-(piperidin-1-yl)ethoxy)chroman-4-one (9)
Intermediate 5 was treated with piperidine according to the general procedure to give the desired product 9 as a yellow solid, yield 69%; 1H NMR (400 MHz, CDCl3) δ 7.81 (d, J = 8.8 Hz, 1H), 7.65 (s, 1H), 7.10 (d, J = 8.2 Hz, 2H), 6.80 (d, J = 8.3 Hz, 2H), 6.49 (d, J = 8.7 Hz, 1H), 6.27 (s, 1H), 5.19 (s, 2H), 3.98 (t, J = 5.7 Hz, 2H), 3.69 (s, 3H), 2.63 (t, J = 5.7 Hz, 2H), 2.36 (s, 4H), 1.51–1.28 (m, 6H). 13C NMR (101 MHz, CDCl3) δ 180.65, 165.07, 162.78, 160.48, 136.30, 131.87, 129.47, 128.78, 127.07, 115.73, 114.15, 110.63, 101.33, 67.95, 66.39, 57.56, 55.28, 54.97, 25.90, 24.12. LC/MS (ESI) m/z: [M + H]+ = 394.2. Purity: 96.2% (by HPLC).
(E)-3-(4-methoxybenzylidene)-7-(3-(piperidin-1-yl)propoxy)chroman-4-one (10)
Intermediate 6 was treated with piperidine according to the general procedure to give the desired product 10 as a yellow solid, yield 74%; 1H NMR (400 MHz, CDCl3) δ 7.89–7.73 (m, 1H), 7.64 (s, 1H), 7.10 (d, J = 8.2 Hz, 2H), 6.80 (d, J = 8.4 Hz, 2H), 6.47 (dd, J = 7.2, 4.8 Hz, 1H), 6.25 (d, J = 3.4 Hz, 1H), 5.18 (d, J = 2.4 Hz, 2H), 3.88 (d, J = 6.3 Hz, 2H), 3.67 (dd, J = 9.2, 5.7 Hz, 3H), 2.42–2.04 (m, 6H), 1.83 (d, J = 5.8 Hz, 2H), 1.54–1.24 (m, 6H). 13C NMR (101 MHz, CDCl3) δ 180.63, 165.33, 162.81, 160.45, 136.23, 131.86, 129.43, 128.79, 127.05, 115.58, 114.14, 110.59, 101.20, 67.92, 66.85, 55.64, 55.25, 54.56, 26.53, 25.91, 24.39. LC/MS (ESI) m/z: [M + H]+ = 408.2. Purity: 98.6% (by HPLC).
(E)-3-(4-methoxybenzylidene)-7-(4-(piperidin-1-yl)butoxy)chroman-4-one (11)
Intermediate 7 was treated with piperidine according to the general procedure to give the desired product 11 as a yellow solid, yield 62%; 1H NMR (400 MHz, CDCl3) δ 7.80 (dd, J = 8.8, 1.4 Hz, 1H), 7.65 (s, 1H), 7.10 (d, J = 7.2 Hz, 2H), 6.84–6.77 (m, 2H), 6.47 (d, J = 8.7 Hz, 1H), 6.24 (s, 1H), 5.19 (s, 2H), 3.85 (dd, J = 6.1, 5.1 Hz, 2H), 3.68 (dd, J = 7.0, 1.7 Hz, 3H), 2.22 (dd, J = 14.7, 6.6 Hz, 6H), 1.70–1.38 (m, 10H). 13C NMR (101 MHz, CDCl3) δ 180.64, 165.33, 162.81, 160.45, 136.24, 131.85, 129.44, 128.79, 127.05, 115.56, 114.13, 110.59, 101.14, 68.17, 67.92, 58.86, 55.25, 54.51, 27.11, 25.89, 24.42, 23.28. LC/MS (ESI) m/z: [M + H]+ = 422.2. Purity: 99.6% (by HPLC).
(E)-3-(4-methoxybenzylidene)-7-(5-(piperidin-1-yl)pentyloxy)chroman-4-one (12)
Intermediate 8 was treated with piperidine according to the general procedure to give the desired product 12 as a yellow solid, yield 58%; 1H NMR (400 MHz, CDCl3) δ 7.94 (d, J = 8.8 Hz, 1H), 7.79 (s, 1H), 7.26 (d, J = 9.0 Hz, 2H), 6.95 (d, J = 8.8 Hz, 2H), 6.60 (dd, J = 8.8, 2.4 Hz, 1H), 6.37 (d, J = 2.3 Hz, 1H), 5.34 (d, J = 1.8 Hz, 2H), 4.02–3.92 (m, 2H), 3.84 (d, J = 10.1 Hz, 3H), 2.36 (s, 4H), 2.32–2.28 (m, 2H), 1.84–1.75 (m, 2H), 1.63–1.54 (m, 6H), 1.50–1.42 (m, 4H). 13C NMR (101 MHz, CDCl3) δ 180.92, 165.47, 162.91, 160.53, 136.41, 131.86, 129.59, 128.98, 127.23, 115.68, 114.21, 110.66, 101.21, 77.35, 77.04, 76.72, 68.29, 68.00, 59.36, 55.36, 54.68, 28.94, 26.67, 26.01, 24.49, 24.12. LC/MS (ESI) m/z: [M + H]+ = 436.3. Purity: 99.0% (by HPLC).
(E)-7-(3-(dimethylamino)propoxy)-3-(4-methoxybenzylidene)chroman-4-one (13)
Intermediate 6 was treated with dimethylamine according to the general procedure to give the desired product 13 as a yellow solid, yield 75%; 1H NMR (400 MHz, CDCl3) δ 7.94 (d, J = 8.8 Hz, 1H), 7.80 (s, 1H), 7.28–7.23 (m, 2H), 6.96 (d, J = 8.7 Hz, 2H), 6.61 (dd, J = 8.8, 2.3 Hz, 1H), 6.39 (d, J = 2.3 Hz, 1H), 5.35 (d, J = 1.8 Hz, 2H), 4.05 (s, 3H), 3.85 (s, 2H), 2.64–2.59 (m, 2H), 2.36 (s, 6H), 1.98 (s, 2H). 13C NMR (101 MHz, CDCl3) δ 180.96, 165.16, 162.91, 160.56, 136.55, 131.89, 129.63, 128.89, 127.18, 115.84, 114.22, 110.57, 101.30, 68.01, 66.34, 55.65, 55.37, 44.48, 26.36, 22.39. LC/MS(ESI) m/z: [M + H]+ = 368.2. Purity: 95.1% (by HPLC).
(E)-7-(3-(diethylamino)propoxy)-3-(4-methoxybenzylidene)chroman-4-one (14)
Intermediate 6 was treated with diethylamine according to the general procedure to give the desired product 14 as a yellow solid, yield 70%; 1H NMR (400 MHz, CDCl3) δ 7.96 (d, J = 8.8 Hz, 1H), 7.82 (s, 1H), 7.31–7.26 (m, 2H), 6.98 (d, J = 8.8 Hz, 2H), 6.64 (dd, J = 8.8, 2.4 Hz, 1H), 6.42 (d, J = 2.3 Hz, 1H), 5.37 (d, J = 1.8 Hz, 2H), 4.07 (s, 2H), 3.87 (s, 3H), 2.56 (dt, J = 21.4, 7.2 Hz, 6H), 1.94 (dt, J = 13.2, 6.4 Hz, 2H), 1.04 (t, J = 7.1 Hz, 6H). 13C NMR (101 MHz, CDCl3) δ 180.95, 165.49, 162.92, 160.53, 136.42, 131.87, 129.58, 128.99, 127.24, 115.70, 114.21, 110.69, 101.26, 68.01, 66.79, 55.37, 49.22, 47.04, 26.94, 11.83. LC/MS (ESI) m/z: [M + H]+ = 396.2. Purity: 99.9% (by HPLC).
(E)-7-(3-(dipropylamino)propoxy)-3-(4-methoxybenzylidene)chroman-4-one (15)
Intermediate 6 was treated with dipropylamine according to the general procedure to give the desired product 15 as a yellow solid, yield 60%; 1H NMR (400 MHz, CDCl3) δ 7.96 (d, J = 8.8 Hz, 1H), 7.82 (s, 1H), 7.32–7.24 (m, 2H), 6.98 (d, J = 8.8 Hz, 2H), 6.63 (dd, J = 8.8, 2.4 Hz, 1H), 6.41 (d, J = 2.3 Hz, 1H), 5.37 (d, J = 1.8 Hz, 2H), 4.07 (d, J = 6.3 Hz, 2H), 3.87 (s, 3H), 2.58 (t, J = 6.9 Hz, 2H), 2.43–2.32 (m, 4H), 1.96–1.88 (m, 2H), 1.46 (dd, J = 14.9, 7.4 Hz, 4H), 0.88 (t, J = 7.4 Hz, 6H). 13C NMR (101 MHz, CDCl3) δ 180.94, 165.55, 162.92, 160.53, 136.40, 131.86, 129.57, 129.01, 127.24, 115.67, 114.21, 110.70, 101.23, 68.01, 66.73, 56.33, 55.37, 50.42, 27.08, 20.39, 11.94. LC/MS (ESI) m/z: [M + H]+ = 424.3. Purity: 99.5% (by HPLC).
(E)-3-(4-methoxybenzylidene)-7-(3-(pyrrolidin-1-yl)propoxy)chroman-4-one (16)
Intermediate 6 was treated with pyrrolidine according to the general procedure to give the desired product 16 as a yellow solid, yield 62%; 1H NMR (400 MHz, CDCl3) δ 7.96 (d, J = 8.8 Hz, 1H), 7.81 (s, 1H), 7.30–7.26 (m, 2H), 6.98 (d, J = 8.8 Hz, 2H), 6.64 (dd, J = 8.8, 2.3 Hz, 1H), 6.42 (d, J = 2.3 Hz, 1H), 5.37 (d, J = 1.7 Hz, 2H), 4.09 (t, J = 6.4 Hz, 2H), 3.87 (s, 3H), 2.66–2.59 (m, 2H), 2.54 (dd, J = 9.2, 3.9 Hz, 4H), 2.06–2.00 (m, 2H), 1.83–1.77 (m, 4H). 13C NMR (101 MHz, CDCl3) δ 180.98, 165.43, 162.92, 160.54, 136.46, 131.88, 129.59, 128.98, 127.23, 115.72, 114.22, 110.69, 101.30, 68.01, 66.85, 55.38, 54.22, 52.90, 28.60, 23.47. LC–MS (ESI + APCI) [M + H]+ = 394.2. Purity: 98.4% (by HPLC).
(E)-3-(4-methoxybenzylidene)-7-(3-morpholinopropoxy)chroman-4-one (17)
Intermediate 6 was treated with morpholine according to the general procedure to give the desired product 17 as a yellow solid, yield 75%; 1H NMR (400 MHz, CDCl3) δ 7.97 (d, J = 8.8 Hz, 1H), 7.82 (s, 1H), 7.31–7.25 (m, 2H), 6.98 (d, J = 8.8 Hz, 2H), 6.64 (dd, J = 8.8, 2.4 Hz, 1H), 6.42 (d, J = 2.3 Hz, 1H), 5.37 (d, J = 1.8 Hz, 2H), 4.09 (t, J = 6.3 Hz, 2H), 3.86 (d, J = 10.7 Hz, 3H), 3.75–3.72 (m, 4H), 2.55–2.51 (m, 2H), 2.50–2.46 (m, 4H), 2.03–1.96 (m, 2H). 13C NMR (101 MHz, CDCl3) δ 180.96, 165.35, 162.92, 160.56, 136.54, 131.89, 129.63, 128.91, 127.20, 115.78, 114.23, 110.65, 101.28, 68.03, 66.96, 66.51, 55.33 (d, J = 10.6 Hz), 53.74, 26.19. LC/MS (ESI) m/z: [M + H]+ = 410.2 Purity: 99.4% (by HPLC).
(E)-7-(3-(cyclohexylamino)propoxy)-3-(4-methoxybenzylidene)chroman-4-one (18)
Intermediate 6 was treated with cyclohexylamine according to the general procedure to give the desired product 18 as a yellow solid, yield 59%; 1H NMR (400 MHz, CDCl3) δ 7.97 (d, J = 8.8 Hz, 1H), 7.82 (s, 1H), 7.31–7.25 (m, 2H), 6.98 (d, J = 8 Hz). 1H NMR (400 MHz, CDCl3) δ 7.96 (d, J = 8.8 Hz, 1H), 7.81 (s, 1H), 7.27 (d, J = 8.9 Hz, 2H), 7.01–6.89 (m, 2H), 6.62 (dd, J = 8.8, 2.4 Hz, 1H), 6.41 (d, J = 2.3 Hz, 1H), 5.36 (d, J = 1.8 Hz, 2H), 4.09 (d, J = 6.2 Hz, 2H), 3.86 (s, 3H), 2.82 (t, J = 7.0 Hz, 2H), 2.45 (s, 1H), 1.97 (t, J = 6.6 Hz, 2H), 1.94–1.86 (m, 2H), 1.78–1.70 (m, 2H), 1.63 (d, J = 11.7 Hz, 2H), 1.32–1.18 (m, 4H), 1.17–1.11 (m, 1H). 13C NMR (101 MHz, CDCl3) δ 180.94, 165.35, 162.91, 160.54, 136.48, 131.88, 129.61, 128.94, 127.21, 115.76, 114.22, 110.64, 101.28, 68.02, 66.88, 56.85, 55.37, 43.66, 33.60, 29.94, 26.15, 25.06. LC/MS (ESI) m/z: [M + H]+ = 422.3. Purity: 98.2% (by HPLC).
Biological activity
In vitro inhibition studies on AChE and BuChE
Acetylcholinesterase (AChE, E.C. 3.1.1.7, from electric eel), butylcholinesterase (BuChE, E.C. 3.1.1.8, from equine serum), 5,5′-dithiobis-(2-nitrobenzoic acid) (Ellman’s reagent, DTNB), acetylthiocholine chloride (ATC) and butylthiocholine chloride (BTC) were purchased from Sigma-Aldrich (St. Louis, MO). Compounds were dissolved in DMSO (10 mM) and diluted in 0.1 M KH2PO4/K2HPO4 buffer (pH 8.0) to the desired final concentration. All the compounds are soluble at the tested concentration. DMSO was diluted to a concentration of less than 0.01%, and no inhibitory action on either AChE or BuChE was detected in separate prior experiments at this concentration. The in vitro AChE assay was performed as follows: all assays were conducted in 0.1 M KH2PO4/K2HPO4 buffer (pH 8.0) using a Shimadzu UV-2450 spectrophotometer. The assay medium (1 mL) consisted of phosphate buffer (pH 8.0), 50 μL of 0.01 M DTNB, 10 μL of enzyme and 50 μL of 0.01 M substrate (ACh chloride solution). Test compounds were added to the assay solution and pre-incubated with the enzyme at 37 °C for 15 min, followed by addition of substrate. The activity was determined by measuring the increase in absorbance at 412 nm at 1 min intervals at 37 °C. Calculations were performed according to the method of the equation in Ellman et al. Each concentration was assayed in triplicate.
The in vitro BuChE assay was performed similarly to the method described above.
Kinetic characterisation of AChE inhibition
Kinetic characterisation of AChE was performed using a reported method. Test compound was added into the assay solution and pre-incubated with the enzyme at 37 °C for 15 min, followed by addition of substrate (the final concentrations of substrate were 0.05, 0.0625, 0.10, 0.125, 0.25, 0.50 mM, respectively). Kinetic characterisation of the hydrolysis of ATC catalyzed by AChE was performed spectrometrically at 412 nm. A parallel control with no inhibitor in the mixture allowed the change in activities to be measured at various times. The plots were assessed by a weighted least square analysis that assumed the variance of V to be a constant percentage of V for the entire data set. The slopes of these reciprocal plots were subsequently plotted against the concentration of the inhibitors in a weighted analysis, and Ki was determined as the ratio of the replot intercept to the replot slope.
Inhibition of MAO activity
The potential effects of the test compounds on hMAO activity were investigated by measuring their effects on the production of H2O2 from p-tyramine, using the Amplex Red MAO assay kit (Molecular Probes, Inc., Eugene, OR) and recombinant human MAO-A or MAO-B (Sigma-Aldrich) according to published proceduresCitation40. Compounds were dissolved in DMSO (10 mM) and diluted in 0.1 M KH2PO4/K2HPO4 buffer (pH 8.0) to the desired final concentration. All the compounds are soluble at the tested concentration. Adequate amounts of recombinant hMAO-A or hMAO-B (Sigma-Aldrich) were acquired and adjusted to 12.5 μg/mL for hMAO-A and 75 μg/mL for hMAO-B. Test drugs (20 μL) and MAO (80 μL) were incubated at 37 °C for 15 min in a flat-black-bottom 96-well microtest plate in dark. The reaction was started by adding 200 μM Amplex Red reagent, 2 U/mL horseradish peroxidase and 2 mM p-tyramine for hMAO-A or 2 mM benzylamine for hMAO-B, at 37 °C for 20 min. The reaction was quantified in a multi-detection microplate fluorescence reader based on the fluorescence generated (excitation, 545 nm; emission, 590 nm).
The specific fluorescence emission was calculated after subtraction of the background activity, which was determined from vials containing all components except the hMAO isoforms, which were replaced by a sodium phosphate buffer solution.
Reversibility and irreversibility experiments
To evaluate whether compound 10 is a reversible or irreversible hMAO-B inhibitor, an effective centrifugation ultrafiltration method (so-called “repeated washing”) was usedCitation40. The experiment was performed according to a reported method. Adequate amounts of recombinant monoamine oxidase with or without test drugs were incubated at 37 °C for 15 min. An aliquot from this incubation was stored at 4 °C for later use in the measurement of MAO activity. Another aliquot was transferred to an Amicon Ultra-0.5 Centrifugal Filter Unit with Ultracel-30 membrane (Millipore) and centrifuged (4 °C, 9000g, 20 min) three times. The enzyme was obtained and used for the subsequent measurement of activity using a method similar to that described above (the section “Inhibition of MAO activity”). The corresponding values of percent hMAO-B inhibition were separately calculated for samples with and without repeated washing.
Docking study
The simulation system was built based on the structure obtained from the Protein Data Bank (PDB: 2V60 for MAO-B, 2CMF for AChE). The heteroatoms and water molecules were removed, and all hydrogen atoms were subsequently added to the protein. Then forcefield was assigned to the enzyme. The ligand binding site was defined as 13 Å from the original ligand. Prior to the docking calculations, the original ligand was removed. The 3D structures of compound 10 were generated and optimized with the Discovery Studio 2.1 package (Accelrys Inc., San Diego, CA).
The CDOCKER program of the Discovery Studio 2.1 software, which allows full flexibility of ligands, was used to perform docking simulations. The docking and subsequent scoring were performed using the default parameters of the CDOCKER program. CDOCKER_INTERACTION_ENERGY is used like a score where a lower value indicates a more favorable binding.
Results and discussion
Chemistry
The homoisoflavonoid derivatives 9–18 were synthesized according to the synthetic protocol outlined in Scheme 1. First, commercial available resorcinol (1) reacted with 3-chloropropionic acid via Fries rearrangement to yield compound 2, and the followed cyclization of 2 in aqueous NaOH gave 7-hydroxychroman-4-one (3). Then, (E)-7-hydroxy-3-(4-methoxybenzylidene)-chroman-4-one (4) was obtained by the condensation of 3 with p-methoxybenzaldehydeCitation39. The reaction of 4 in butanone with α,ω-dibromo alkane in the presence of K2CO3 provided responding intermediates 5–8. Finally, target products 9–18 were got by the reactions of 5–8 with commercially available secondary amines (dimethylamine, pyrrolidine, etc.) in 53–87% yields. All the compounds were characterized using analytical and NMR spectroscopic data (see Supplementary data).
Scheme 1. Reagents and conditions: (a) 3-chloropropionic acid trifluoromethanesulfonic acid; (b) NaOH, 0 °C; (c) p-methoxybenzaldehyde, piperidine, 80 °C; (d) α,ω-dibromo alkane, K2CO3, KI, butanone, rt; (e) amines, acetonitrile, reflux.
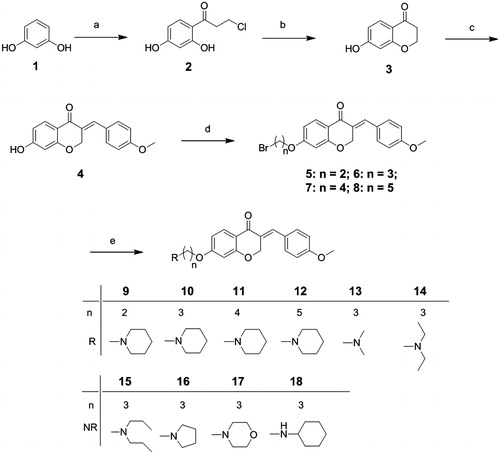
Compounds 5–18 were synthesized by the following procedures. Intermediates 2, 3 and 4 were synthesized according to referenceCitation39.
In vitro inhibition of AChE and BuChE
The AChE (E.C.3.1.1.7) inhibitory effects of the homoisoflavonoid derivatives were determined by the spectroscopic method described by Ellman et al. using rivastigmine as the standardCitation41. As shown in , most of the hybrids demonstrated potent inhibitory activity against AChE and BuChE with IC50 values in the sub-micromolar to micromolar range. Compound 9, a piperidine moiety linked with homoisoflavonoid by two carbon spacers, gave the most potent inhibitory activity (IC50 = 0.74 μM), which was about 135-fold more potent than lead compound 4 (IC50 > 100 μM). A simple structure–activity relationship analysis showed that the AChE inhibitory potency closely related to the length of the alkylene chain. When the length of the alkylene chain increased to 5, the AChE inhibitory activity decreased dramatically (12, IC50 = 14.1 μM). From the IC50 values of the tested compounds, it appears that a 3-carbon spacer between homoisoflavonoid moiety and amino group seems to be the proper length for balancing the inhibition activity against AChE and MAO-B. Moreover, amino group is also important for the inhibitory activities. Among the three aliphatic substituted amino derivatives, the N,N-dimethyl amino group (13, IC50 = 3.78 μM) and N,N-diethyl amino group (14, IC50 = 2.45 μM) gave better results than N,N-dipropyl amino group (15, IC50 = 7.47 μM). On the other hand, among the cyclic substituted amino derivatives, compounds 10 and 16, piperidine and pyrrolidine linked with berberine by three carbon spacers, gave IC50 values of 3.94 and 2.83 μM, respectively. Compound 17 (IC50 = 7.47 μM), morpholine linked with homoisoflavonoid by three carbon spacers, afforded poor activities compared with compounds 10 and 16. Surprisingly, the replacement of the piperidine group by a cyclohexylamine group markedly decreased the inhibitory activity (10, IC50 = 3.94 μM versus 18, IC50 = 27.86 μM).
Table 1. In vitro inhibition IC50 (μM) and selectivity of compounds 4, 9–18 on ChEs and MAOs.
The in vitro BuChE (E.C. 3.1.1.8) inhibition activities were also determined using the same method. Most of the tested compounds demonstrated much higher inhibitory potency against BuChE than homoisoflavonoid 4. Compound 10, which exhibited a good balance of AChE/MAO-B inhibitory activity, was chosen for further studies.
Kinetic study of AChE
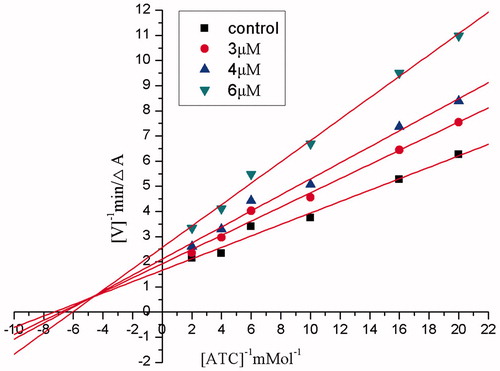
In order to investigate the mechanism of homoisoflavonoid derivatives against AChE, kinetic study was performed using compound 10 and the steady-state inhibition data for AChE are shown in . Lineweaver–Burk reciprocal plots revealed that there was an increasing slope and an increasing intercept at higher inhibitor concentrations, indicating a mixed inhibition. Based on the kinetic analysis, we concluded that homoisoflavonoid derivatives exhibited as dual binding sites AChE inhibitors, which target catalytic and peripheral sites of AChE simultaneously.
Figure 4. Steady state inhibition by compound 10 of AChE hydrolysis of ACh; the plots show mixed-type inhibition for compound 10 on AChE.
Molecular modeling studies of AChE
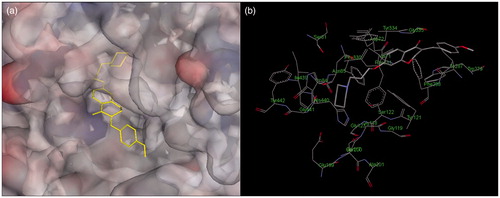
To explore the interaction mode of homoisoflavonoid derivatives with AChE and the structure–activity relationships, molecular docking simulations were performed with the Discovery studio 2.1 software based on the structure of the complex of Torpedo californica enzyme (TcAChE) (PDB entry 2CMF). As shown in , compound 10 could simultaneously interact with the CAS and the PAS of AChE, which is consistent with the result of kinetic study. Near the bottom of the gorge, the protonated piperidine moiety of compound 10 interacts with Phe330 and Trp84 via hydrophobic interactions to form a cation–π interaction (4.76 and 4.32 Å, respectively). The homoisoflavonoid moiety of 10 reached the top of the cavity and interacted with PAS aromatic residues Trp279, phe331, tyr121 (5.48, 4.40 and 5.36 Å, respectively) through π–π stacking. The homoisoflavonoid moiety snaked along the gorge, interacting with Tyr334, Ser122 and Val71 by hydrophobic interactions.
Figure 5. Docking models of the compound–enzyme complex. (a) stereoviews looking down the gorge of TcAChE binding with 10 (yellow). (b) 10 docked into the catalytic gorge of TcAChE, highlighting the protein residues belonging to ACS and PAS that establish the main interactions.
In vitro inhibition studies of hMAO-A and hMAO-B
To confirm the multipotent biological profile of homoisoflavonoid derivatives, inhibitory activity against MAOs was determined using ladostigil as reference compound and the results are summarized in . Most of these target compounds exhibited high selective inhibition to hMAO-B in the micromolar range. Among them, compound 12 presented the most potent inhibition for hMAO-B with IC50 value of 0.65 μM, which is 2-fold and 57-fold more potent than lead compound 4 and ladostigil, respectively. A structure–activity relationship analysis showed that the length of carbon spacer also played an important role in inhibition activity. However, unlike that of AChE, a longer linker resulted in better inhibitory activity, the inhibition activity against MAO-B increased as the increasing of the carbon spacer length. For example, compound 9, in which piperidine group linked with homoisoflavonoid via a 2-carbon spacer, provides IC50 value of more than 25 μM. Its homologous compound 12, with 5-carbon spacer, gave 0.65 μM of the IC50 value, which was 40 times as that of compound 9. On the other hand, all of the target compounds showed very weak inhibition activity against MAO-A. Overall, among the synthesized compounds, compound 10 exhibited best balance of inhibition for both AChE and MAO-B.
Reversibility and irreversibility study of hMAO-B
The reversibility and irreversibility of inhibitory activities of 10 was assessed according to a method of referenceCitation40. Pargyline, known as irreversible MAO-B inhibitor, was used as a reference compound. Data are listed in . Similarly with pargyline, MAO-B inhibition of 10 was irreversible for the enzyme activity, which did not restore after repeated washing.
Table 2. Reversibility and irreversibility of hMAO-B inhibition of hybrid 10 and pargylinea.
Molecular modeling studies of MAO-B
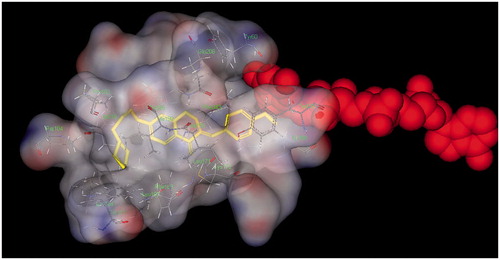
To evaluate the binding modes of the homoisoflavonoid derivatives with MAO-B, docking simulation of compound 10 was carried out using the CDOCKER program in the Discovery studio 2.1 software based on the X-ray crystal structure of human MAO-B (PDB entry 2V60). As shown in , the benzyl group of homoisoflavonoid moiety is properly oriented to the flavin adenine dinucleotide (FAD) cofactor adopting between Tyr398 (4.25 Å) and Tyr435 (4.33 Å) form face-to-face p–p stacking interactions in a “sandwich” form. Besides, the pyridine moiety and carbon linker are embedded into a hydrophobic pocket delimited by Pro102, Pro104, Trp119, Leu164, Leu167, Phe186, Ile316.
Figure 6. Docking pose of derivative 10 (yellow) into human MAO-B (2V60) highlighting the protein residues that establish the main interactions with 10. The FAD cofactor was depicted using space fill representation.
Conclusion
In summary, we have designed, synthesized and evaluated a series of homoisoflavonoid derivatives as dual functional cholinesterase and MAO inhibitors for the treatment of AD. Most of these compounds were potent inhibitors of ChE, which are more active than lead compound 4. Meanwhile, compound 12 presented the most potent inhibition for hMAO-B with IC50 value of 0.65 μM. Compound 10, with a pyridine group linked with homoisoflavonoid moiety by a three carbon linker, provided a good balance of AChE and hMAO-B inhibition activities, with IC50 value of 3.94 and 3.44 μM, respectively. Kinetic studies revealed that compound 10 was a mixed-type inhibitor of AChE and an irreversible inhibitor of hMAO-B. Molecular modeling studies indicated that 10 could interact with both CAS and PAS of AChE, which is consistent with kinetic studies. Based on these results, compound 10 is a promising “one-compound-multi-targets” candidate for AD treatment and needs further study.
Supplementary material available online
Supplemental Material.pdf
Download PDF (375.7 KB)Declaration of interest
The authors declare no conflicts of interest.
We thank the National Natural Science Foundation of China (No. 21302235, 20972198), the Opening Project of Guangdong Provincial Key Laboratory of New Drug Design and Evaluation (2011A060901014) and Ph.D. Programs Foundation of Ministry of Education of China (20120171120045) for financial support of this study.
References
- Blennow K, De Leon M, Zetterberg H. Alzheimer's disease. Lancet 2006;368:387–403
- Goedert M. Century of Alzheimer's disease. Science 2006;314:777–81
- Mucke L. Neuroscience: Alzheimer's disease. Nature 2009;461:895–7
- Bartus R, Dean R, Beer B, et al. The cholinergic hypothesis of geriatric memory dysfunction. Science 1982;217:408–14
- Talesa VN. Acetylcholinesterase in Alzheimer's disease. Mech Ageing Dev 2001;122:1961–9
- Hardy J, Higgins G. Alzheimer's disease: the amyloid cascade hypothesis. Science 1992;256:184–5
- Hardy J, Selkoe DJ. Medicine – the amyloid hypothesis of Alzheimer's disease: progress and problems on the road to therapeutics. Science 2002;297:353–6
- Hardy J. Alzheimer's disease: the amyloid cascade hypothesis: an update and reappraisal. J Alzheimers Dis 2006;9:151–3
- Barnham KJ, Masters CL, Bush AI. Neurodegenerative diseases and oxidative stress. Nat Rev Drug Discov 2004;3:205–14
- Beal MF. Aging, energy, and oxidative stress in neurodegenerative diseases. Ann Neurol 1995;38:357–66
- Simonian NA, Coyle JT. Oxidative stress in neurodegenerative diseases. Ann Rev Pharmacol 1996;36:83–106
- Lin MT, Beal MF. Mitochondrial dysfunction and oxidative stress in neurodegenerative diseases. Nature 2006;443:787–95
- Grundke-Iqbal I, Iqbal K, Tung YC, et al. Abnormal phosphorylation of the microtubule-associated protein tau (tau) in Alzheimer cytoskeletal pathology. Nat Acad Sci USA 1986;83:4913–17
- Goedert M, Spillantini MG, Jakes R, et al. Multiple isoforms of human microtubule-associated protein tau: sequences and localization in neurofibrillary tangles of Alzheimer's disease. Neuron 1989;3:519–26
- Colombres M, Sagal J, PInestrosa NC. An overview of the current and novel drugs for Alzheimer's disease with particular reference to anti-cholinesterase compounds. Curr Pharm Design 2004;10:3121–30
- Contestabile A. The history of the cholinergic hypothesis. Behav Brain Res 2011;221:334–40
- Harel M, Schalk I, Ehret-Sabatier L, et al. Quaternary ligand binding to aromatic residues in the active-site gorge of acetylcholinesterase. Nat Acad Sci USA 1993;90:9031–5
- Raves ML, Harel M, Pang Y-P, et al. Structure of acetylcholinesterase complexed with the nootropic alkaloid, (−)-huperzine A. Nat Struct Biol 1997;4:57–63
- Kryger G, Silman I, Sussman JL. Structure of acetylcholinesterase complexed with E2020 (Aricept (R)): implications for the design of new anti-Alzheimer drugs. Struct Fold Des 1999;7:297–307
- Inestrosa NC, Alvarez A, Perez CA, et al. Acetylcholinesterase accelerates assembly of amyloid-beta-peptides into Alzheimer's fibrils: possible role of the peripheral site of the enzyme. Neuron 1996;16:881–91
- Bourne Y, Taylor P, Radic Z, et al. Structural insights into ligand interactions at the acetylcholinesterase peripheral anionic site. Embo J 2003;22:1–12
- Piazzi L, Rampa A, Bisi A, et al. 3-(4-{benzyl(methyl)amino methyl}-phenyl)-6,7-dimethoxy-2H-2-chromenone (AP2238) inhibits both acetylcholinesterase and acetylcholinesterase-induced beta-amyloid aggregation: a dual function lead for Alzheimer's disease therapy. J Med Chem 2003;46:2279–82
- Carlier PR, Han YF, Chow ESH, et al. Evaluation of short-tether bis-THA AChE inhibitors. A further test of the dual binding site hypothesis. Bioorg Med Chem 1999;7:351–7
- Bartolini M, Bertucci C, Cavrini V, et al. Beta-amyloid aggregation induced by human acetylcholinesterase: inhibition studies. Biochem Pharm 2003;65:407–16
- Walker WH, Kearney EB, Seng RL, et al. The covalently-bound flavin of hepatic monoamine oxidase. 2. Identification and properties of cysteinyl riboflavin. Eur J Biochem/FEBS 1971;24:328–31
- Chiba K, Trevor A, Castagnoli N Jr. Metabolism of the neurotoxic tertiary amine, MPTP, by brain monoamine oxidase. Biochem Biophys Res Comm 1984;120:574–8
- Binda C, Newton-Vinson P, Hubalek F, et al. Structure of human monoamine oxidase B, a drug target for the treatment of neurological disorders. Nat Struct Biol 2002;9:22–6
- Edmondson DE, DeColibus L, Binda C, et al. New insights into the structures and functions of human monoamine oxidases A and B. Neural Transm (Vienna, Austria: 1996) 2007;114:703–5
- Weinreb O, Amit T, Bar-Am O, et al. Rasagiline: a novel anti-Parkinsonian monoamine oxidase-B inhibitor with neuroprotective activity. Prog Neurobiol 2010;92:330–44
- Carreiras MC, Marco JL. Recent approaches to novel anti-Alzheimer therapy. Curr Pharm Des 2004;10:3167–75
- Sterling J, Herzig Y, Goren T, et al. Novel dual inhibitors of AChE and MAO derived from hydroxy aminoindan and phenethylamine as potential treatment for Alzheimer's disease. J Med Chem 2002;45:5260–79
- Perjesi P, Das U, De Clercq E, et al. Design, synthesis and antiproliferative activity of some 3-benzylidene-2,3-dihydro-1-benzopyran-4-ones which display selective toxicity for malignant cells. Eur J Med Chem 2008;43:839–45
- Desideri N, Olivieri S, Stein ML, et al. Synthesis and anti-picornavirus activity of homo-isoflavonoids. Antiviral Chem Chemoth 1997;8:545–55
- Quaglia MG, Desideri N, Bossu E, et al. Enantioseparation and anti-rhinovirus activity of 3-benzylchroman-4-ones. Chirality 1999;11:495–500
- Siddalah V, Rao CV, Venkateswarlu S, et al. Synthesis, stereochemical assignments, and biological activities of homoisoflavonoids. Bioorg Med Chem 2006;14:2545–51
- Hung TM, Cao Van T, Nguyen Tien D, et al. Homoisoflavonoid derivatives from the roots of Ophiopogon japonicus and their in vitro anti-inflammation activity. Bioorg Med Chem Lett 2010;20:2412–16
- Desideri N, Bolasco A, Fioravanti R, et al. Homoisoflavonoids: natural scaffolds with potent and selective monoamine oxidase-B inhibition properties. J Med Chem 2011;54:2155–64
- Lu C, Zhou Q, Yan J, et al. A novel series of tacrine-selegiline hybrids with cholinesterase and monoamine oxidase inhibition activities for the treatment of Alzheimer's disease. Eur J Med Chem 2013;62:745–53
- Sun Y, Chen J, Chen X, et al. Inhibition of cholinesterase and monoamine oxidase-B activity by Tacrine–Homoisoflavonoid hybrids. Bioorg Med Chem 2013;21:7406–17
- Secci D, Bolasco A, Carradori S, et al. Recent advances in the development of selective human MAO-B inhibitors: (Hetero)arylidene-(4-substituted-thiazol-2-yl)hydrazines. Eur J Med Chem 2012;58:405–17
- Ellman GL, Courtney KD, Andres BJ, et al. A new and rapid colorimetric determination of acetylcholinesterase activity. Biochem Pharmacol 1961;7:88–95