
Abstract
Carbonic anhydrase type IX (CA IX) enzyme is mostly over expressed in different cancer cell lines and tumor tissues. Potent CA IX inhibitors can be effective for adjusting the pH imbalance in tumor cells. In the present work, we represented the successful application of high throughput virtual screening (HTVS) of large dataset from ZINC database included of ∼7 million compounds to discover novel inhibitors of CA IX. HTVS and molecular docking were performed using consequence Glide/standard precision (SP), extra precision (XP) and induced fit docking (IFD) molecular docking protocols. For each compound, docking code calculates a set of low-energy poses and then exhaustively scans the binding pocket of the target with small compounds. Novel CA IX inhibitor candidates were suggested based on molecular modeling studies and a few of them were tested using in vitro analysis. These compounds were determined as good inhibitors against human CA IX target with Ki in the range of 0.85–1.58 μM. In order to predict the pharmaceutical properties of the selected compounds, ADME (absorption, distribution, metabolism and excretion) analysis was also carried out.
Introduction
Carbonic anhydrase (CA) family includes different isomers that display different level of enzyme activity and kinetic characteristicsCitation1–11. Only two of the CA isomers are distinguished as targets that are related to cancer: CA IX and CA XIICitation12. While CA IX is commonly represented in a narrow number of ordinary tissues, its over expression is identified on solid tumors, where it is always connected with the hypoxic phenotype, mediated by the transcription factor HIF-1Citation13. Over expression of CA IX is mostly affiliated with a weak responsiveness to classical chemotherapyCitation14. CA IX has been the subject of several studies as an enzyme responsible for the pH adjustment of tumor cells, as well as for the cell reproduction and cell adhesionCitation15,Citation16. It is an important target for cancer diagnostics and therapyCitation17. CA IX has an N-terminal domain, an extracellular CA catalytic domain, a trans-membrane (TM) helical domain and a short intra-cytoplasmic tail. The first domain plays an important role in the protein-mediated cell adhesion events. The catalytic domain catalyzes the reversible hydration of CO2, thus it contributes to pH regulationCitation18. In this work, a large ligand database (∼7 million) from ZINC database is used to detect novel CA IX inhibitors using high throughput virtual screening (HTVS) approach. A few proposed compounds based on virtual screening analysis are then tested by the in vitro and experimental data confirmed simulation results. Absorption, distribution, metabolism and excretion (ADME) profiles of studied compounds are also predicted using molecular modeling techniques.
Methods
Structural preparation of enzyme
The crystal structure of carbonic anhydraseCitation19 IX (CA IX) (PDB ID: 3IAI) has been taken from the Protein Data BankCitation20. This structure is a dimer, which involves two identical inhibitors () inside the binding sites. In this work only one chain is used for docking simulations. Hydrogen atoms were added using Protein Preparation WizardCitation21 integrated in the Maestro molecular modeling Suite. The disulfide bond between Cys23 and Cys203 was constrained utilizing the patch DISU command during preparationCitation22. The PROPKACitation23–25 program is used to locate protonation states at the physiological pH of 7.4. Hydrogen atoms were then relaxed using the energy minimization.
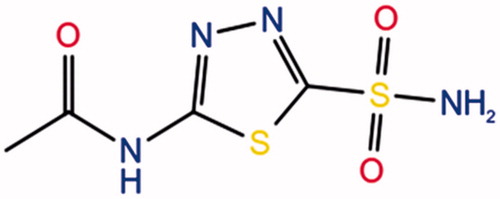
Figure 1. Chemical structure of acetazolamide (AZM) exists in each monomer.
MD simulations protocol
In order to relax the side chains of the CA IX complex, 10 ns MD simulations was performed before the docking simulations. Ligand–protein complex was hydrated with explicit water molecules using TIP3P water models, and neutralized by 0.15 M KCI using VMD (Visual Molecular Dynamics)Citation26 program. MD simulations were then carried out using the parallel MD softwareCitation27 NAMD 2.9 with CHARMM36 force-field parametersCitation28. SHAKE algorithm was used to restrain the hydrogen atoms. Langevin piston method with Langevin thermostat was used to maintain constant temperature at 310 K and at a constant pressure of 1 atm during simulations. Periodic Boundary Conditions (PBC) and Particle Mesh Ewalds (PME) methods were utilized for treating the long-range electrostatic interactions. The non-bonded cut-off radius was set as 10 Å for the nonbonding (Lennard-Jones (LJ 6-12) and Coulomb) interactions. Water molecules around the CA IX were equilibrated with 500 ps MD simulations at fixed coordinates of the atoms of the CA IX. The production MD run was then carried out for 10 ns for all the studied system () without any constraints.
Figure 2. A snapshot picture taken from the simulation box: Protein (CA IX-dimer in yellow, inhibitor (inside the protein), water (as quick surface) and K+, Cl− ions (cyan and brown colored spheres).
Co-crystallized ligand docking
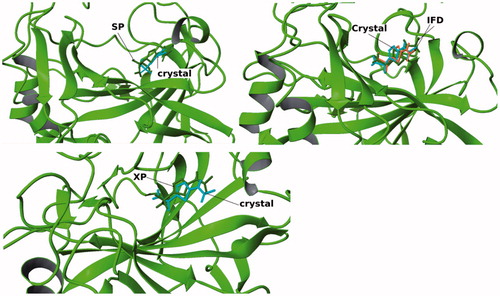
In an initial step, we tested the capability of Glide/induced fit docking (IFD)Citation29, Glide/SP and Glide/XPCitation30 protocols to predict the ligand position of reference ligand () in the CA IX X-ray structure. Co-crystallized inhibitor is used at the ligand preparation module LigPrepCitation31 and then it is docked into the active site of the protein using the Glide docking protocols with different approaches. The binding cavity was assigned as an outer box of (30 × 30 × 30) ÅCitation3 which is determined by the position of the reference ligand. We employed the SP, XP and IFD methods of the Glide docking. The conformation of co-crystallized X-ray structure is compared with docking poses. RMSD values for all used docking methods were found as less than 1.5 Å ().
Figure 3. Cognate docking poses alignment with the co-crystallized ligand conformation using different methods.
Virtual docking screening
It has become possible to perform HTVS on large libraries (i.e. ZINC database) with the enormous increase in computer facilities. Chemical structures of around 7 million ligands are obtained from the ZINC database in the present studyCitation32. The 3D structures were prepared using LigPrep module of Maestro. The physiological medium and ionization states were determined for all the ligands and the pH was set to 7.4. The docking screening process was carried out in two steps: (i) Glide/HTVS is performed for accelerated docking simulations of 7 million ligands, (ii) the ligands with top- docking scores from HTVS were selected for further detailed analyses by Glide/XP docking.
ADME analysis
The absorption, distribution, metabolism and excretion (ADME) characteristics of all selected compounds were predicted using QikPropCitation33 software, which calculates physicochemical properties of the compounds. This software may validate the drug-likeliness of the ligands based on Lipinski's criteriaCitation34.
In vitro analysis
Adherent epithelium renal cell adenocarcinoma cell line (ACHN, ATCC CRL-1611) was grown at Dulbecco's modified Eagle's medium (Sigma-Aldrich, Germany) containing 10% (v/v) FBS, 100 units/mL streptomycin and 100 µg/mL penicillin. Cells were maintained 80% confluency at 5% CO2 at 37 °C into T75 flasks and then transferred into four T125 flask and maintained 90% confluency. Cells were quickly washed three times with 50 mM KxPO4 buffer. Cells were scratched from T125 flasks and transferred into falcon tubes (4 °C) and centrifuged 1500 rpm, 4 °C, 5 min. The cell pellet was maintained at −80 °C for 4 h and then lyophilized. About 1 mL dry ACHN cell pellet was obtained. CA IX activity was assayed by following the change in absorbance at 348 nm of 4-nitrophenylacetate (NPA) to 4-nitrophenylate ion over a period of 3 min at 25 ºC using a spectrophotometer (Shimadzu UV-VIS) according to the method described by Verpoorte et al.Citation35 The enzymatic reaction, in a total volume of 3.0 mL, contained 1.4 mL 0.05 M tris-SO4 buffer (pH 7.4), 1.0 mL 3 mM NPA, 0.5 mL H2O and 0.1 mL enzyme solution. A reference measurement was obtained by preparing the same cuvette without enzyme solution. The Lineweaver–Burk curves were used to determine kinetic parameters and inhibition constantsCitation36.
Results and discussions
MD simulations
It is necessary to prepare the used X-ray structure and to relax the target before starting the drug screening in order to get successful results. For this aim, we performed 10 ns MD simulations to relax the CA IX complex. Figure S1 shows the RMSD of the Cα atoms of the target during the MD stimulations. It is clear that system reached to the acceptable stability after the 3.0 ns simulations. Average structure from generated trajectories is constructed and representative structure selected from the derived trajectories, which is the smallest RMSD with the average structure, for docking simulations.
Structure-based virtual screening
The ZINC ligand library consists of commercially available ligand structures for docking screening. It involves almost 20 million ligands that can be used for virtual screening. In order to determine novel CA-IX inhibitors from the ZINC database, Glide/HTVS docking simulations for 7 million drug-like ligands were performed. Out of these ligands, 70 compounds were selected with the high-binding scores at the target site (see supplementary Figure S2). Glide/XP protocol is then utilized to identify the final CA-IX inhibitor candidates. Their chemical structures and binding energies are shown in . There are two criteria for choosing the potent ligands. The screened ligands were ranked according to their docking scores and ligands forming critical hydrogen bonding interactions with key-residues such as Thr199 and Thr200 at active sites are also considered. All the reported selected compounds have docking scores of less than −8 kcal/mole (). In order to better understand the molecular mechanisms of inhibition profiles of the selected CA IX inhibitors, their docking poses at the binding site are analyzed (Figure S3). Figure S3 shows the ligand interaction diagram for each selected compound at the binding pocket of CA IX. All selected ligands were superimposed in the active pocket in order to show the population of the ligand poses (). As it is shown, functional groups of similar chemical properties were positioned in similar modes with analogous bindings with the CA IX residues and encourage for an acceptable protein–ligand interactions, that was indicated the same inhibitory treatment of these ligands (). The docked poses of selected ligands display reasonable hydrogen-bonding network including the hydroxyl group in most of the selected compounds and polar residue of the active cavity.
Figure 4. Superimposition of selected ligands from docking simulations in the active site.
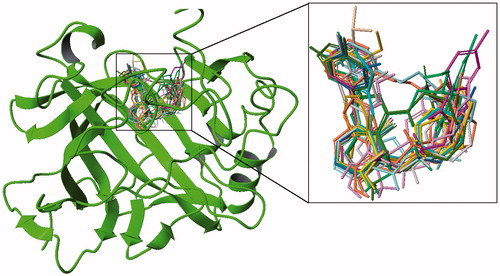
Table 1. 2D chemical structures and their corresponding docking scores for selected top-docking scored compounds.
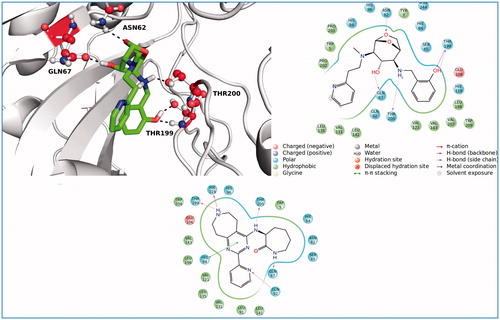
The top docking pose of compound from the ZINC database (ZINC20464003) was shown in . This ligand fits into the CA IX active cavity. The ligand–protein complex was stabilized by establishing four polar interactions via residues Asn62, Gln67, Thr199 and Thr200 at the target. The other main residues that participated in hydrophobic interactions were represented as 2D ligand interaction diagram using docking poses of ZINC20464003 and ZINC72421916 compounds in . Thr200, His94 and Thr199 were suggested as key amino acid residues in CA IX inhibitory mechanismCitation19. All these amino acids formed strong interactions with docked compounds in our study.
Figure 5. 3D and 2D ligand interaction diagrams for the ligands, ZINC20464003 (upper panel) and ZINC72421916 (lower panel).
Physicochemical predictions of docked compounds
ADME analysis was also performed for the selected compounds. Physicochemical and pharmaceutical properties of selected ligands which involve molecular weight, octanol/water partition coefficient, apparent Caco-2 cell permeability in nm/s, blockage of hERG K channel, percentage of human oral absorption and Lipinski's rule of five were listed in . As shown, all of the predicted values are in the acceptable ranges. Lipinski's rule of 5 is a rule of thumb to predict drug like compounds and, selected compounds were positioned in good range of Lipinski's rule.
Table 2. Selected compounds with their ADME properties.
In addition, a similarity search toward standard drugs was carried out. All the discovered ligands displayed more than 80% similarity towards known drugs are listed in Table S1. As shown in Table S1, beta-adrenergic antagonists (Propranolol, Timolol, Nebivolol, Tretoquinol, Carazolol, Bucumolol, Tertatolol, Penbutolol and Xamoterol) have more similarity with proposed selected compounds in this study. It is known that beta-adrenergic inhibitors block the carbonic anhydrase receptorsCitation37. These experimental reports can validate the results of present virtual screening study. In order to compare docking scores of known CA-IX inhibitors with selected compounds from our study, we docked 14 known CA-IX inhibitorsCitation38–44 at the active site of the target. Docking scores and chemical structures of these compounds are found in the range of −5.66 to −6.79 kcal/mole (Table S2).
In vitro analysis
In order to test and validate the predicted results of molecular simulations three compounds are ordered from available compound databanks (ChemBridge Corporation, San Diego, CA) and tested by in vitro analysis. Results showed that all compounds are inhibited CA IX in the range of 0.85–1.58 μM Ki values. Interestingly docking scores of these compounds are in correct order with their inhibition data (). 3D docking poses and 2D ligand interaction diagrams of these selected compounds are represented in and .
Figure 6. 3D docking poses of selected compounds at the active site of CA-IX.
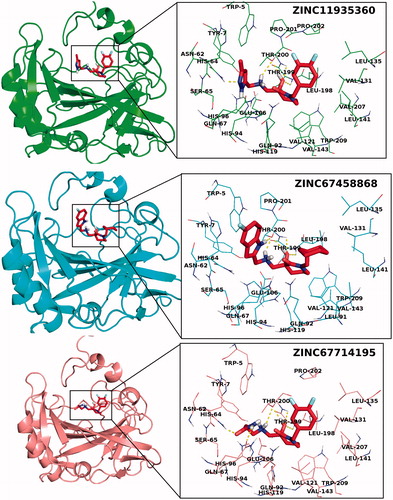
Figure 7. 2D ligand interaction diagrams of selected compounds into the active site of CA-IX.
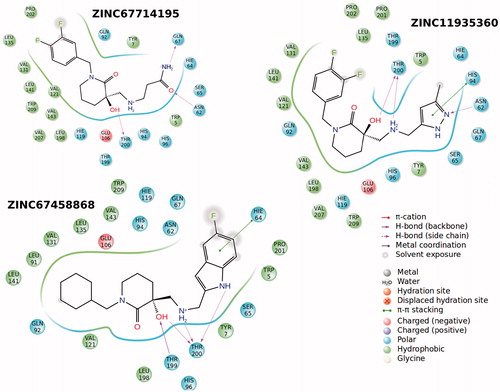
Table 3. hCA IX inhibition data of selected compounds.
Conclusions
We performed a low-cost and accelerated structure-based virtual screening methodCitation45–48 to discover novel inhibitor candidates of hCA IX. In summary, ∼7 million compounds from the ZINC database were virtually screened against CA IX to discover novel ligands with high affinity toward this target. 70 ligands showed a high-docking score using Glide/HTVS method. These compounds are then docked with Glide/XP method and 19 of them showed high-docking scores at the binding cavities of the CA IX target. All selected compounds displayed drug-like properties and their properties are similar to standard drugs. A few of these ligands are ordered from available provider of the ligands and tested by in vitro analysis in order to validate the molecular simulations results. In vitro analysis showed that ZINC67714195, ZINC11935360, and ZINC67458868 compounds inhibited hCA IX by Ki values of 0.85, 1.03, and 1.58 μM, respectively. These discovered novel compounds may be guided as candidate inhibitors of CA IX.
Supplementary material available online
Supplementary Figures S1–S3 and Tables S1–S2
Supplemental Material.pdf
Download PDF (2 MB)Acknowledgments
Part of computations for the work described in this paper was supported by Turkish Scientific and Technical Research Council (TUBITAK) ULAKBIM High Performance Computing Center. SD acknowledges support from Bilim Akademisi - The Science Academy, Turkey, under the BAGEP program
Declaration of interest
The authors report no conflicts of interest. The authors alone are responsible for the content and writing of this article.
References
- Thiry A, Dogné JM, Masereel B, Supuran CT. Targeting tumor-associated carbonic anhydrase IX in cancer therapy. Trends Pharmacol Sci 2006;27:566–73
- Pastorek J, Pastoreková S, Callebaut I, et al. Cloning and characterization of MN, a human tumor-associated protein with a domain homologous to carbonic anhydrase and a putative helix-loop-helix DNA binding segment. Oncogene 1994;9:2877–88
- Balaydin HT, Durdagi S, Ekinci D, et al. Inhibition of human carbonic anhydrase isozymes I, II and VI with a series of bisphenol, methoxy and bromophenol compounds. J Enzyme Inhib Med Chem 2012;27:467–75
- Ekinci D, Kurbanoglu NI, Salamci E, et al. Carbonic anhydrase inhibitors: inhibition of human and bovine isoenzymes by benzenesulphonamides, cyclitols and phenolic compounds. J Enzyme Inhib Med 2012;27:845–8
- Ekinci D, Al-Rashida M, Abbas G, et al. Chromone containing sulfonamides as potent carbonic anhydrase inhibitors. J Enzyme Inhib Med Chem 2012;27:744–7
- Cavdar H, Ekinci D, Talaz O, et al. alpha-Carbonic anhydrases are sulfatases with cyclic diol monosulfate esters. J Enzyme Inhib Med Chem 2012;27:148–54
- Senturk M, Ekinci D, Goksu S, Supuran CT. Effects of dopaminergic compounds on carbonic anhydrase isozymes I, II, and VI. J Enzyme Inhib Med Chem 2012;27:365–9
- Ekinci D, Karagoz L, Ekinci D, et al. Carbonic anhydrase inhibitors: in vitro inhibition of α isoforms (hCA I, hCA II, bCA III, hCA IV) by flavonoids. J Enzyme Inhib Med Chem 2013;28:283–8
- Ozdemir ZO, Senturk M, Ekinci D. Inhibition of mammalian carbonic anhydrase isoforms I, II and VI with thiamine and thiamine-like molecules. J Enzyme Inhib Med Chem 2013;28:316–19
- Abdel-Aziz AA-M, El-Azab AS, Ekinci D, et al. Investigation of arenesulfonyl-2-imidazolidinones as potent carbonic anhydrase inhibitors. J Enzyme Inhib Med Chem 2015;30:81–4
- Korkmaz N, Obaidi OA, Senturk M, et al. Synthesis and biological activity of novel thiourea derivatives as carbonic anhydrase inhibitors. J Enzyme Inhib Med Chem 2015;30:75–80
- Pastorekova S, Parkkila S, Zavada J. Tumor-associated carbonic anhydrases and their clinical significance. Adv Clin Chem 2006;42:167–216
- Wykoff CC, Beasley NJP, Watson PH, et al. Hypoxia-inducible expression of tumor-associated carbonic anhydrases. Cancer Res 2000;60:7075–83
- Potter C, Harris AL. Hypoxia inducible carbonic anhydrase IX, marker of tumor hypoxia, survival pathway and therapy target. Cell Cycle 2004;3:164–7
- Supuran CT, Scozzafava A, Casini A. Carbonic anhydrase inhibitors. Med Res Rev 2003;23:146–89
- Swietach P, Wigfield S, Cobden P, et al. Tumor-associated carbonic anhydrase 9 spatially coordinates intracellular pH in three-dimensional multicellular growths. J Biol Chem 2008;283:20473–83
- Supuran CT. Carbonic anhydrases: novel therapeutic applications for inhibitors and activators. Nat Rev Drug Discov 2008;7:168–81
- Supuran CT. Carbonic anhydrases as drug targets – an overview. Curr Top Med Chem 2007;7:825–33
- Alterio V, Hilvo M, Di Fiore A, et al. Crystal structure of the catalytic domain of the tumor-associated human carbonic anhydrase IX. Proc Natl Acad Sci USA 2009;106:16233–8
- Berman HM, Westbrook J, Feng Z, et al. The Protein Data Bank. Nucleic Acids Res 2000;28:235–42
- (a) Schrödinger Suite 2012 Protein Preparation Wizard; Epik version 2.3. New York (NY): Schrödinger, LLC; 2012. (b) Impact version 5.8. New York (NY): Schrödinger, LLC; 2012. (c) Prime version 3.1. New York (NY): Schrödinger, LLC; 2012
- Mårtensson L-G, Karlsson M, Carlsson U. Dramatic stabilization of the native state of human carbonic anhydrase II by an engineered disulfide bond. Biochemistry 2002;41:15867–75
- Li H, Robertson AD, Jensen JH. Very fast empirical prediction and rationalization of protein pK a values. Proteins Struct Funct Genet 2005;61:704–21
- Bas DC, Rogers DM, Jensen JH. Very fast prediction and rationalization of pKa values for protein-ligand complexes. Proteins Struct Funct Genet 2008;73:765–83
- Olsson MHM, SØndergaard CR, Rostkowski M, Jensen JH. PROPKA3: Consistent treatment of internal and surface residues in empirical p K a predictions. J Chem Theory Comput 2011;7:525–37
- Humphrey W, Dalke A, Schulten K. VMD: Visual molecular dynamics. J Mol Graph 1996;14:33–8
- Phillips JC, Braun R, Wang W, et al. Scalable molecular dynamics with NAMD. J Comput Chem 2005;26:1781–802
- Zhu X, Lopes PEM, Mackerell AD. Recent developments and applications of the CHARMM force fields. Wiley Interdisciplinary Rev 2012;2:167–85
- Friesner RA, Banks JL, Murphy RB, et al. Glide: a new approach for rapid, accurate docking and scoring. 1. Method and assessment of docking accuracy. J Med Chem 2004;47:1739–49
- Suite 2012: Glide, version 5.8. New York (NY): Schrödinger, LLC; 2012
- Suite 2012: LigPrep, version 2.5. New York (NY): Schrödinger, LLC; 2012
- Irwin JJ, Shoichet BK. ZINC – a free database of commercially available compounds for virtual screening. J Chem Inf Model 2005;45:177–82
- QikProp, version 3.5. New York (NY): Schrödinger, LLC; 2012
- Lipinski CA, Lombardo F, Dominy BW, Feeney PJ. Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Adv Drug Deliv Rev 2001;46:3–26
- Verpoorte JA, Mehta S, Edsall JT. Esterase activities of human carbonic anhydrases B and C. J Biol Chem 1967;242:4221–9
- Lineweaver H, Burk D. The determination of enzyme dissociation constants. J Am Chem Soc 1934;56:658–66
- Puşcas I, Reznicek A, Moldovan A, et al. Activation of carbonic anhydrase by beta-adrenergic agonists and inhibition by beta-adrenergic blockers. Med Interne 1985;23:185–9
- Turkmen H, Durgun M, Yilmaztekin S, et al. Carbonic anhydrase inhibitors. Novel sulfanilamide/acetazolamide derivatives obtained by the tail approach and their interaction with the cytosolic isozymes I and II, and the tumor-associated isozyme IX. Bioorganic Med Chem Lett 2005;15:367–72
- Maresca A, Temperini C, Pochet L, et al. Deciphering the mechanism of carbonic anhydrase inhibition with coumarins and thiocoumarins. J Med Chem 2010;53:335–44
- Innocenti A, Gülçin I, Scozzafava A, Supuran CT. Carbonic anhydrase inhibitors. Antioxidant polyphenols effectively inhibit mammalian isoforms I-XV. Bioorganic Med Chem Lett 2010;20:5050–3
- Winum JY, Pastorekova S, Jakubickova L, et al. Carbonic anhydrase inhibitors: synthesis and inhibition of cytosolic/tumor-associated carbonic anhydrase isozymes I, II, and IX with bis-sulfamates. Bioorganic Med Chem Lett 2005;15:579–84
- Winum JY, Innocenti A, Scozzafava A, et al. Carbonic anhydrase inhibitors. Inhibition of the human cytosolic isoforms I and II and transmembrane, tumor-associated isoforms IX and XII with boronic acids. Bioorganic Med Chem 2009;17:3649–52
- Durdagi S, Sentürk M, Ekinci D, et al. Kinetic and docking studies of phenol-based inhibitors of carbonic anhydrase isoforms I, II, IX and XII evidence a new binding mode within the enzyme active site. Bioorganic Med Chem 2011;19:1381–9
- Pacchiano F, Carta F, McDonald PC, et al. Ureido-substituted benzenesulfonamides potently inhibit carbonic anhydrase IX and show antimetastatic activity in a model of breast cancer metastasis. J Med Chem 2011;54:1896–902
- Mavromoustakos T, Durdagi S, Koukoulitsa C, et al. Strategies in the rational drug design. Curr Med Chem 2011;18:2517–30
- Politi A, Durdagi S, Moutevelis-Minakakis P, et al. Development of accurate binding affinity predictions of novel renin inhibitors through molecular docking studies. J Mol Graph Model 2010;29:425–35
- Durdagi S, Scozzafava GL, Vullo D, et al. Inhibition of mammalian carbonic anhydrases IXIV grayanotoxin III: solution and in silico studies. J Enzyme Inhib Med Chem 2013;29:469–75
- Ekinci D, Fidan I, Durdagi S, et al. Kinetic and in silico analysis of thiazolidin-based inhibitors of α-carbonic anhydrase isoenzymes. J Enzyme Inhib Med Chem 2013;28:370–4