
Abstract
The ubiquitin proteasome pathway is crucial in regulating many processes in the cell. Modulation of proteasome activities has emerged as a powerful strategy for potential therapies against much important pathologies. In particular, specific inhibitors may represent a useful tool for the treatment of tumors. Here, we report studies of a new series of peptide-based analogues bearing a naphthoquinone pharmacophoric unit at the C-terminal position. Some derivatives showed inhibition in the µM range of the post-acidic-like and chymotrypsin-like active sites of the proteasome.
Introduction
The ubiquitin proteasome system is the major pathway for intracellular protein degradation that plays a central role in regulating cellular function and maintaining homeostasisCitation1,Citation2. Polyubiquitinated proteins are degraded by proteasome 26S composed of a core 20S particle capped with two 19S structures. The 20S is assembled in a four stacked rings, two outer rings composed of seven α subunits and two inner rings shaped of seven different β subunitsCitation3–5. In eukaryotes, three major proteolytic activities are associated with different subunits: chymotrypsin-like (CT-L) in β5, trypsin-like (T-L) in β2 and caspase-like (C-L) in β1 subunits, respectivelyCitation6,Citation7. Proteolysis by the β subunits of the 20S core particle occur by γ-hydroxyl function of the N-terminal threonine residue as a nucleophileCitation8. The most of cellular proteins are degraded through this pathway involving a broad array of processes such as cell cycle, apoptosis, transcription, DNA repair, protein quality control and antigen presentation. Defects within this pathway are associated with much important pathologies, including cancerCitation9. As the essential role of the proteasome in cell function, inhibitors of this multicatalytic complex are potential drugs suitable in various therapeutic applications. Many natural and synthetic molecules have been studied as 20S catalytic subunits inhibitorsCitation10–19, among those the boronic pseudodipeptide bortezomib was the first FDA approved proteasome inhibitor for the treatment of multiple myeloma (MM) and recently carfilzomib became the second FDA-approved to treat MM in advanced stateCitation20,Citation21.
In previous studies, we have reported the design, synthesis and activity of certain classes of peptide molecules able to inhibit the proteasome, having different pharmacophoric units, consisting of electrophilic groups potentially able to interact with the catalytic threonineCitation22–25. In this work, we have dealt with the development of a new series of peptide-based derivatives bearing at the C-terminal a unit formed by a 2-chloronaphtoquinone pharmacophoric group. The naphtoquinone is present in molecules with biological activity, e.g. in powerful antidiabetic agents and inhibitors of protein-tyrosine phosphataseCitation26–28. Recently, through library screenings, compounds bearing 2-cloronaphtoquinonic unit named PI-083 (NSC-45382), able to express a good inhibition against chymotryptic activity of the proteasome have been identified (compound a in ). These compounds showed, in vitro and in vivo, the capacity to selectively inhibit tumor cell proliferationCitation29. Other non-peptide analogues with increased activity compared to the initial prototype, were also developed (general structure b in )Citation30,Citation31.
Figure 1. PI-083 (a), schematic structure of the naftoquinone non-peptidic compounds (b) and general structure of the naftoquinone dipeptide derivatives (c).
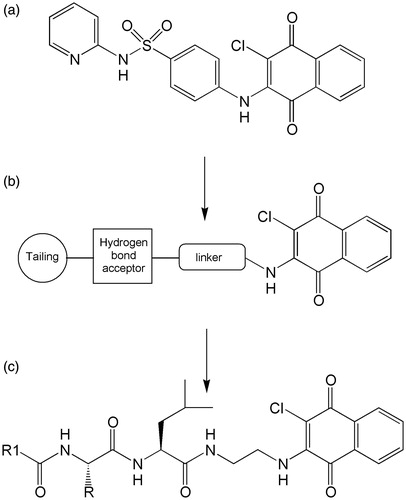
On the basis of these studies, we have designed pseudopeptide compounds having the general structure c shown in . The 2-chloronaphtoquinone (ClNafQ), which is the pharmacophoric unit able to function as a potential substrate for the threonine catalytic electrophile, is located at the C-terminal of the dipeptidic sequences. Cited studiesCitation29–31 suggest the proximity of the hydroxyl group of catalytic threonine to the 2-chloronaphtoquinone unit potentially able to attack carbon 2 of the pharmacophoric unit. Our docking studies suggest the same interaction for our compounds making reasonable the nucleophilic substitution of chlorine in position 2. The link between the peptide portion and the pharmacophore is obtained by a spacer consisting of ethylenediamine, chosen for its flexibility and to maintain the minimum distance between the pharmacophoric unit and the P1 position. While the N-terminal end is functionalized with 2-methyl,3-hydroxybenzoyl (Hmb) or p-nitrobenzoyl moiety (NBz). Hmb was chosen as N-terminal group since we have previously shown that favors the activity of peptide-based proteasome inhibitorsCitation23–25, while p-nitrobenzoyl was chosen because it is a suitable substituent in the non-peptidic naphtoquinone derivatives of the phenotypic structure PI-083Citation30. The central dipeptidic sequences have the constant presence of a leucine residue at C-terminal, while the N-terminal position provides residues with different characteristics such as serine, phenylalanine, leucine or asparagine.
Materials and methods
Chemistry – general
Amino acids, amino acid derivatives and chemicals were purchased from Bachem, Novabiochem and Fluka (Switzerland). Crude products were purified by preparative reversed-phase HPLC using a Water Delta Prep 4000 system with a Waters PrepLC 40 mm Assembly column C18 (30 cm × 4 cm, 300 Å, 15 μm spherical particle size column). The column was perfused at a flow rate of 30 ml/min, with a mobile phase-containing solvent A (10%, v/v, acetonitrile in 0.1% TFA), and a linear gradient from 0% to 100% of solvent B (60%, v/v, acetonitrile in 0.1% TFA); 30 min was the time adopted for elution of the compounds. HPLC analysis was performed using a Beckman System Gold with a Hypersil BDS C18 column (5 μm; 4.6 mm × 250 mm). Analytical determination and retention time (Tr) of the peptides were assayed via HPLC conditions in the above solvent system (solvents A and B), programmed at flow rates of 1 ml/min, using the following linear gradients: (a) from 0% to 90% B for 25 min and (b) from 30% to 100% B for 25 min. No pseudo-dipeptide showed >1% impurity when monitored at 220 and 254 nm. The molecular weights of the compounds were determined by electrospray ionization (ESI) (MICROMASS ZMD 2000), and the values are expressed as [M + H]+. HR-MS spectra were recorded by Agilent ESI-Q-TOF 6520 instrument. TLC was performed on pre-coated plates of silica gel F254 (Merck, Darmstadt, Germany), exploiting the following solvent systems: (c) AcOEt/n-hexane (1:1, v/v), (d) CH2Cl2/methanol (9.5:0.5, v/v), (e) CH2Cl2/methanol (9:1, v/v) and (f) CH2Cl2/methanol/toluene (17:2:1, v/v/v). Ninhydrin (1%) or chlorine iodine spray reagents were employed to detect the peptides. Melting points were determined by a Kofler apparatus and are uncorrected. Optical rotations were determined by a Perkin–Elmer 141 polarimeter with a 10-cm water-jacketed cell. 1H NMR spectroscopy was obtained using a Varian 400 MHz spectrometer.
Synthesis
To obtain all the compounds, a synthetic strategy, allowing the lowest number synthetic steps, was used (Scheme 1). Specifically, the ethylenediamine was mono-protected on an amine functionality by reaction with di-tert-butyl dicarbonate. Fmoc-leucine was condensed using 1-ethyl-3-(3-dimethylaminopropyl)-carbodiimide (WSC) and N-hydroxybenzotriazole (HOBt) as acylating agents, to give the intermediate functionalized with the appropriate linker. After fluorenylmethoxycarbonyl remotion with piperidine, the sequence is completed by condensation of the selected Fmoc-protected residue (Leu, Phe, Ser and Asn), always for activation of the carboxyl group by WSC/HOBt. After removal of the Fmoc, the N-functionalization is carried out by acylation with 2-methyl-3-hydroxybenzoic acid or p-nitrobenzoic acid using WSC/HOBt as a coupling agent. After the Boc deprotection with TFA, the synthesis proceed to the final condensation with 2,3-dichloro-1,4-naphthoquinone in EtOH (95%) at reflux, for an average duration of 3 days.
Scheme 1. Synthesis of naphthoquinone dipeptide molecules 1–8.
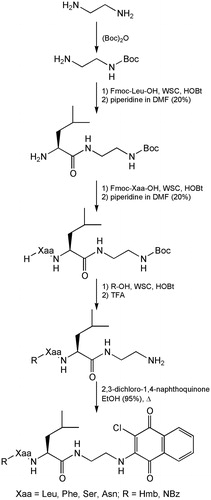
General synthetic procedures
Condensation with Fmoc-amino acids, methyl-2-3-hydroxybenzoic acid or p-nitrobenzoic acid
To 1 mmol of carboxylic component dissolved in 10 ml of DMF were added 1.1 mmol of HOBt, 1.1 mmol of WSC and 1.1 mmol of the amine component. The reaction mixture is put under stirring for 1 h at 0 °C and overnight at room temperature. The DMF is evaporated, the residue is taken with AcOEt and washed successively with 10% citric acid, 5% NaHCO3 and again with brine. The organic phase is dried with Na2SO4, filtered and evaporated.
Removal of the Fmoc
The Fmoc is removed with piperidine solution in DMF 20%. After 1 h of treatment, the solvent is evaporated and proceeds with precipitation by use of ethyl ether. The precipitate is then separated by centrifugation and collected.
Removal of the Boc
The Boc are removed with 90% aqueous TFA for 50 min (1 ml for mmol of Boc-protected compound). After evaporation, the residue is triturated with ethyl ether several times, and separated by centrifugation.
Condensation with 2,3-dichloro-1,4-naphthoquinone
About 0.3 mmol of amine are dissolved in ∼15 ml 95% EtOH and treated with NMM (N-methyl-morpholine), then were added 0.6 mmol of 2,3-dichloro-1,4-naphthoquinone and allowed to react at reflux for 3 days at 115 °C. The solvent is evaporated and precipitated with ethyl ether and then isolate the solid by centrifugation.
Preparation of Boc-ethylenediamine
At 10 mmol of ethylenediamine dissolved in 20 ml of t-ButOH/H2O (2:1) were added 7 mmol of (Boc)2O and allowed to react for 2 h under stirring at RT. Water (20 ml) was added and extracted for 2 times with 10 ml of n-pentane in a separatory funnel. The aqueous phase was focused and extracted with 50 ml of AcOEt. The organic phase is dried with anhydrous Na2SO4 and evaporated to yield the title compound as a colorless oil (0.96 g, 85%); 1H NMR (CDCl3) δ: 5.98 (bs, 2H), 3.08 (m, 2H), 2.69 (m, 2H), 1.75 (bs, 2H), 1.39 (s, 9H),; MS (M + H+) 161.2; HPLC (Tr) 6.54 min. Spectroscopic data are consisted with to previously reportedCitation32.
H-Leu-NH-(CH2)2-NH-Boc
The common intermediate Fmoc-Leu-NH-(CH2)2-NH-Boc was prepared for acylation of the Boc-ethylenediamine by Fmoc-Leu-OH following general procedure: (a) to give the title compound. Fmoc-protected intermediate was then treated on the basis of method and (b) to give H-Leu-NH-(CH2)2-NH-Boc as a white solid (yield 77%). MP = 78–81 °C; −34.8 (c = 1, MeOH); 1H NMR, (CDCl3) δ: 3.49–3.41 (m, 2H), 3.15 (m, 1H), 3.03 (m, 1H), 2.90 (m, 1H), 1.77 (m, 1H), 1.39 (s, 9H), 1.34 (m, 2H), 0.94 (d, 3H, J = 7.4 Hz), 0.89 (d, 3H, J = 7.4 Hz); MS (M + H+) 274.3; HPLC (Tr) 13.87 min.
H-Xaa-Leu-NH-(CH2)2-NH-Boc
The dipeptides Fmoc-Xaa-Leu-NH-(CH2)2-NH-Boc was prepared for acylation of the H-Leu-NH-(CH2)2-NH-Boc by Fmoc-Xaa-OH following general procedure: (a) to give the title compound. Fmoc-protected intermediate was then treated on the basis of method and (b) to give H-Xaa-Leu-NH-(CH2)2-NH-Boc.
H-Leu-Leu-NH-(CH2)2-NH-Boc (yield 65%). MP = 118–122 °C; −19.4 (c = 1, MeOH); 1H NMR (CDCl3) δ: 4.06, (m, 1H), 3.66, (m, 2H), 3.31–3.24 (m, 1H), 3.20 (m, 1H), 3.12–3.00 (m, 2H), 2.68 (m, 2H), 1.81–1.73 (m, 3H), 1.39 (s, 3H), 1.28 (m, 1H), 0.95–0.85 (m, 12H); MS (M + H+) 387.6; HPLC (Tr) 18.37 min.
H-Phe-Leu-NH-(CH2)2-NH-Boc (yield 71%). MP = 111–114 °C; −17.7 (c = 1, MeOH); 1H NMR (CDCl3) δ: 7.22–7.18 (m, 5H), 4.25 (m, 1H), 3.86–3.75 (m, 2H), 3.56 (m, 1H), 3.37 (m, 1H), 2.74–2.61 (m, 2H), 2.49 (m, 1H), 1.74 (m, 2H), 1.50 (m, 1H), 1.37, (s, 9H), 0.93 (d, 3H, J = 7.6 Hz), 0.87 (d, 3H, J = 7.6 Hz); MS (M + H+) 421.5; HPLC (Tr) 19.16 min.
H-Ser-Leu-NH-(CH2)2-NH-Boc (yield 75%).MP = 145–150 °C; −21.5 (c = 1, MeOH);1H NMR (CDCl3) δ: 4.75 (bs, 1H), 4.30 (m, 1H), 4.10–4.05 (m, 2H), 3.71–3.64 (m, 2H), 3.49 (m, 1H), 3.13–3.07 (m, 2H), 2.97 (m, 1H), 1.74 (m, 2H), 1.47 (m, 1H), 1.39 (s, 9H), 0.92 (d, 3H, J = 7.2 Hz), 0.86 (d, 3H, J = 7.2 Hz); MS (M + H+) 361.4; HPLC (Tr) 13.08 min.
H-Asn-Leu-NH-(CH2)2-NH-Boc (yield 63%). MP = 121–126 °C; −23.8 (c = 1, MeOH); 1H NMR (CDCl3) δ: 4.16 (m, 1H), 4.02 (m, 1H), 3.67–3.60 (m, 1H), 3.02–2.95 (m, 2H), 2.62–2.56 (m, 1H), 2.46–2.34 (m, 2H), 1.74 (m, 2H), 1.50 (m, 1H), 1.39 (s, 9H), 0.93 (d, 3H, J = 7.6 Hz), 0.87 (d, 3H, J = 7.6 Hz); MS (M + H+) 388.4; HPLC (Tr) 10.14 min.
R-Xaa-Leu-NH-(CH2)2-NH2
N-terminal functionalized dipeptides R-Xaa-Leu-NH-(CH2)2-NH-Boc was prepared for acylation of the H-Xaa-Leu-NH-(CH2)2-NH-Boc by Hmb-OH or NBz-OH following general procedure: (a) to give the title compound. Boc protection was removed by described procedure (b) to give R-Xaa-Leu-NH-(CH2)2-NH2.
Hmb-Leu-Leu-NH-(CH2)2-NH2 (yield 61%). MP = 149–152 °C; −25.7 (c = 1, MeOH); 1H NMR (CDCl3) δ: 7.13–7.08 (m, 2H), 6.92 (m, 1H), 4.24 (m, 1H), 4.12 (m, 1H), 3.26 (m, 1H), 2.83–2.74 (m, 2H), 2.39 (m, 1H), 2.25 (s, 3H),1.80–1.63 (m, 5H), 1.50 (m, 1H), 0.94–0.86 (m, 12H); MS (M + H+) 421.3; HPLC (Tr) 13.22 min.
Hmb-Phe-Leu-NH-(CH2)2-NH2 (yield 64%). MP = 119–121 °C; −19.1 (c = 1, MeOH); 1H NMR (CDCl3) δ: 7.19 (m, 5H), 7.06–6.98 (m, 2H), 6.88–6.84 (m, 1H), 4.88 (m, 1H), 4.21 (m, 1H), 3.24–3.14 (m, 2H), 3.02–2.96 (m, 1H), 2.79–2.57 (m, 3H), 2.21 (s, 3H), 1.74 (m, 2H), 1.45 (m, 1H), 0.93 (d, 3H), 0.84 (d, 3H). MS (M + H+) 439.4; HPLC (Tr) 17.41 min.
Hmb-Ser-Leu-NH-(CH2)2-NH2 (yield 71%). MP = 167–170 °C; −28.4 (c = 1, MeOH); 1H NMR (CDCl3) δ: 7.25–7.22 (m, 1H), 7.12 (m, 1H), 6.89 (m, 1H), 4.62 (m, 1H), 4.26 (m,1H), 3.66–3.60 (dd, 1H, J = 6.8 Hz, J = 3.2 Hz), 3.55–3.49 (dd, 1H, J = 6.8 Hz, J = 3.2 Hz), 3.35 (m, 1H), 2.64 (m, 3H), 2.27 (s, 3H), 1.87–1.72 (m, 3H), 0.94–0.85 (m, 6H); MS (M + H+) 395.3; HPLC (Tr) 8.83 min.
Hmb-Asn-Leu-NH-(CH2)2-NH2 (yield 54%). MP = 131–135 °C; −28.1 (c = 1, MeOH); 1H NMR (CDCl3) δ: 7.23–7.12 (m, 3H, Ar), 6.90 (m, 1H, Ar), 4.18–4.08 (m, 2H), 3.04 (m, 1H), 2.86 (m, 1H), 2.79–2.62 (m, 2H), 2.39–2.29 (m, 2H), 2.27 (s, 3H), 1.74 (m, 2H), 1.50 (sept, 1H, J = 7.4 Hz), 0.93 (d, 3H, J = 7.4 Hz), 0.84 (d, 3H, J = 7.4 Hz); MS (M + H+) 422.5; HPLC (Tr) 6.67 min.
NBz-Leu-Leu-NH-(CH2)2-NH2 (yield 71%). MP = 143–145 °C; −29.1 (c = 1, MeOH); 1H NMR (CDCl3) δ: 7.87 (m, 2H), 7.59 (m, 1H), 7.48 (m, 1H), 4.29 (m, 1H), 4.20 (m, 1H), 3.00 (m, 1H), 2.44–2.26 (m, 3H), 1.80–1.72 (m, 4H), 1.58–1.45 (m, 2H), 0.94–0,80 (m, 12H); MS (M + H+) 391.4; HPLC (Tr) 15.38 min.
NBz-Phe-Leu-NH-(CH2)2-NH2 (yield 73%). MP = 131–135 °C; −20.5 (c = 1, MeOH); 1H NMR (CDCl3) δ: 7.86 (m, 2H), 7.61 (m, 1H), 7.50 (m, 2H), 7.19 (s, 5H), 4.86 (m, 1H), 4.17 (m, 1H), 3.22 (m, 1H), 3.06 (m, 1H), 2.98–2.82 (m, 2H), 2.68 (m,1H), 2.41 (m, 1H), 1.74 (m, 2H), 1.54 (sept, 1H, J = 7.4 Hz), 0.95 (d, 3H, J = 7.4 Hz), 0.88 (d, 3H, J = 7.4 Hz); MS (M + H+) 440.3; HPLC (Tr) 16.24 min.
NBz-Ser-Leu-NH-(CH2)2-NH2 (yield 75%). MP = 154–158 °C; −27.1 (c = 1, MeOH); 1H NMR, (CDCl3) δ: 7.89 (m, 2H), 7.65 (m, 1H), 7.54 (m, 2H), 4.59 (m, 1H), 4.25 (m, 1H), 3.53–3.43 (m, 2H), 2.98 (m, 1H), 2.66 (m, 2H), 2.35 (m, 1H), 1.75 (m, 2H), 1.62 (sept, 1H, J = 7.4 Hz), 0.94 (d, 3H, J = 7.4 Hz), 0.87 (d, 3H, J = 7.4 Hz); MS (M + H+) 365.3; HPLC (Tr) 12.94 min.
NBz-Asn-Leu-NH-(CH2)2-NH2 (yield 62%). MP = 119–124 °C; −23.6 (c = 1, MeOH); 1H NMR (CDCl3) δ: 7.83 (m, 2H), 7.59 (m, 1H), 7.48 (m, 2H), 4.23–4.14 (m, 2H), 3.08–2.89 (m, 2H), 2.85–2.64 (m, 2H), 2.38–2.29 (m, 2H), 1.72 (m, 2H), 1.53 (m, 1H), 0.89 (m, 6H); MS (M + H+) 392.2; HPLC (Tr) 9.38 min.
R-Xaa-Leu-NH-(CH2)2-NH-ClNafQ
The compounds 1–8 were obtained for reaction by R-Xaa-Leu-NH-(CH2)2-NH2 with 2,3-dichloro-1,4-naphthoquinone following general procedure (d). All the products, after purification by RP-HPLC, were analyzed by mass spectrometry (HRMS) and NMR.
Hmb-Leu-Leu-NH-(CH2)2-NH-ClNafQ (1) (yield 32%). MP = 97–102 °C; −23.6 (c = 1, MeOH); 1H NMR (CDCl3) δ: 7.24–7.18 (m, 2H), 7.12–7.07 (m, 2H), 6.91 (m, 1H), 6.67 (s, 2H), 4.28 (m, 1H), 4.13 (m, 1H), 3.55 (m, 1H), 3.20–3.07 (m, 2H), 2.51 (m, 1H), 2.10 (s, 3H), 1.80–1.72 (m, 2H), 1.38 (m, 2H), 0.93 (m, 6H), 0.87 (m, 6H); HRMS (M + H+) Calc: 561.2474, found: 561.2485; HPLC (Tr) 18.63 min.
Hmb-Phe-Leu-NH-(CH2)2-NH-ClNafQ (2) (yield 44%). MP = 116–121 °C; −16.2 (c = 1, MeOH); 1H NMR (CDCl3) δ: 7.26–7.15 (m, 8H), 6.94 (m, 1H), 6.85 (m, 2H), 6.65 (d, 2H, J = 1.2 Hz), 5.00 (m, 1H), 4.16 (m, 1H), 3.34–3.15 (m, 2H), 2.98–2.76 (m, 4H), 2.26 (s, 3H), 1.74 (m, 1H), 1.54 (m, 2H), 0.94 (d, 3H, J = 7.4 Hz), 0.89 (d, 3H, J = 7.4 Hz); HRMS (M + H+) Calc: 595.2317, found: 595.2302; HPLC (Tr) 19.08 min.
Hmb-Ser-Leu-NH-(CH2)2-NH-ClNafQ (3) (yield 41%). MP = 129–132 °C; −18.5 (c = 1, MeOH); 1H NMR (CDCl3) δ: 0.82 (d 6H); 1:49 (m 1H); 1.62 (1H d); 1.64–1.79 (m 1H); 2:28 (s 3H); 3:31 (m 1H); 3.21–3.88 (m 1H); 4:22 (m 1H); 4:55 (m1H); 4.68 (s 1H); 4.81 (m 1H); 5.61 (bs 1H); 5.78 (bs 1H); 6:21 (bs 1H); 7:01 (dd 1H); 7:30 (dd 1H); 7:41 (m 1H); 7.62 (m 2H); 7.71 (m 2H); 7.97 (bs 1H); HRMS (M + H+) Calc: 535.1954, found: 535.1941; HPLC (Tr) 9.09 min.
Hmb-Asn-Leu-NH-(CH2)2-NH-ClNafQ (4) (yield 34%). MP = 122–126 °C; −20.5 (c = 1, MeOH); 1H NMR (CDCl3): δ 0.82 (d 6H); 1:49 (m 1H); 1.25–1.98 (m 1H); 2:28 (s 3H); 2.80–3.26 (m 1H); 3:47 (m 1H); 3:31 (m 1H); 4.72 (m 1H); 4.80 (m 1H); 5:28 (bs 1H); 5.61 (bs 1H); 6:27 (bs 1H); 6.99 (dd 1H); 7:30 (dd 1H); 7:40 (m 1H); 7:59 (m 2H); 7.68 (m 2H); 7.94 (bs 1H); HRMS (M + H+) Calc: 562.2063, found: 562.2074; HPLC (Tr) 6.89 min.
NBz-Leu-Leu-NH-(CH2)2-NH-ClNafQ (5) (yield 33%). MP = 92–95 °C; −24.2 (c = 1, MeOH); 1H NMR (CDCl3) δ: 0.85 (d 6H); 1:47 (m 1H); 1.62(m 2H); 2:09 (m 2H); 3.48 (m 2H); 3.86 (m 2H); 4:32(m1H); 4.50 (m 1H); 5:21 (bs 1H); 5.66 (bs 1H); 6:32 (bs 1H); 7:17 (bs 1H); 7.62 (m 2H); 7.71 (m 2H); 8.62 (d 2H); 8.87 (d 2H); HRMS (M + H+) Calc: 576.2219, found: 576.2234;; HPLC (Tr) 19.77 min.
NBz-Phe-Leu-NH-(CH2)2-NH-ClNafQ (6) (yield 40%). MP = 93–97 °C; −22.3 (c = 1, MeOH); 1H NMR (CDCl3) δ: 0.83 (d 6H); 1:49 (m 1H); 1.97 (m 2H); 3.11–3.71 (m 6H); 4:55 (m 1H); 5:56 (m 1H); 6:41 (bs 1H); 6.88 (bs 1H); 7:22 (m 5H); 7:28 (bs 1H);7.66 (m 2H);7.75 (m 2H), 8:13 (d 2H), 8:28 (d 2H); HRMS (M + H+) Calc: 610.2063, found: 610.2069; HPLC (Tr) 23.44 min.
NBz-Ser-Leu-NH-(CH2)2-NH-ClNafQ (7) (yield 37%). MP = 124–128 °C; −18.5 (c = 1, MeOH); 1H NMR, (CDCl3) δ: 8.30–8.10 (m, 5H), 6.70 (d, 2H, J = 1.2 Hz), 4.61 (m, 1H), 4.37 (m, 1H), 3.94 (m, 1H), 3.40 (m, 1H), 3.26–2.92 (m, 3H), 2.46 (m, 1H), 1.75–1.62 (m, 2H), 0.90 (m, 6H); HRMS (M + H+) Calc: 550.1699, found: 550.1705; HPLC (Tr) 19.67 min.
NBz-Asn-Leu-NH-(CH2)2-NH-ClNafQ (8) (yield 29%). MP = 106–111 °C; −24.2 (c = 1, MeOH); 1H NMR (CDCl3) δ: 8.30 (d, 2H, J = 7.2 Hz), 8.11 (d, 2H, J = 7.2 Hz), 6. 88 (bs, 2H), 6.61 (s, 2H), 4.30 (m, 1H), 4.12 (m, 1H), 3.15 (m, 1H), 3.02–2.82 (m, 2H), 2.68 (m, 1H), 2.53–2.42 (m, 2H), 1.74 (m, 1H), 1.50 (sept, 1H, J = 7.2 Hz), 0.93 (d, 3H, J = 7.6 Hz), 0.87 (d, 3H, J = 7.6 Hz); HRMS (M + H+) Calc: 577.1808, found: 577.1815; HPLC (Tr) 17.02 min.
Biological assays
Proteasome purification proteasome subunit inhibition
Proteasomes were isolated from lymphoblastoid cell lines (LCL) as previously describedCitation33.
Suc-LLVY-AMC, Boc-LRR-AMC and Z-LLE-AMC (Sigma) were used to measure CT-L, T-L and post-acidic proteasome activities, respectively. Substrates were incubated at 37 °C for 30 min with proteasomes, untreated or pre-treated with 0.1–100 μM of test compounds, in activity buffer. Fluorescence was determined by a fluorimeter (Spectrafluor plus, Tecan, Salzburg, Austria), using an excitation of 360 nm and emission of 465 nm. Activity was evaluated in fluorescence units and the inhibitory activity of the compounds is expressed as IC50.
Growth inhibition assays
Cell growth inhibition assays were carried out using the breast cancer cell line MDA and ovarian cancer cell line A2780. Cell lines were obtained from ATCC and maintained in DMEM and RPMI, respectively, supplemented with 10% fetal bovine serum (FBS), penicillin (100 U ml−1), streptomycin (100 U ml−1) and glutamine (2 mM); incubation was performed at 37 °C in a 5% CO2 atmosphere. Cells were routinely passaged every 3 days at 70% confluence; 0.05% trypsin-EDTA was used. The antiproliferative activity of compounds was tested with the MTT assay. The cells were seeded in triplicate in 96-well trays at a density of 25 × 103 in 50 μl of complete medium. Stock solutions (10 mM) of each compound were made in DMSO and diluted in complete medium to give final concentrations of 10 and 100 μM. MG-132 was employed as a control. Untreated cells were placed in every plate as a negative control. The cells were exposed to the compounds, in 100 μl total volume, for 72 h, and then 25 μl of a 12-mM solution of 3-(4,5-dimethylthiozol-2-yl)-2,5-diphenyltetrazolium bromide solution (MTT) was added. After 2 h of incubation, 100 μl of lysing buffer (50% DMF + 20% SDS, pH 4.7) was added to convert the MTT solution into a violet-colored formazan. After an additional 18 h, the solution absorbance, proportional to the number of live cells, was measured by spectrophotometer at 570 nm and converted into % of growth inhibitionCitation34.
Docking
The equilibrium geometry of molecule 5 was obtained using semi-empirical PM3 calculations and subsequently used in a docking simulation to both β1 and β5 binding sites of 20S proteasome. The simulation was performed utilizing the crystal structures of PDB codes 1G65Citation35 and 4LQICitation36 for β1 and β5 binding sites, respectively. Before the simulation, hydrogen atoms were added to the inner part of the enzyme and the energy of the structure was minimized keeping fixed the atoms of the mainframe and using the MMFF94 molecular mechanics forcefieldCitation37. Molecule 5 was placed in β1 and β5 binding sites using a pharmacophore query derived from the bound inhibitors epoxomicin (structure 1G65) and omuralide (structure 4LQI), respectively, as a filter for docking placement. A pharmacophore is a set of structural features in a ligand that are directly related to the ligand’s recognition at a receptor site and its biological activity. Out of 50 unique poses obtained, the 10 having the highest score on the base of the value assumed by the enthalpic contribution to the free energy of binding were retained. These poses were in turn rescored on the base of the estimation of the free energy of binding of the ligand, i.e. the sum of the electrostatic and dispersive interaction energy between the ligand and the target as well as the intramolecular energy of the ligand due to changes of its conformation. All the calculations were performed using MOE-Dock integrated in the MOE system of programs [MOE, Chemical Computing Group, release 2013.0802].
Results and discussion
Naphtoquinone pseudodipeptides were synthesized following the strategy reported in Scheme 1 and tested as proteasome inhibitors.
Inhibition of β1, β2 and β5 active sites of the 20S proteasome, purified from lymphoblastoid cell linesCitation25, was determined using fluorogenic substrates specific for the three main proteolytic activities of the enzymatic complex. Suc-LLVY-AMC, Boc-LRR-AMC and Z-LLE-AMC were used to measure CT-L, T-L and C-L proteasome activities, respectively. Substrates were incubated, at 37 °C for 30–180 min, with the proteasome, pre-treated with incremented concentrations (from 0.1 to 100 µM) of the new naphthoquinone derivatives and compared with the well-known proteasome inhibitor MG132 in activity buffer. Substrate degradation was evaluated in fluorescence units, and the inhibitory activity of all the compounds is expressed here as IC50. From the data reported in , after 30 min of incubation it can observed that some of the naphtoquinone derivatives present an interesting biological profile. The inhibition of the post-acidic (PGPH) and chymotryptic (ChT-L) activities is significant for some of the derivatives. In particular, the activity against the β1 subunit is remarkable for the analogs 1, 5 and 8 with IC50 values around µM unit. In addition, compounds 5 and 8, but not compound 1, were able to some extent to inhibit the chymotryptic activity. None of the compounds were able to inhibit the tryptic activity. The most efficient compounds of the series are 5 and 8, bearing the central Leu-Leu and Asn-Leu dipeptide sequence, respectively. The different N-terminal functionalizations appear not determinant for biological response.
Table 1. Inhibition of the proteasome subunits by naphthoquinone dipeptides 1–8 and reference compound MG132.
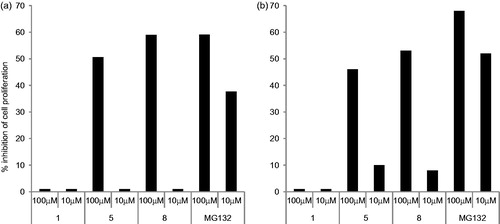
Since proteasomes play a key role in cell viability and proliferation, we evaluated the anti-proliferative activities of three selected compounds (1, 5 and 8) on MDA and A2780 tumor cells. To this end, cells were treated with 10 or 100 µM concentrations of compounds 1, 5 or 8. After 3 days, cell proliferation was evaluated and as shown in , only compounds 5 and 8 at 100 µM demonstrated the capacity to inhibit cell proliferation at levels similar to the proteasome inhibitor MG132. No anti-proliferative activity was observed at 10 µM. Of note, compound 1 that inhibits only the post-acidic activity of the proteasome did not show any anti-proliferative activity.
Figure 2. Effect of compounds 1, 5 and 8 on cell proliferation. MDA (panel a) and A2780 (panel b) tumor cells were cultured for 3 days in the presence or absence of the indicated concentrations of compounds 1, 5 and 8. Results are expressed as % inhibition proliferation calculated in respect to untreated cells. The means of three independent experiments performed in duplicate are shown.
Structure–activity relationship provided suggests that the C-terminal pharmacophoric is a favorable electrophilic substrate able to undergo nucleophilic attack by the enzymatic threonine. The secondary interactions that stabilize the enzyme-inhibitor binding are most effective when the dipeptide sequence has features more hydrophobic. As shown by docking studies, the enzyme-inhibitors interaction should be not covalent, this data suggest a reversible inhibition, confirmed by the decreasing of IC50 values during the time of incubation of the naphtoquinone derivatives with the isolated proteasome.
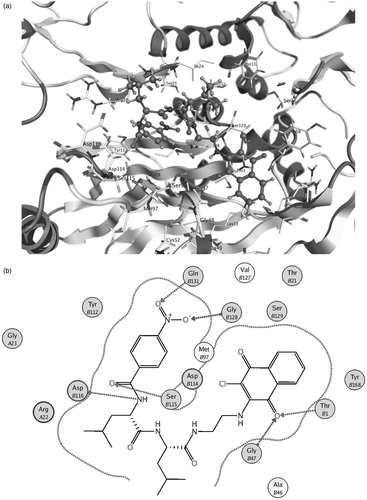
The results of the docking study of compound 5 to both β1 and β5 binding sites of 20S proteasome seem to support the previous considerations; actually, in both sites, the naphtoquinonic unit is positioned near the active Thr1 residue. In the β1 pocket, the O2 quinonic oxygen is located near Thr1 and Gly47 residues at a quite short distance of some 2.6–2.7 Å, compatible with the formation of hydrogen bonding interactions (). Conversely, in the β5 site the O1 oxygen of the naphtoquinonic unit is found at a higher distance with respect to the active Thr1 residue (some 3.1 Å) but a number of additional interactions can be observed between the functional groups of the N-terminal fragment and Ser115, Gln131, Gly128 and Asp116 residues () at ligand–protein distances in the range 3.0–3.5 Å which contribute to the binding stability.
Figure 3. Molecule 5 in the β1 binding pocket (a) and a schematic diagram of the inhibitor–protein interactions (b).
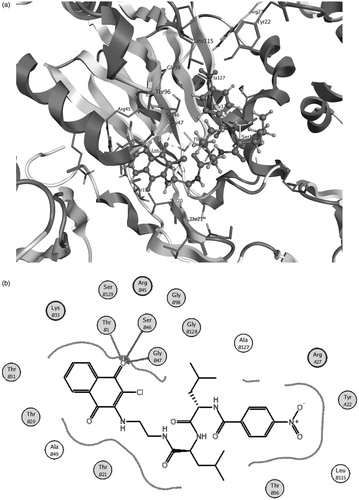
Figure 4. Molecule 5 in the β5 binding pocket (a) and a schematic diagram of the inhibitor–protein interactions (b).
Conclusions
This work reports the design, synthesis, biological activities and molecular modeling analysis of pseudo-dipeptide molecules assayed as inhibitors of three major catalytic activities of the proteasome. These compounds have at the C-terminal position a 2-chloronaftoquinone group pharmacophoric unit primary responsible for the interaction with the active subunits of the enzymatic complex. The same scaffold is present in non-peptidic molecules previously reported as inhibitors of the 20 S proteasome. Data on the biological response of some analogs of this new series showed an interesting inhibition of the proteasome. Derivatives 5 and 8 were the most active against the β1 and β5 subunits, with a biological profile that makes them potentially capable of operating such as antineoplastic agents. The molecular structure of the new inhibitors allows further studies on the structure-activity relationship, maintaining the C-terminus naphtoquinonic unit as electrophilic substrate.
Declaration of interest
The authors report no conflicts of interest. The authors alone are responsible for the content and writing of this article.
References
- Ciechanover A. The ubiquitin-proteasome proteolytic pathway. Cell 1994;79:13–21
- Reed SI. The ubiquitin-proteasome pathway in cell cycle control. Results Probl Cell Differ 2006;42:147–81
- Löwe J, Stock D, Jap B, et al. Crystal structure of the 20S proteasome from the archaeon T. acidophilum at 3.4 A resolution. Science 1995;268:533–9
- Coux O, Tanaka K, Goldberg AL. Structure and functions of the 20S and 26S proteasomes. Annu Rev Biochem 1996;65:801–47
- Groll M, Ditzel L, Löwe J, et al. Structure of 20S proteasome from yeast at 2.4 A resolution. Nature 1997;386:463–71
- Groll M, Heinemeyer W, Jäger S, et al. The catalytic sites of 20S proteasomes and their role in subunit maturation: a mutational and crystallographic study. Proc Natl Acad Sci USA 1999;96:10976–83
- Heinemeyer W, Fischer M, Krimmer T, et al. The active sites of the eukaryotic 20S proteasome and their involvement in subunit precursor processing. J Biol Chem 1997;272:25200–9
- Nussbaum AK, Dick TP, Keilholz W, et al. Cleavage motifs of the yeast 20S proteasome beta subunits deduced from digests of enolase 1. Proc Natl Acad Sci USA 1998;95:12504–9
- Borissenko L, Groll M. 20S proteasome and its inhibitors: crystallographic knowledge for drug development. Chem Rev 2007;107:687–717
- Kisselev AF, Goldberg AL. Proteasome inhibitors: from research tools to drug candidates. Chem Biol 2001;8:739–58
- Nazif T, Bogyo M. Global analysis of proteasomal substrate specificity using positional-scanning libraries of covalent inhibitors. Proc Natl Acad Sci USA 2001;98:2967–72
- Kisselev AF, van der Linden WA, Overkleeft HS. Proteasome inhibitors: an expanding army attacking a unique target. Chem Biol 2012;19:99–115
- Groll M, Huber R, Moroder L. The persisting challenge of selective and specific proteasome inhibition. J Pept Sci 2009;15:58–66
- Genin E, Reboud-Ravaux M, Vidal J. Proteasome inhibitors: recent advances and new perspectives in medicinal chemistry. Curr Top Med Chem 2010;10:232–56
- Micale N, Scarbaci K, Troiano V, et al. Peptide-based proteasome inhibitors in anticancer drug design. Med Res Rev 2014;34:1001–69
- Dorsey BD, Iqbal M, Chatterjee S, et al. Discovery of a potent, selective, and orally active proteasome inhibitor for the treatment of cancer. J Med Chem 2008;51:1068–72
- Mroczkiewicz M, Winkler K, Nowis D, et al. Studies of the synthesis of all stereoisomers of MG-132 proteasome inhibitors in the tumor targeting approach. J Med Chem 2010;53:1509–18
- Bazzaro M, Anchoori RK, Mudiam MK, et al. α,β-Unsaturated carbonyl system of chalcone-based derivatives is responsible for broad inhibition of proteasomal activity and preferential killing of human papilloma virus (HPV) positive cervical cancer cells. J Med Chem 2011;54:449–56
- Desvergne A, Genin E, Maréchal X, et al. Dimerized linear mimics of a natural cyclopeptide (TMC-95A) are potent noncovalent inhibitors of the eukaryotic 20S proteasome. J Med Chem 2013;56:3367–78
- Adams J. The development of proteasome inhibitors as anticancer drugs. Cancer Cell 2004;5:417–21
- Demo SD, Kirk CJ, Aujay MA, et al. Antitumor activity of PR-171, a novel irreversible inhibitor of the proteasome. Cancer Res 2007;67:6383–91
- Marastoni M, Baldisserotto A, Canella A, et al. Arecoline tripeptide inhibitors of proteasome. J Med Chem 2004;47:1587–90
- Marastoni M, Baldisserotto A, Trapella C, et al. P3 and P4 position analysis of vinyl ester pseudopeptide proteasome inhibitors. Bioorg Med Chem Lett 2006;16:3125–30
- Baldisserotto A, Ferretti V, Destro F, et al. Alpha,beta-unsaturated N-acylpyrrole peptidyl derivatives: new proteasome inhibitors. J Med Chem 2010;53:6511–15
- Franceschini C, Trapella C, Calia R, et al. C-terminal trans, trans-muconic acid ethyl ester partial retro-inverso pseudopeptides as proteasome inhibitors. J Enzyme Inhib Med Chem 2013;28:1034–9
- Ahn JH, Cho SY, Ha JD, et al. Synthesis and PTP1B inhibition of 1,2-naphthoquinone derivatives as potent anti-diabetic agents. Bioorg Med Chem Lett 2002;12:1941–6
- Urbanek RA, Suchard SJ, Steelman GB, et al. Potent reversible inhibitors of the protein tyrosine phosphatase CD45. J Med Chem 2001;44:1777–93
- Brun MP, Braud E, Angotti D, et al. Design, synthesis, and biological evaluation of novel naphthoquinone derivatives with CDC25 phosphatase inhibitory activity. Bioorg. Med Chem 2005;13:4871–9
- Lawrence HR, Kazi A, Luo Y, et al. Synthesis and biological evaluation of naphthoquinone analogs as a novel class of proteasome inhibitors. Bioorg Med Chem 2010;18:5576–92
- Xu K, Xiao Z, Tang YB, et al. Design and synthesis of naphthoquinone derivatives as antiproliferative agents and 20S proteasome inhibitors. Bioorg Med Chem Lett 201;22:2772–4
- Ge Y, Kazi A, Marsilio F, et al. Discovery and synthesis of hydronaphthoquinones as novel proteasome inhibitors. J Med Chem 2012;55:1978–98
- Aubineau T, Cossy J. Chemoselective alkynylation of N-sulfonylamides versus amides and carbamates – synthesis of tetrahydropyrazine. Chem Commun 2013;49:3303–5
- Gavioli R, Vertuani S, Masucci MG. Proteasome inhibitors reconstitute the presentation of cytotoxic T-cell epitopes in Epstein-Barr virus-associated tumors. Int J Cancer 2002;101:532–8
- Hansen MB, Nielsen SE, Berg K. Re-examination and further development of a precise and rapid dye method for measuring cell growth/cell kill. J Immunol Methods 1989;119:203–10
- Groll M, Kim KB, Kairies N, et al. Crystal structure of epoxomicin: 20S proteasome reveals a molecular basis for selectivity of α′, β′-epoxyketone proteasome inhibitor. J Am Chem Soc 2000;122:1237–8
- List A, Zeiler E, Gallastegui N, et al. Omuralide and vibralactone: differences in the proteasome-β-lactone-γ-lactam binding scaffold alter target preferences. Angew Chem Int Ed Engl 2014;53:571–4
- Halgren TA. Merck molecular force field. I. Basis, form, scope, parameterization, and performance of MMFF94. J Comput Chem 1996;17:490–519