
Abstract
A study concerning design, synthesis, structure and in vitro antimycobacterial and anticancer evaluation of new fused derivatives with pyrrolo[2,1-c][4,7]phenanthroline skeleton is described. The strategy adopted for synthesis involves a [3 + 2] dipolar cycloaddition of several in situ generated 4,7-phenanthrolin-4-ium ylides to different substituted alkynes and alkenes. Stereo- and regiochemistry of cycloaddition reactions were discussed. The structure of the new compounds was proven unambiguously, single-crystal X-ray diffraction studies including. The antimycobacterial and anticancer activity of a selection of new synthesized compounds was evaluated against Mycobacterium tuberculosis H37Rv under aerobic conditions and 60 human tumour cell line panel, respectively. Five of the tested compounds possess a moderate antimycobacterial activity, while two of the compounds have a significant antitumor activity against renal cancer and breast cancer.
Introduction
Phenanthroline derivatives attracted attention in the last years especially due to their biological effectsCitation1–3, materials science applicationsCitation4, crystal engineeringCitation5,Citation6, their unique π-electrons delocalizationCitation7,Citation8 and complexation propertiesCitation9.
Phenanthrolines polycyclic skeletons are also present in sterols, sex hormones, cardiac glycosides, bile acids and morphine alkaloidsCitation10. Recently, compounds with phenathroline skeleton have been synthesized and biologically evaluatedCitation11–15 as analogues of benzo[c]phenanthridine alkaloids (e.g. nitidine, fagaronine, sanguinarine) that attracted attention due to their interesting biological propertiesCitation16–19.
Compared to 1,10-phenanthroline that has been widely studied both for synthesis and applications, 4,7-phenanthroline received much less interest due to difficulties in its synthesis. However, there are several reports regarding biological properties of 4,7-phenanthroline derivatives and their analogues as antibacterial activityCitation20, amoebicideCitation21 or antiviral propertiesCitation22. Besides, 4,7-phenathroline derivatives were found to exhibit staining properties to HeLa cells and can be used as fluorophores which can bind with proteinsCitation23. Other derivatives were reported as high affinity triple-helix DNA stabilizing agentsCitation24.
As part of our ongoing research in the field of design and synthesis of new anti-TBCitation25–28 and anticancer derivativesCitation29–32 with azaheterocycles skeleton, we report here the design, synthesis, structure and in vitro antimycobacterial and anticancer evaluation of new class of compounds with pyrrolo[2,1-c][4,7]phenanthroline scaffold. To the best of our knowledge, there is no previous report, concerning the synthesis of pyrrolo[2,1-c][4,7]-phenanthroline derivatives.
Methods
General
Melting points were recorded on an A. Krüss Optronic Melting Point Meter KSPI and are uncorrected. Proton and carbon nuclear magnetic resonance (δH, δC) spectra were recorded on a DRX-500 Bruker (500 MHz). All chemical shifts are quoted on the δ-scale in ppm. Coupling constants are given in Hz. IR spectra were recorded on a FTIR Shimadzu or Jasco 660 plus FTIR spectrophotometer. Thin layer chromatography (TLC) was carried out on Merck silica gel 60F254 plates. Column chromatography was carried out on silica gel (Roth 60, 0.04–0.063 mm). Visualization of the plates was achieved using a UV lamp (λmax = 254 or 365 nm). All commercially available products were used without further purification unless otherwise specified.
X-ray crystallography
Crystallographic measurements were carried out with an Oxford-Diffraction XCALIBUR E CCD diffractometer equipped with graphite-monochromated Mo Kα radiation. Single crystals were positioned at 40 mm from the detector and 211 and 248 frames were measured each for 60 and 180 s over 1 ° scan width for 2259 and 2340, respectively. The unit cell determination and data integration were carried out using the CrysAlis package of Oxford DiffractionCitation33. The structures were solved by direct methods using Olex2Citation34 software with the SHELXS structure solution program and refined by full-matrix least-squares on F2 with SHELXL-97Citation35. Atomic displacements for non-hydrogen atoms were refined using an anisotropic model. Hydrogen atoms were placed in fixed, idealized positions and refined as rigidly bonded to the corresponding atoms. The molecular plots were obtained using the Olex2 program. The main crystallographic data together with refinement details are summarized in Supplemental Table S1. CCDC-1048147 (for 7a) and 1048148 (for 11f) contain the supplementary crystallographic data for this contribution. These data can be obtained free of charge via www.ccdc.cam.ac.uk/conts/retrieving.html (or from the Cambridge Crystallographic Data Centre, 12 Union Road, Cambridge CB2 1EZ, UK; fax: (+44) 1223-336-033; or [email protected]).
General procedure for synthesis of compounds 4, 6, 7, 8, 9, 10 and 11
The cycloimmonium salt 1a–e (1 mmol, 1 equiv.) and dipolarophile (DMAD, EP, N-phenylmaleimide or acrylonitrile, 1.1 mmol, 1.1 equiv.) were added to 5 mL of dichloromethane and the obtained suspension is stirred at room temperature (rt) under N2 atmosphere. Triethylamine (TEA, 3 mmol, 3 equiv.) was added drop-wise over 1 h (magnetic stirring) and the resulting mixture was then stirred for 24 h at rt under nitrogen. Methanol (5 mL) was added and the resulting mixture is kept for 24 h without stirring. The formed precipitate was collected by filtration to give a powder which was washed with few mL of methanol. The product was crystallized from an appropriate solvent or purified by column chromatography.
Dimethyl 9-(4-methoxybenzoyl)pyrrolo[2,1-c][4,7]phenanthroline-7,8-dicarboxylate (4a)
Crystallized from ethanol–chloroform 1:1, (v/v). Yellow powder (51% yield), mp = 244–247 °C. IR [KBr, ν (cm−1)]: 2953, 1739, 1702, 1596, 1252, 1211, 1161, 1086, 803. 1H NMR (500 MHz, CDCl3, 25 °C): δ 3.53 (s, 3H, 21-H), 3.90 (s, 3H, OMe), 3.91 (s, 3H, 19-H), 6.99 (d, J = 8.5 Hz, 2H, phenyl-H), 7.57 (dd, J = 8.0, 4.0 Hz, 1H, 3-H), 7.92 (d, J = 9.5 Hz, 1H, 11-H), 8.02 (d, J = 8.5 Hz, 2H, phenyl-H), 8.07 (d, J = 9.5 Hz, 1H, 12-H), 8.29 (d, J = 9.5 Hz, 1H, 5-H), 8.37 (d, J = 9.5 Hz, 1H, 6-H), 8.80 (d, J = 8.0 Hz, 1H, 4-H), 8.95 (d, J = 4.0 Hz, 1H, 2-H). 13C NMR (125 MHz, CDCl3): δ 51.9 C-19, 52.6 C-21, 55.8 OMe, 105.0 C-7, 114.2 2 × CH-Ph, 118.9 C-6, 120.9 C-14, 121.7 C-5, 122.0 C-11, 122.5 C-3, 125.0 C-13, 126.3 C-9, 128.6 C-8, 130.4 Cq-Ph, 130.9 C-12, 131.0 C-16, 131.3 C-4, 132.6 2 × CH-Ph, 137.0 C-15, 146.0 C-17, 150.6 C-2, 163.6 C-18, 164.7 Cq-Ph, 165.4 C-20, 186.2 CO ppm. Anal. Calcd. for C27H20N2O6: C, 69.22; H, 4.30; N, 5.98. Found: C, 69.39, H, 4.28; N, 6.12.
Dimethyl 9-(4-nitrobenzoyl)pyrrolo[2,1-c][4,7]phenanthroline-7,8-dicarboxylate (4b)
Crystallized from ethanol–chloroform 1:1, (v/v). Orange powder (50% yield), mp = 237–238 °C. IR [KBr, ν(cm−1)): 1736, 1718, 1699, 1488, 1340, 1235, 1203, 1091, 805. 1H NMR (500 MHz, CDCl3, 25 °C): δ 3.48 (s, 3H, 21-H), 3.94 (s, 3H, 19-H), 7.68 (dd, J = 8.5, 4.5 Hz, 1H, 3-H), 7.87 (d, J = 9.5 Hz, 1H, 11-H), 8.16 (d, J = 8.5 Hz, 3H, phenyl-H, 12-H), 8.36 (d, J = 8.5 Hz, 2H, phenyl-H), 8.51 (d, J = 9.5 Hz, 1H, 5-H), 8.57 (d, J = 9.5 Hz, 1H, 6-H), 8.91 (d, J = 8.5 Hz, 1H, 4-H), 9.05 (d, J = 4.5 Hz, 1H, 2-H). 13C NMR (125 MHz, CDCl3): δ 52.2 C-19, 52.8 C-21, 105.9 C-7, 118.9 C-6, 121.4 C-14, 122.1 C-11, 122.8 C-3, 123.3 C-5, 123.9 2 × CH-Ph, 125.0 C-9, 125.1 C-13, 130.0 C-8, 130.9 2 × CH-Ph, 131.0 C-16, 131.0 C-4, 131.5 C-12, 137.7 C-15, 142.6 Cq-Ph, 146.2 C-17, 150.8 Cq-Ph, 151.0 C-2, 163.3 C-18, 165.2 C-20, 184.5 CO ppm. Anal. Calcd. for C26H17N3O7: C, 64.60; H, 3.54; N, 8.69. Found: C, 64.69, H, 3.48; N, 8.83.
Dimethyl 9-(4-chlorobenzoyl)pyrrolo[2,1-c][4,7]phenanthroline-7,8-dicarboxylate (4c)
Crystallized from ethanol–chloroform 1:1, (v/v). Yellow powder (55% yield), mp = 245–247 °C. IR [KBr, ν (cm−1)]: 2947, 1726, 1710, 1644, 1223, 1212, 1086. 1H NMR (500 MHz, CDCl3, 25 °C): δ 3.51 (s, 3H, 21-H), 3.93 (s, 3H, 19-H), 7.50 (d, J = 8.0 Hz, 2H, phenyl-H), 7.65 (dd, J = 8.0, 4.0 Hz, 1H, 3-H), 7.89 (d, J = 9.5 Hz, 1H, 11-H), 7.96 (d, J = 8.0 Hz, 2H, phenyl-H), 8.12 (d, J = 9.5 Hz, 1H, 12-H), 8.44 (d, J = 9.5 Hz, 1H, 5-H), 8.55 (d, J = 9.5 Hz, 1H, 6-H), 8.88 (d, J = 8.0 Hz, 1H, 4-H), 9.03 (as, 1H, 2-H). 13C NMR (125 MHz, CDCl3): δ 52.0 C-19, 52.7 C-21, 105.4 C-7, 118.9 C-6, 121.2 C-14, 122.1 C-11, 122.5 C-5, 122.7 C-3, 125.1 C-13, 125.6 C-9, 129.2 2 × CH-Ph, 130.1 C-8, 131.0 C-16, 131.2 C-4, 131.3 C-12, 131.4 2 × CH-Ph, 136.1 Cq-Ph, 137.7 C-15, 140.7 Cq-Ph, 146.2 C-17, 150.9 C-2, 163.5 C-18, 165.3 C-20, 185.8 CO ppm. Anal. Calcd. for C26H17ClN2O5: C, 66.04; H, 3.62; N, 5.92. Found: C, 66.20, H, 3.53; N, 5.99.
Dimethyl 9-cyanopyrrolo[2,1-c][4,7]phenanthroline-7,8-dicarboxylate (4d)
Crystallized from ethanol–chloroform 1:1, (v/v). Brown powder (50% yield), mp = 261–263 °C. IR [KBr, ν (cm−1)]: 2955, 2216, 1740, 1694, 1225, 1099. 1H NMR (500 MHz, CDCl3, 25 °C): δ 3.97 (s, 3H, 19-H), 4.07 (s, 3H, 21-H), 7.71 (dd, J = 8.5, 4.0 Hz, 1H, 3-H), 8.42 (d, J = 10.0 Hz, 1H, 12-H), 8.46 (d, J = 10.0 Hz, 1H, 5-H), 8.51 (d, J = 10.0 Hz, 1H, 6-H), 8.92 (d, J = 8.5 Hz, 1H, 4-H), 9.11 (ad, J = 4.0 Hz, 1H, 2-H), 9.51 (d, J = 10.0 Hz, 1H, 11-H). 13C NMR (125 MHz, CDCl3): δ 52.3 C-19, 53.4 C-21, 98.3 C-9, 106.9 C-7, 114.7 CN, 118.1 C-11, 118.9 C-6, 121.1 C-14, 122.5 C-5, 123.0 C-3, 125.1 C-13, 131.4 C-4, 132.0 C-16, 133.4 C-8, 132.7 C-12, 136.8 C-15, 146.4 C-17, 151.4 C-2, 162.7 C-18, 163.3 C-20 ppm. Anal. Calcd. for C20H13N3O4: C, 66.85; H, 3.65; N, 11.69. Found: C, 66.81, H, 3.58; N, 11.72.
Trimethyl pyrrolo[2,1-c][4,7]phenanthroline-7,8,9-tricarboxylate (4e)
Crystallized from ethanol–chloroform 1:1, (v/v). Pale pink powder (55% yield), mp = 215–218 °C. IR [KBr, ν (cm−1)]: 2953, 1741, 1704, 1677, 1263, 1161, 1099, 802. 1H NMR (500 MHz, CDCl3, 25 °C): δ 3.94 (s, 3H, 19-H), 4.00 (s, 3H, OMe), 4.03 (s, 3H, 21-H), 7.76 (dd, J = 8.0, 4.0 Hz, 1H, 3-H), 8.36 (d, J = 10.0 Hz, 1H, 11-H), 8.38 (d, J = 9.5 Hz, 1H, 5-H), 8.43 (d, J = 10.0 Hz, 1H, 12-H), 8.49 (d, J = 9.5 Hz, 1H, 6-H), 9.01 (d, J = 8.5 Hz, 1H, 4-H), 9.05 (dd, J = 4.0, 1.0 Hz, 1H, 2-H). 13C NMR (125 MHz, CDCl3): δ 52.1 C-19, 52.9 OMe, 53.2 C-21, 105.4 C-7, 117.6 C-9, 119.3 C-6, 121.1 C-14, 122.2 C-5, 122.5 C-3, 124.1 C-11, 125.2 C-13, 128.3 C-12, 131.6 C-16, 132.3 C-8, 133.6 C-4, 137.9 C-15, 144.1 C-17, 148.7 C-2, 161.1 COOMe, 163.2 C-18, 165.9 C-20 ppm. Anal. Calcd. for C21H16N2O6: C, 64.28; H, 4.11; N, 7.14. Found: C, 64.20, H, 4.03; N, 7.20.
Ethyl 9-(4-methoxybenzoyl)pyrrolo[2,1-c][4,7]phenanthroline-7-carboxylate (6a)
Purified by column chromatography: CH2Cl2→ CH2Cl2: MeOH (98:2, v:v), then crystallized from ethanol–chloroform 1:1, (v/v). Orange powder (50% yield), mp = 252–253 °C. IR [KBr, ν (cm−1)]: 2982, 1694, 1232, 1166, 1080, 800. 1H NMR (500 MHz, CDCl3, 25 °C): δ 1.41 (t, J = 7.0 Hz, 3H, 20-H), 3.95 (s, 3H, OMe), 4.40 (q, 2H, J = 7.0 Hz, 19-H), 7.07 (d, J = 8.0 Hz, 2H, phenyl-H), 7.60 (dd, J = 8.0, 4.0 Hz, 1H, 3-H), 7.73 (s, 1H, 8-H), 8.20 (m, 4H, 11-H, 12-H, phenyl-H), 8.40 (d, J = 9.5 Hz, 1H, 5-H), 8.51 (d, J = 9.5 Hz, 1H, 6-H), 8.85 (d, J = 8.0 Hz, 1H, 4-H), 8.99 (d, J = 4.0 Hz, 1H, 2-H). 13C NMR (125 MHz, CDCl3): δ 14.7 C-20, 55.8 OMe, 60.4 C-19, 107.4 C-7, 114.1 2 × CH-Ph, 118.6 C-6, 120.6 C-14, 122.3 C-3, 122.4 C-5, 123.3 C-11, 125.0 C-13, 128.0 C-9, 129.1 C-8, 130.8 Cq-Ph, 130.6 C-12, 131.2 C-4, 131.7 C-16, 132.6 2 × CH-Ph, 139.8 C-15, 146.3 C-17, 150.4 C-2, 163.9 Cq-Ph, 164.2 C-18, 184.0 CO ppm. Anal. Calcd. for C26H20N2O4: C, 73.57; H, 4.57; N, 6.60. Found: C, 73.52, H, 4.49; N, 6.62.
Ethyl 9-(4-nitrobenzoyl)pyrrolo[2,1-c][4,7]phenanthroline-7-carboxylate (6b)
Crystallized from ethanol–chloroform 1:1, (v/v). Orange powder (49% yield), mp = 332–335 °C. IR [KBr, ν (cm−1)]: 3070, 2968, 1694, 1638, 1516, 1495, 1344, 1237, 1081, 801. 1H NMR (500 MHz, CDCl3, 25 °C): δ 1.42 (t, J = 7.0 Hz, 3H, 20-H), 4.41 (q, 2H, J = 7.0 Hz, 19-H), 7.68 (dd, J = 8.5, 4.0 Hz, 1H, 3-H), 7.75 (s, 1H, 8-H), 8.29 (m, 4H, 11-H, 12-H, phenyl-H), 8.44 (d, J = 8.5 Hz, 2H, phenyl-H), 8.59 (d, J = 9.5 Hz, 1H, 5-H), 8.63 (d, J = 9.5 Hz, 1H, 6-H), 8.94 (d, J = 8.5 Hz, 1H, 4-H), 9.06 (d, J = 4.0 Hz, 1H, 2-H). 13C NMR (125 MHz, CDCl3): δ 14.7 C-20, 60.7 C-19, 108.4 C-7, 118.6 C-6, 121.1 C-14, 122.6 C-3, 123.4 C-11, 124.0 2 × CH-Ph, 124.1 C-5, 125.0 C-13, 127.4 C-9, 131.0 2 × CH-Ph, 131.4 C-12, 131.5 C-4, 131.6 C-8, 131.9 C-16, 141.2 C-15, 143.8 Cq-Ph, 146.5 C-17, 150.4 Cq-Ph, 150.8 C-2, 163.7 C-18, 181.8 CO ppm. Anal. Calcd. for C25H17N3O5: C, 68.33; H, 3.90; N, 9.56. Found: C, 68.35, H, 3.80; N, 9.63.
Ethyl 9-(4-chlorobenzoyl)pyrrolo[2,1-c][4,7]phenanthroline-7-carboxylate (6c)
Crystallized from ethanol–chloroform 1:1, (v/v). Yellow powder (59% yield), mp = 278–280 °C. IR [KBr, ν (cm−1)]: 3049, 2977, 1697, 1229, 1211, 1077, 803. 1H NMR (500 MHz, CDCl3, 25 °C): δ 1.42 (t, J = 7.0 Hz, 3H, 20-H), 4.40 (q, 2H, J = 7.0 Hz, 19-H), 7.57 (d, J = 8.0 Hz, 2H, phenyl-H), 7.62 (dd, J = 8.0, 4.0 Hz, 1H, 3-H), 7.73 (s, 1H, 8-H), 8.09 (d, J = 8.0 Hz, 2H, phenyl-H), 8.22 (as, 2H, 11-H, 12-H), 8.46 (d, J = 9.5 Hz, 1H, 5-H), 8.54 (d, J = 9.5 Hz, 1H, 6-H), 8.88 (d, J = 8.0 Hz, 1H, 4-H), 9.01 (d, J = 4.0 Hz, 1H, 2-H). 13C NMR (125 MHz, CDCl3): δ 14.7 C-20, 60.5 C-19, 107.8 C-7, 118.5 C-6, 120.8 C-14, 122.4 C-3, 123.2 C-5, 123.4 C-11, 125.0 C-13, 127.6 C-9, 129.1 2 × CH-Ph, 130.4 C-8, 130.8 C-12, 131.3 C-4, 131.6 2 × CH-Ph, 131.8 C-16, 136.7 Cq-Ph, 139.6 Cq-Ph, 140.5 C-15, 146.3 C-17, 150.6 C-2, 164.0 C-18, 185.3 CO ppm. Anal. Calcd. for C25H17ClN2O3: C, 70.01; H, 4.00; N, 6.53. Found: C, 69.99, H, 3.93; N, 6.60.
Ethyl 9-cyanopyrrolo[2,1-c][4,7]phenanthroline-7-carboxylate (6d)
Crystallized from ethanol–chloroform 1:1, (v/v). Brown powder (55% yield), mp = 224–226 °C. IR [KBr, ν (cm−1)]: 3075, 2992, 2213, 1698, 1248, 1080, 799. 1H NMR (500 MHz, CDCl3, 25 °C): δ 1.45 (t, J = 7.0 Hz, 3H, 20-H), 4.43 (q, 2H, J = 7.0 Hz, 19-H), 7.67 (dd, J = 8.5, 4.0 Hz, 1H, 3-H), 7.96 (s, 1H, 8-H), 8.37 (d, J = 9.5 Hz, 1H, 12-H), 8.40 (d, J = 10.0 Hz, 1H, 5-H), 8.58 (d, J = 10.0 Hz, 1H, 6-H), 8.90 (d, J = 8.5 Hz, 1H, 4-H), 9.07 (ad, J = 4.0 Hz, 1H, 2-H), 9.46 (d, J = 9.5 Hz, 1H, 11-H). 13C NMR (125 MHz, CDCl3): δ 14.6 C-20, 60.7 C-19, 97.7 C-9, 108.3 C-7, 115.8 CN, 118.2 C-11, 118.9 C-6, 120.5 C-14, 121.6 C-5, 122.8 C-3, 125.1 C-13, 128.7 C-8, 131.3 C-4, 132.3 C-12, C-16, 137.5 C-15, 146.2 C-17, 151.1 C-2, 163.2 C-18 ppm. Anal. Calcd. for C19H13N3O2: C, 72.37; H, 4.16; N, 13.33. Found: C, 72.35, H, 4.09; N, 13.39.
7-Ethyl 9-methyl pyrrolo[2,1-c][4,7]phenanthroline-7,9-dicarboxylate (6e)
Crystallized from ethanol–chloroform 1:1, (v/v). Pale pink powder (49% yield), mp = 173–174 °C. IR [KBr, ν (cm−1)]: 3076, 2993, 2213, 1726, 1689, 1248, 1080, 799. 1H NMR (500 MHz, CDCl3, 25 °C): δ 1.44 (t, J = 7.0 Hz, 3H, 20-H), 4.00 (s, 3H, OMe), 4.41 (q, 2H, J = 7.0 Hz, 19-H), 7.57 (dd, J = 8.5, 4.0 Hz, 1H, 3-H), 8.05 (s, 1H, 8-H), 8.21 (d, J = 10.0 Hz, 1H, 12-H), 8.29 (d, J = 9.5 Hz, 1H, 5-H), 8.45 (d, J = 9.5 Hz, 1H, 6-H), 8.54 (d, J = 10.0 Hz, 1H, 11-H), 8.98 (d, J = 8.5 Hz, 1H, 4-H), 8.98 (ad, J = 4.0 Hz, 1H, 2-H). 13C NMR (125 MHz, CDCl3): δ 14.7 C-20, 52.4 OMe, 60.4 C-19, 107.4 C-7, 118.6 C-6, 119.7 C-9, 120.7 C-14, 121.6 C-5, 122.2 C-3, 123.2 C-11, 124.8 C-13, 127.5 C-8, 129.9 C-12, 131.3 C-4, 131.8 C-16, 139.5 C-15, 146.0 C-17, 150.3 C-2, 162.2 COOMe, 164.0 C-18 ppm. Anal. Calcd. for C20H16N2O4: C, 68.96; H, 4.63; N, 8.04. Found: C, 68.90, H, 4.57; N, 8.10.
Ethyl 9-(4-methoxybenzoyl)-8,9-dihydropyrrolo[2,1-c][4,7]phenanthroline-7-carboxylate (7a)
Purified by column chromatography: CH2Cl2→ CH2Cl2: MeOH (98:2, v:v), then crystallized from ethanol–chloroform 1:1, (v/v). Red crystals (25% yield), mp = 243–246 °C. IR [KBr, ν (cm−1)]: 1684, 1650, 1266, 1222, 1163, 1060, 1033, 803. 1H NMR (500 MHz, CDCl3, 25 °C): δ 1.28 (t, J = 7.0 Hz, 3H, 20-H), 3.09 (dd, J = 15.5, 5.5 Hz, 1H, 8-H), 3.64 (at, J = 9.5 Hz, 8-H), 3.93 (s, 3H, OMe), 4.18 (q, 2H, J = 7.0 Hz, 19-H), 6.88 (d, J = 9.5 Hz, 11-H), 6.99 (dd, J = 14.0, 5.0 Hz, 1H, 9-H), 7.04 (d, J = 8.5 Hz, 2H, phenyl-H), 7.48 (dd, J = 8.5, 4.0 Hz, 1H, 3-H), 7.73 (d, J = 10.0 Hz, 1H, 5-H), 7.92 (d, J = 10.0 Hz, 1H, 6-H), 7.97 (d, J = 9.5 Hz, 12-H), 8.04 (d, J = 8.5 Hz, 2H, phenyl-H), 8.49 (d, J = 8.5 Hz, 1H, 4-H), 8.77 (d, J = 4.0 Hz, 1H, 2-H). 13C NMR (125 MHz, CDCl3): δ 14.9 C-20, 33.7 C-8, 55.8 OMe, 59.0 C-19, 64.3 C-9, 90.1 C-7, 114.6 2 × CH-Ph, 117.3 C-11, 119.2 C-5, 115.2 C-14, 122.6 C-3, 124.8 C-13, 125.8 Cq-Ph, 129.5 C-6, 130.0 C-4, 130.1 C-16, 131.5 2 × CH-Ph, 132.8 C-12, 138.0 C-15, 144.0 C-17, 148.2 C-2, 164.7 Cq-Ph, 166.1 C-18, 191.5 CO ppm. Anal. Calcd. for C26H22N2O4: C, 73.23; H, 5.20; N, 6.57. Found: C, 73.22, H, 5.09; N, 6.62.
12-(4-Nitrobenzoyl)-10-phenyl-11a,12-dihydro-8aH-pyrrolo[3′,4′:3,4]pyrrolo[2,1-c][4,7]phenanthroline-9,11(8bH,10H)-dione (8b)
Crystallized from ethanol-chloroform 1:1, (v/v). Yellow powder (58% yield), mp = 177–180 °C. IR [KBr, ν (cm−1)]: 1718, 1596, 1523, 1497, 1382, 1343, 1195. 1H NMR (500 MHz, CDCl3, 25 °C): δ 3.72 (t, J = 8.0 Hz, 1H, 8-H), 3.84 (ad, J = 8.0 Hz, 1H, 18-H), 5.39 (d, J = 8.0, Hz, 1H, 7-H), 5.99 (s, 1H, 12-H), 6.28 (ad, J = 10.0 Hz, 1H, 6-H), 6.79 (d, J = 9.0 Hz, 1H, 13-H), 7.16 (ad, J = 8.0 Hz, 3H, 5-H, 2 × phenyl-CH), 7.35-7.43 (m, 4H, 3 × phenyl-CH, 3-H), 7.86 (d, J = 9.0 Hz, 1H, 14-H), 8.29 (ad, J = 9.0 Hz, 1H, 4-H), 8.45 (d, J = 8.5 Hz, 2H, 2 × CH-(p)-NO2-Ph, 8.54 (d, J = 8.5 Hz, 2H, 2 × CH-(p)-NO2-Ph, 8.69 (ad, J = 3.0 Hz, 1H, 2-H). 13C NMR (125 MHz, CDCl3): δ 46.7 C-18, 48.1 C-8, 61.1 C-7, 65.5 C-12, 114.0 C-17, 116.9 C-13, 120.1 C-6, 121.7 C-5, 121.9 C-3, 124.6 2 × CH-(p)-NO2-phenyl, 125.8 C-16, 126.3 2 × CH-Ph, 129.3 CH-Ph, 129.4 2 × CH-Ph, 129.7 C-4, 130.5 2 × CH-(p)-NO2-phenyl, 130.9 C-14, 131.4 Cq-Ph, 137.6 Cq-(p)-NO2-phenyl, 139.5 C-19, 142.0 C-15, 147.4 C-2, 151.3 C-NO2, 174.4 C-9, 176.2 C-11, 194.1 C-20 ppm. Anal. Calcd. for C30H20N4O5: C, 69.76; H, 3.90; N, 10.85. Found: C, 69.77, H, 3.82; N, 10.95.
12-(3-Methoxybenzoyl)-10-phenyl-11a,12-dihydro-8aH-pyrrolo[3′,4′:3,4]pyrrolo[2,1-c][4,7]phenanthroline-9,11(8bH,10H)-dione (8f)
Crystallized from ethanol–chloroform 1:1, (v/v). Orange powder (82% yield), mp = 173–176 °C. IR [KBr, ν (cm−1)]: 1706, 1582, 1490, 1377, 1263, 1169. 1H NMR (500 MHz, CDCl3, 25 °C): δ 3.69 (t, J = 8.0 Hz, 1H, 8-H), 3.92 (s, 3H, OMe), 3.87 (ad, J = 8.0 Hz, 1H, 18-H), 5.45 (d, J = 8.0, Hz, 1H, 7-H), 5.96 (s, 1H, 12-H), 6.26 (ad, J = 10.0 Hz, 1H, 6-H), 6.80 (d, J = 9.0 Hz, 1H, 13-H), 7.13 (ad, J = 8.5 Hz, 1H, 5-H), 7.20 (ad, J = 7.5 Hz, 2H, 2 × phenyl-CH), 7.25 [overlapped signal, 1H, (m)-OMe-phenyl-H], 7.36–7.43 (m, 3H, 3 × phenyl-CH), 7.32 (dd, J = 4.0, 9,0 Hz, 1H, 3-H), 7.51 (t, J = 8.0 Hz, 1H, (m)-OMe-phenyl-H), 7.82 (d, J = 9.0 Hz, 1H, 14-H), 7.84 (s, 1H, (m)-OMe-phenyl-H), 7.98 (d, J = 7.5 Hz, 1H, (m)-OMe-phenyl-H), 8.26 (ad, J = 9.0 Hz, 1H, 4-H), 8.66 (ad, J = 3.0 Hz, 1H, 2-H). 13C NMR (125 MHz, CDCl3): δ 47.3 C-18, 48.1 C-8, 55.8 OMe, 61.1 C-7, 65.0 C-12, 112.8 CH-(m)-OMe-phenyl, 113.8 C-17, 117.2 C-13, 120.0 C-6, 121.7 C-5, 121.8 C-3, 122.0 CH-(m)-OMe-phenyl, 125.6 C-16, 126.5 2 × CH-Ph, 129.2 CH-Ph, 129.3 2 × CH-Ph, 129.4 C-4, 130.5 CH-(m)-OMe-phenyl, 130.8 CH-(m)-OMe-phenyl, 130.8 C-14, 131.5 Cq-Ph, 134.4 Cq-(m)-OMe-phenyl, 140.0 C-19, 143.1 C-15, 147.2 C-2, 160.4 C-OMe, 174.9 C-9, 176.5 C-11, 195.1 C-20 ppm. Anal. Calcd. for C31H23N3O4: C, 74.24; H, 4.62; N, 8.38. Found: C, 74.27, H, 4.52; N, 8.45.
Dimethyl 8-(phenylcarbamoyl)-8,9-dihydropyrrolo[2,1-c][4,7]phenanthroline-7,9-dicarboxylate (9e)
Purified by column chromatography: CH2Cl2: MeOH (99.6:0.4, v:v). Orange red powder (30% yield), mp = 166–168 °C. IR [KBr, ν (cm−1)]: 2951, 1751, 1721, 1678, 1615, 1597, 1549, 1211, 1186. 1H NMR (500MHz, CDCl3, 25 °C): δ 3.81 (s, 3H, OMe), 3.89 (s, 3H, OMe), 3.36 (d, J = 3.0 Hz, 1H, 8-H), 6.28 (dd, J = 3.0 Hz, 1H, 9-H), 7.09 (t, J = 7.5 Hz, 1H, Ph-H), 7.32 (t, J = 7.5 Hz, 2H, Ph-H), 7.41 (d, J = 9.0 Hz, 1H, 11-H), 7.52 (dd, J = 8.5, 4.0 Hz, 1H, 3-H), 7.60 (ad, J = 8.0 Hz, 3H, 6-H, Ph-H), 8.09 (d, J = 10.0 Hz, 1H, 5-H), 8.15 (d, J = 9.0 Hz, 1H, 12-H), 8.50 (d, J = 8.5 Hz, 1H, 4-H), 8.86 (dd, J = 4.0, 1.0 Hz, 1H, 2-H), 10.19 (s, 1H, NH). 13C NMR (125 MHz, CDCl3): δ 51.3 OCH3, 51.4 C-8, 53.4 OCH3, 62.2 C-9, 115.9 C-14, 117.5 C-11, 118.5 C-7, 119.7 2 × CH-Ph, 6-H, 122.8 C-3, 124.1 CH-Ph, 125.0 C-13, 129.1 2 × CH-Ph, 129.9 C-4, 131.3 C-5, 133.8 C-12, 137.5 C-16, 151.1 C-15, 144.3 C-17, 149.0 C-2, 168.32 CO, 170.1 CO. Anal. Calcd. for C26H21N3O5: C, 68.56; H, 4.65; N, 9.23. Found: C, 68.50, H, 4.58; N, 8.28.
9-(4-Nitrobenzoyl)-6a,7,8,9-tetrahydropyrrolo[2,1-c][4,7]phenanthroline-7-carbonitrile (10b)
Crystallized from ethanol–chloroform 1:1, (v/v). Dark red powder (45% yield), mp = 265–268 °C. IR [KBr, ν (cm−1)]: 2911, 2176, 1708, 1630, 1499, 1472, 1344, 792. 1H NMR (500 MHz, CDCl3, 25 °C): δ 2.20–2.26 (m, 1H, 8a-H), 2.88–2.93 (m, 1H, 8b-H), 3.50 (at, J = 5.5 Hz,1H, 7-H), 5.31 (at, J = 2.5 Hz, 1H, 15-H), 5.57 (dd, J = 9.0, 6.5 Hz, 1H, 9-H), 5.82 (ad, J = 10.5 Hz, 1H, 6-H), 6.53 (d, J = 9.0 Hz, 1H, 11-H), 7.26 (overlapped signal, 1H, 5-H), 7.33 (dd, J = 4.0, 9.0 Hz, 1H, 3-H), 7.77 (d, J = 9.0 Hz, 1H, 12-H), 8.25 (ad, J = 9.0 Hz, 3H, 2 × phenyl-H, 4-H), 8.44 (d, J = 8.5 Hz, 2H, 2 × phenyl-H), 8.65 (ad, J = 3.0 Hz, 1H, 2-H). 13C NMR (125 MHz, CDCl3): δ 32.3 C-8, 35.9 C-7, 61.9 C-9, 62.2 C-15, 112.0 C-14, 116.6 C-11, 117.9 C-6, 122.0 C-3, 119.1 CN, 123.6 C-5, 124.7 2 × CH-Ph, 125.8 C-13, 129.1 C-4, 129.9 2 × CH-Ph, 131.4 C-12, 138.3 Cq-Ph, 139.6 C-16, 143.2 C-17, 147.3 C-2, 151.2 Cq-Ph, 196.2 C-18 ppm. Anal. Calcd. for C23H16N4O3: C, 69.69; H, 4.07; N, 14.13. Found: C, 69.78, H, 4.03; N, 14.19.
Methyl 7-cyano-8,9-dihydropyrrolo[2,1-c][4,7]phenanthroline-9-carboxylate (11e)
Crystallized from ethanol–chloroform 1:1, (v/v). Dark red powder (45% yield), mp = 230–232 °C. IR [KBr, ν (cm−1)]: 2948, 2175, 1723, 1630, 1276, 833, 792. 1H NMR (500 MHz, CDCl3, 25 °C): δ 3.15 (dd, J = 16.0, 4.0 Hz, 1H, 8b-H), 3.48 (t, J = 15.0 Hz, 1H, 8a-H), 3.84 (s, 3H, OMe), 5.22 (dd, J = 13.0, 4.5 Hz, 1H, 9-H), 6.96 (d, J = 10.0 Hz, 1H, 6-H), 7.28 (d, J = 9.5 Hz, 1H, 11-H), 7.70 (d, J = 10.0 Hz, 1H, 5-H), 7.90 (dd, J = 8.5, 2.5 Hz, 1H, 3-H), 8.82 (ad, J = 4.0 Hz, 1H, 2-H), 8.89 (d, J = 9.0 Hz, 1H, 12-H), 8.95 (d, J = 8.5 Hz, 1H, 4-H). 13C NMR (125 MHz, CDCl3): δ 33.3 C-8, 53.1 OCH3, 61.2 C-9, 72.1 C-7, 119.7 C-6, 121.2 C-11, 114.7 C-14, 119.1 CN, 122.7 C-3, 125.4 C-12, 127.6 C-5, 126.4 C-13, 138.3 C-4, 135.0 C-17, 140.2 C-16, 140.4 C-2, 153.0 C-15, 169.3 CO. Anal. Calcd. for C18H13N3O2: C, 71.28; H, 4.32; N, 13.85. Found: C, 71.30, H, 4.29; N, 13.88.
9-(3-Methoxybenzoyl)-8,9-dihydropyrrolo[2,1-c][4,7]phenanthroline-7-carbonitrile (11f)
Crystallized from ethanol–chloroform 1:1, (v/v). Dark red powder (40% yield), mp = 221–224 °C. IR [KBr, ν (cm−1)]: 3072, 2916, 2168, 1689, 1633, 1268, 1136, 794. 1H NMR (500 MHz, CDCl3, 25 °C): δ 3.02 (dd, J = 15.0, 5.0 Hz, 1H, 8b-H), 3.63 (t, J = 14.0 Hz, 1H, 8a-H), 3.88 (s, 3H, OMe), 6.03 (dd, J = 14.0, 5.0 Hz, 1H, 9-H), 6.81 (d, J = 9.5 Hz, 1H, 11-H), 6.89 (d, J = 10.5 Hz, 1H, 5-H), 7.25 (overlapped signal, 1H, 3-H), 7.48–7.52 (m, 2H, 2 × phenyl-H), 7.54–7.58 (m, 2H, 2 × phenyl-H), 7.87 (d, J = 10.0 Hz, 1H, 6-H), 7.98 (d, J = 9.0 Hz, 1H, 12-H), 8.45 (d, J = 8.5 Hz, 1H, 4-H), 8.80 (dd, J = 4.0, 1.0 Hz, 1H, 2-H). 13C NMR (125 MHz, CDCl3): δ 33.6 C-8, 55.7 OCH3, 64.6 C-9, 66.9 C-7, 113.6 CH-Ph, 116.8 C-5, C-11, 115.1 C-14, 119.6 CN, 121.3 C-3, CH-Ph, 122.8 CH-Ph, 129.7 C-4, 129.9 C-6, 130.5 CH-Ph, 133.3 C-12, 133.9 Cq-Ph, 138.1 C-16, 125.3 C-13, 148.5 C-2, 155.1 C-15, 143.7 C-17, 160.5 Cq-Ph, 191.9 CO. Anal. Calcd. for C24H17N3O2: C, 75.97; H, 4.52; N, 11.08. Found: C, 76.02, H, 4.49; N, 11.13.
Microbiology
Antimycobacterial activities of the compounds were performed by Center of Tuberculosis Antimicrobial Acquisition and Coordinating Facility (TAACF) at Southern Research InstituteCitation36–38.
The Primary Cycle High Throughput Screening (HTS). Determination of 90% inhibitory concentration (IC90), 50% inhibitory concentration (IC50) and Minimum Inhibitory Concentration (MIC)
The MIC of compound was determined by measuring bacterial growth after 5 days in the presence of test compounds. Compounds were prepared as 10-point two-fold serial dilutions in DMSO and diluted into 7H9-Tw-OADC medium in 96-well plates with a final DMSO concentration of 2%. The highest concentration of compound was 200 µM where compounds were soluble in DMSO at 10 mM. For compounds with limited solubility, the highest concentration was 50 × less than the stock concentration, e.g. 100 µM for 5 mM DMSO stock, 20 µM for 1 mM DMSO stock. For potent compounds, assays were repeated at lower starting concentrations. Each plate included assay controls for background (medium/DMSO only, no bacterial cells), zero growth (100 µM Rifampicin) and maximum growth (DMSO only), as well as a rifampicin dose response curve. Plates were inoculated with M. tuberculosis and incubated for 5 days: growth was measured by OD590 and fluorescence (Ex 560/Em 590) using a BioTek™ Synergy 4 plate reader. Growth was calculated separately for OD590 and RFU. To calculate the MIC, the 10-point dose response curve was plotted as % growth and fitted to the Gompertz model using GraphPad Prism 5. Data points obtained from a dose response growth inhibition assay are curve-fitted using the Gompertz model to calculate MIC () and the Levenberg–Marquardt algorithm to calculate IC50 and IC90 (). (A) The MIC is the concentration at which complete inhibition growth is seen and is derived from the point of inflection at which the curve meets the lower asymptote (zero growth). (B) IC50 and IC90 are points at which growth is inhibited by 50% and 90% respectively. Orange line = MIC; Green line = IC50; Blue line = IC90.
Figure 1. Dose response curves used to calculate MIC, IC50 and IC90.
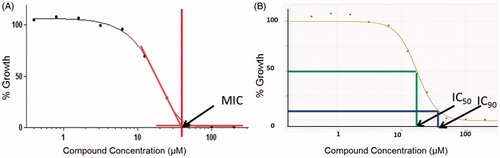
MIC values were reported when the following quality control criteria were satisfied:
For each plate
No growth in the background (un-inoculated) control wells.
OD590 > 0.3 in maximum growth wells.
Rifampicin MIC within 3-fold of the expected value.
For each compound curve. MICs were reported if
There were two points with growth >75%.
There were two points with growth <75%.
If only one point was >75% inhibition then the MIC value was reported as the maximum concentration tested.
If no point reached 75% inhibition, the MIC was reported as > maximum concentration tested.
Results and discussion
Chemistry
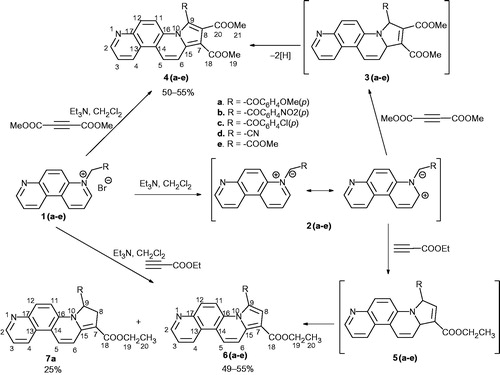
All monoquaternary 4,7-phenanthrolin-4-ium bromides 1 were prepared following our previously reported proceduresCitation39. Then, a variety of functionalized alkynes and alkenes were used as dipolarophiles for 3 + 2 cycloaddion of the in situ generated N-ylides 2 in order to synthesize a series of different substituted pyrrolo[2,1-c][4,7]phenanthroline derivatives. Thus, the dipolar cycloaddition reactions of the ylides 2(a–e) [generated under triethylamine treatment from the corresponding salts 1] to dimethylacetylene dicarboxylate (DMAD) and ethyl propiolate (EP) gave moderate yields of the expected aromatic pyrrolo[2,1-c][4,7]phenanthroline derivatives 4(a–e) and 6(a–e) (Scheme 1). From the reactions of ylides 2 with EP, only a single regioisomer has been isolated in accordance with the electronic effects in both reactants. The reaction of ylide 2a and EP gave a mixture consisting of 8,9-dihydropyrrolo[2,1-c][4,7]phenanthroline derivative 7a and aromatized compound 6a. We assume that all cycloadditions in Scheme 1, result initially in formation of the unisolable 9,15-dihydropyrrolo[2,1-c][4,7]phenanthroline intermediates 3 and 5 that undergo an oxidative aromatization, converting into more stable aromatic compounds 4 and 6. The formation of dihydroderivative 7a takes place probably via a regioselective prototropic rearrangement.
Scheme 1. Synthesis of fused pyrrolo derivatives 4, 6 and 7.
The structures of the newly compounds were proven by elemental and spectral (IR, NMR, X-ray) methods.
The 1H and 13C NMR spectra of compound 4 and 6 prove the pyrrolo ring formation. Interestingly, the signal of proton 11 that appear in the region of 7.87–8.54 ppm in the spectra of cycloadducts 4a–c, 4e, 6a–c and 6e shifts to the lower fields at 9.51 ppm in the spectrum of 4d, and 9.46 ppm in the spectrum of 6d, compounds in which the position 9 is substituted with a cyano group. We suppose that shifting is due to the positioning of the proton 11 in a deshielding anisotropy region of cyano group rather than a possible H-bond involving.
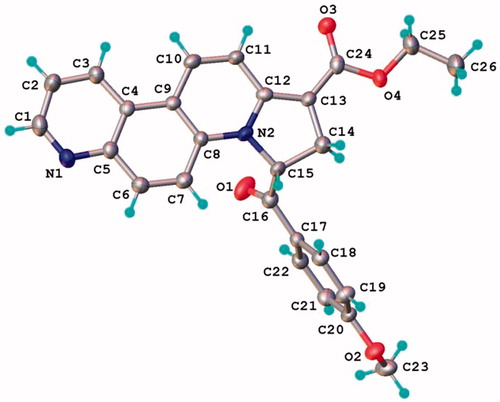
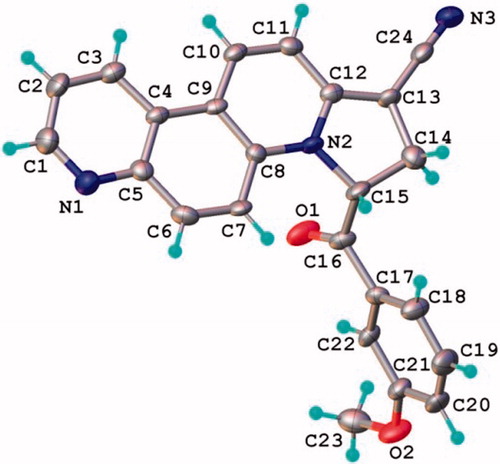
The position of the two protons on the dihydropyrrolo ring of 7a were established on the basis of NMR and confirmed by single-crystal X-ray study. Thus, the 1H-NMR spectrum of 7a shows in the aliphatic region two signals at 3.09 and 3.64 ppm corresponding to the two geminal protons 8 coupled with the deshielded proton 9 that furnishes a signal at 6.99 ppm. As well, the chemical shift of the carbonyl ester groups shifts in 13C-NMR spectrum shows an attachment to a Csp2 (166.1 ppm). The result of single crystal X-ray study of compound 7a (crystallized from CH2Cl2:MeOH 1:1 (v:v)) is shown in , while the bond lengths and angles are summarized in Supplemental Table S2. The X-ray data show that dihydropyrrolo[2,1-c][4,7]phenanthroline core is almost planar, the maximal deviation of the atoms from the mean plane being 0.082 Å.
Figure 2. X-ray molecular structure of compound 7a with thermal ellipsoids at 50% probability level.
In the crystal structure, molecules 7a are stocked through the CH···O hydrogen bonding into a columnar array aligned parallel to the c crystallographic axis, as shown in . Significant π–π stacking interactions are also present within the column. The ring-centroid separation between inversion molecules are of 3.732 Å, and shift distance of 1.711 Å. The crystal structure of compound 7a clearly demonstrated that it crystallizes without any solvent molecules.
Figure 3. View of supramolecular columnar architecture in the crystal structure of 7a. Only H atoms involved in hydrogen bonding are shown. H-bonds parameters: C10–H⋯O3 [C10–H 0.93 Å, H⋯O3 2.38 Å, C10⋯O3(2 − x, 1 − y, −z) 3.308(4) Å, C10–H⋯O3 175.8 °]; C3–H⋯O3 [C3–H 0.93 Å, H⋯O3 2.49 Å, C3⋯O3(2 − x, 1 − y, −z) 3.419(4) Å, C10–H⋯O3 173.0°]; C14–H⋯O3 [C14–H 0.97 Å, H⋯O3 2.44 Å, C14⋯O3(−1 + x, y, z) 3.299(4) Å, C10–H⋯O3 148.0°].
![Figure 3. View of supramolecular columnar architecture in the crystal structure of 7a. Only H atoms involved in hydrogen bonding are shown. H-bonds parameters: C10–H⋯O3 [C10–H 0.93 Å, H⋯O3 2.38 Å, C10⋯O3(2 − x, 1 − y, −z) 3.308(4) Å, C10–H⋯O3 175.8 °]; C3–H⋯O3 [C3–H 0.93 Å, H⋯O3 2.49 Å, C3⋯O3(2 − x, 1 − y, −z) 3.419(4) Å, C10–H⋯O3 173.0°]; C14–H⋯O3 [C14–H 0.97 Å, H⋯O3 2.44 Å, C14⋯O3(−1 + x, y, z) 3.299(4) Å, C10–H⋯O3 148.0°].](/cms/asset/218d1528-e6f5-437c-8737-207ad727215f/ienz_a_1039530_f0003_c.jpg)
Next step in our research plan was the study of 3 + 2 cycloaddition of ylides 2b, e, f to symmetrical (N-phenylmaleimide) and unsymmetrical (acrylonitrile) substituted alkenes. Thus, N-ylides 2 were in situ generated and reacted with N-phenylmaleimide giving dihydropyrrolo[3′,4′:3,4]pyrrolo[2,1-c][4,7]phenanthroline derivatives 8 (Scheme 2).
Scheme 2. Synthesis of fused pyrrolo derivatives 8, 9, 10 and 11.
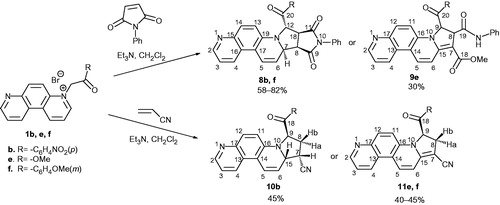
The cycloaddition reactions with N-phenylmaleimide occur highly stereoselective, a single isomer (8) being obtained. Interestingly, from the reaction of 1e in the same conditions, we isolated compound 9e formed by hydrolysis of unstable intermediate 8 during column chromatography separation process. Thus, using CH2Cl2/MeOH (99.6:0.4; v/v) as eluent under acidic conditions on silica gel, the imidic cycle opened forming compound 9e. As far, for the cycloaddition with acrylonitrile, the reactions are as expected highly regioselective. The reaction pathway with acrylonitrile occurs differently according the starting ylide we used: a single compound with tetrahydropyrrolo structure 10b was obtained in the case of ylide type b, while for ylides type e and f, a dihydroderivative (11e and 11f, respectively) have been obtained. The formation of dihydroderivatives 11e and 11f could be explained by an oxidative dehydrogenation of tetrahydro intermediates 10, leading to thermodynamically more stable product 11.
In 1H NMR of compounds 8, there are four distinct signals in the aliphatic region corresponding to the four hydrogen atoms on the pyrrolo ring. The signals delivered by H12 appear as singlets, the lack of coupling for H12 proton being a proof for a trans position relative to proton 18. On the other hand, the big values for the coupling constants J18,8 (8.0 Hz) and J8,7 (8.0 Hz) show us that hydrogen atoms H18, H8 and H7 lay on the same side of the tetrahydropyrrolic ring, but on the opposite side of H12.
The structure of compound 9e is proved by the existence in the 1H NMR spectrum, of two singlets in the aliphatic region (3.81 and 3.89 ppm) corresponding to the methyl ester groups, two coupled doublets (at 4.36 and 6.28 ppm) corresponding to the dihydropyrrolo hydrogens and the singlet at low field (10.19 ppm) furnished by the amidic proton.
The presence of five signals in the aliphatic region of the 1H NMR of compound 10b, corresponds to the expected tetrahydropyrrolo ring hydrogen atoms, the most deshielded being H9 (dd, 5.57 ppm).
The similarity of 1H NMR spectra of compound 11e, 11f and 7a diminished the work for elucidation of structure 11, these compounds being probably obtained by an oxidative process in which the intermediate compounds 10 lose two hydrogen atoms. The single crystal X-ray diffraction investigation has demonstrated that the structure of compound 11f [crystallized from CH2Cl2:MeOH 1:1 (v:v)] is built up from molecular entities depicted in , which confirmed the conclusions made on the base of above-mentioned methods.
Figure 4. The X-ray molecular structure of compound 11f with the thermal ellipsoids at 50% probability level.
The crystal structure of compound 11f resembles that of compound 7a, since the core is planar (the maximal deviation of the atoms from the mean plane is 0.079 Å) and similar columnar supramolecular architecture represents the main packing motif. The molecules 11f are interacting within the column through the CH···O hydrogen-bonding donation to the oxygen atom O1 and π–π stacking interactions, as shown in . The ring-centroid distance between parallel packed aromatic systems exhibits the value of 3.757 Å and shift distance of 1.740 Å. No molecules of solvent are present in the crystal structure of compound 11f.
Figure 5. View of supramolecular columnar architecture in the crystal structure of 11f. Only H atoms involved in hydrogen bonding are shown. H-bonds parameters: C7–H⋯O1 [C7–H 0.93 Å, H…O1 2.48 Å, C7⋯O1(1 + x, y, z) 3.158(5) Å, C10–H⋯O3 129.8 °].
![Figure 5. View of supramolecular columnar architecture in the crystal structure of 11f. Only H atoms involved in hydrogen bonding are shown. H-bonds parameters: C7–H⋯O1 [C7–H 0.93 Å, H…O1 2.48 Å, C7⋯O1(1 + x, y, z) 3.158(5) Å, C10–H⋯O3 129.8 °].](/cms/asset/10234eb5-549d-46c0-adfe-8067ee6951e7/ienz_a_1039530_f0005_c.jpg)
Design and biological activity
We have recently discovered that 1-[2-(4-methoxyphenyl)-2-oxoethyl]-4-[[1-(ethoxycarbonyl)-3-benzoyl]-indolizin-7-yl]pyridinium bromide (Scheme 3) is a potent compound against both replicating and non-replicating M. tuberculosisCitation25. On the other hand, phenanthroline, azaindolizine and phenanthridine derivatives were successful identified as DNA intercalating agents or possessing antimycobacterial activityCitation26,Citation30,Citation40–43. In the same time, benzo[c]phenanthridinium alkaloids as fagaronine and nitidine which showed antileukemic activity on rodentsCitation44 and act as topoisomerase I and II inhibitorsCitation45–47, played the role of model compound for the development of new anticancer agentsCitation14. Having all these above consideration in mind and encouraged by our previous promising results in the field of anti-TBCitation25–28 and anticancerCitation29,Citation30,Citation32 derivatives with indolizine and/or phenanthroline skeleton, we have focused on the design of novel structures that contain four or five fused (hetero)cycles with pyrrolo[2,1-c][4,7]phenanthroline skeleton (Scheme 3). In equal measure, we were interested to see the influence of the substituents at pyrrolo ring (especially the pharmacophoric moiety p-chloro-benzoylCitation26) on the pharmacological properties, and if the increasing of number of fused rings from four to five will affect these properties (Scheme 3).
Scheme 3. Design in the class of pyrrolo[2,1-c][4,7]phenanthroline derivatives.
![Scheme 3. Design in the class of pyrrolo[2,1-c][4,7]phenanthroline derivatives.](/cms/asset/d518a9d2-430f-4ca0-8294-8e984b346505/ienz_a_1039530_sch0003_c.jpg)
The pyrrolo[2,1-c][4,7]phenanthroline derivatives 4 and 6 have been evaluated for in vitro antimycobacterial activity against M. tuberculosis H37Rv (grown under aerobic conditions), as a part of the TAACF TB screening program under direction of the US National Institute of Health, the NIAID division. Initially, the relative solubility of compounds in microbiological medium was measured using turbidity as a measure (). A serial dilution of compounds was prepared in DMSO and then transferred to microbiological medium at pH 6.8. Turbidity was measured and compared to a control; the lowest concentration at which compounds are insoluble (defined as turbidity > 300% of control) was recordedCitation48. The data from illustrate that all tested indolizine with phenanthroline skeleton have a good solubility in microbiological medium, which is quite promising from the pharmacological properties point of view. Then, the IC50, IC90 and MIC of compound were determined by measuring bacterial growth after 5 days in the presence of test compoundsCitation36–38,Citation49. The obtained results are listed in .
Table 1. Solubility in microbiological medium and antimycobacterial activity of compounds 4a–d and 6a–d against M. tuberculosis H37Rv under aerobic conditions.
As can be seen, five from the eight tested compounds (4a–d and 6a–d) had activity against M. tuberculosis H37Rv under aerobic conditions, with an IC50 in a range of 20–50 μM. We may also notice that the presence of p-chloro-benzoyl moiety at position 9 did not affect the antimycobacterial activity of compounds 4c and 6c comparing with the other derivatives type 4 and 6. Unfortunately, we are not able at the moment to comment the influence of the number of rings on the biological activity because the antimycobacterial evaluation of compounds 8, 9, 10 and 11 is underway.
Four indolizines with phenanthroline skeleton, 4a, 4c, 6c and 6d, were selected and tested in vitro for anticancer activity by the National Cancer Institute (NCI, USA), under the Developmental Therapeutics Program (DTP), at a single high dose (10−5 M) cell assay. This assay was performed in a 60 human tumor cell line panel, representing leukemia, melanoma and cancers of lung, colon, brain, breast, ovary, kidney and prostate, in accordance with the protocol of the NCI [US National Cancer Institute (NCI), Bethesda; http://dtp.nci.nih.gov/]Citation50–52. The results are expressed as “percentage growth inhibition” (PGI) term, and represent growth relative to the no-drug control, and relative to the time zero number of cells (). This allows detection of both growth inhibition (values between 0 and 100) and lethality (values less than 0)Citation51. Supplementary information concerning the in vitro anticancer assay could be found in a research overview by Michael R. BoydCitation52 and in the ESI of this article.
Table 2. Percentage growth inhibition (PGI, μM) data of compounds 4a, 4c, 6c and 6d against an NCI 60 human tumour cell lines (selection).
The results from indicate that pyrrolo [2,1-c][4,7]phenanthroline derivatives 6c and 6d exhibit a significant antitumor growth inhibitory activity (around 50%) against renal cancer (UO-31) and breast cancer (MCF7), respectively. We may also notice a weaker antitumor growth inhibitory activity (around 25%) of 6c against CNS Cancer (SF-SNB-19), 4a against Melanoma (M14) and 6d against Breast Cancer (T-47D).
As to the mechanism, taking into account, the above consideration concerning biological activity, as well as the fact that the X-ray structure of the new indolizines with phenanthroline skeleton show coplanar cores of these compounds, we assert that an interaction with DNA via an intercalation mechanism would be reasonable.
Conclusions
A new class of compounds with pyrrolo[2,1-c][4,7]phenanthroline skeleton has been synthesized by [3 + 2] dipolar cycloaddition of 4,7-phenanthrolin-4-ium ylides generated in situ from the corresponding monoquaternary salts, to activated symmetrical and unsymmetrical substituted alkynes or alkenes. The cycloaddition reactions to symmetrically substituted alkenes (N-phenylmaleimide) occur highly stereoselective (a single isomer being obtained), whereas the cycloadditions to unsymmetrically substituted dipolarophiles (acrylonitrile and ethyl propiolate) occur highly regioselective (a single regioisomer being formed), under charge control. The formation of aromatized and dihydro-fused polycyclic indolizines (in some cases) was explained by an oxidative dehydrogenation of intermediate tetrahydro-fused derivatives, process that leads to thermodynamically more stable compounds. The structure of the new compounds was proved by elemental and spectral analysis, including X-ray diffraction. The antimycobacterial activity of eight compounds was investigated against Mycobacterium tuberculosis H37Rv under aerobic conditions, five of the tested compounds showing moderate activity. Four new compounds have been evaluated by NCI for anticancer activity. Two of the compounds exhibiting a significant antitumor growth inhibitory activity (around 50%) against renal cancer (UO-31) and breast cancer (MCF7), respectively.
Supplementary material available online
Supplemental Tables S1 and S2.
Supplemental Material.pdf
Download PDF (778 KB)Acknowledgements
The authors gratefully acknowledge the National Institutes of Health and the National Institute of Allergy and Infectious Diseases (Contract No. HHSN272201100009I / HHSN27200001A08) for antimycobacterial tests and, to National Cancer Institute (NCI) for biological evaluation of compounds on their 60-cell panel: the testing was performed by the Developmental Therapeutics Program, Division of Cancer Treatment and Diagnosis (the URL to the Program's website: http://dtp.cancer.gov/). We also thank to the POSCCE-O 2.2.1, SMIS-CSNR 13984-901, Project no. 257/28.09.2010, CERNESIM, for the NMR experiments.
Declaration of interest
The authors report no conflict of interest. C. M. Al Matarneh is thankful for the financial support of grant POSDRU/159/1.5/S/137750, Project “Doctoral and Postdoctoral programs support for increased competitiveness in Exact Sciences research” co-financed by the European Social Found within the Sectorial Operational Program Human Resources Development 2007–2013.
References
- Sall C, Yapi A-D, Desbois N, et al. Design, synthesis, and biological activities of conformationally restricted analogs of primaquine with a 1,10-phenanthroline framework. Bioorg Med Chem Lett 2008;18:4666–9
- Nielsen MC, Larsen AF, Abdikadir FH, Ulven T. Phenanthroline-2,9-bistriazoles as selective G-quadruplex ligands. Eur J Med Chem 2014;72:119–26
- Wesselinova D, Neykov M, Kaloyanov N, et al. Antitumour activity of novel 1,10-phenanthroline and 5-amino-1,10-phenanthroline derivatives. Eur J Med Chem 2009;44:2720–3
- Accorsi G, Listorti A, Yoosaf K, Armaroli N. 1,10-Phenanthrolines: versatile building blocks for luminescent molecules, materials and metal complexes. Chem Soc Rev 2009;38:1690–700
- Kurth DG, Fromm KM, Lehn JM. Hydrogen-bonding and metal-ion-mediated self-assembly of a nanoporous crystal lattice. Eur J Inorg Chem 2001;6:1523–6
- Beauchamp DA, Loeb SJ. Hydrogen-bonded networks through second-sphere coordination. Chemistry 2002;8:5084–8
- Stępień BT, Krygowski TM, Cyrański MK, et al. How far is the π-electron delocalization of the phenanthrene moiety modified in the aza-analogues and their N-oxides? ARKIVOC 2004;3:185–201
- Pavlopoulos TG. Search for vibronic spin-orbit interactions in the triplet-manifold of phenanthrene N-heterocyclic analogues. J Photochem Photobiol A Chem 2002;149:45–54
- Bencini A, Lippolis V. 1,10-Phenanthroline: a versatile building block for the construction of ligands for various purposes. Coord Chem Rev 2010;254:2096–180
- Castedo L, Tojo G. Phenanthrene alkaloids. In: Brossi A, ed. The alkaloids. San Diego: Academic Press Inc; 1990:99–139
- Serbetci T, Genes C, Depauw S, et al. Synthesis and biological evaluation of dialkylaminoalkylamino benzo[c][1,7] and [1,8]phenanthrolines as antiproliferative agents. Eur J Med Chem 2010;45:2547–58
- Le TN, Gang SG, Cho W-J. A facile synthesis of benzo [c] phenanthridine alkaloids: oxynitidine and oxysanguinarine using lithiated toluamide–benzonitrile cycloaddition. Tetrahedron Lett 2004;45:2763–6
- Wang X-S, Yin M-Y, Wang S-L, et al. An efficient method for the synthesis of 4-arylfuro[2,3-a][4,7]phenanthroline derivatives catalyzed by iodine. J Heterocyclic Chem 2012;49:585–8
- Prado S, Michel S, Tillequin F, et al. Synthesis and cytotoxic activity of benzo[c][1,7] and [1,8]phenanthrolines analogues of nitidine and fagaronine. Bioorg Med Chem 2004;12:3943–53
- Kozlov NG, Tereshko AB, Gusak KN. Condensation of quinolin-5-amine with aromatic aldehydes and cyclohexane-1,3-dione. Russ J Org Chem 2007;43:1371–8
- Ishikawa T. Benzo[c]phenanthridine bases and their antituberculosis activity. Med Res Rev 2001;21:61–72
- Fleury F, Sukhanova A, Ianoul A, et al. Molecular determinants of site-specific inhibition of human DNA topoisomerase I by fagaronine and ethoxidine. J Biol Chem 2000;275:3501–9
- Chang Y-C, Hsieh P-W, Chang F-R, et al. Two new protopines argemexicaines A and B and the anti-HIV alkaloid 6-acetonyldihydrochelerythrine from formosan Argemone mexicana. Planta Med 2003;69:148–52
- Nakanishi T, Masuda A, Suwa M, et al. Synthesis of derivatives of NK109, 7-OH benzo[c]phenanthridine alkaloid, and evaluation of their cytotoxicities and reduction-resistant properties. Bioorg Med Chem 2000;10:2321–3
- Husseini R, Stretton RJ. Studies on the antibacterial activity of phanquone: effect on metabolic activities of Escherichia coli and Staphylococcus aureus. Microbios 1981;30:7–18
- Kradolfer F, Neipp L. Experimental studies on amebicidal, antibacterial, and antiparasitic phenanthroline compounds. Antibiot Chemother 1985;8:297–308
- Carta A, Loriga M, Paglietti G, et al. Design, synthesis, and preliminary in vitro and in silico antiviral activity of [4,7]phenantrolines and 1-oxo-1,4-dihydro-[4,7]phenantrolines against single-stranded positive-sense RNA genome viruses. Bioorg Med Chem 2007;15:1914–27
- Majumdar KJ, Ponra S, Ghosh T, et al. Synthesis of novel pyrano[3,2-f]quinoline, phenanthroline derivatives and studies of their interactions with proteins: an application in mammalian cell imaging. Eur J Med Chem 2014;71:306–15
- Strekowski L, Hojjat M, Wolinska E, et al. New triple-helix DNA stabilizing agents. Bioorg Med Chem Lett 2005;15:1097–100
- Danac R, Managalagiu II. Antimycobacterial activity of nitrogen heterocycles derivatives: bipyridine derivatives. Part III. Eur J Med Chem 2014;74:664–70
- Danac R, Daniloaia T, Antoci V, Mangalagiu II. Design, synthesis and antimycobacterial activity of some new azaheterocycles: phenanthroline with p-halogeno-benzoyl skeleton. Part V. Lett Drug Des Discov 2015;12:14–19
- Mantu D, Antoci V, Mangalagiu II. Design, synthesis and antimycobacterial activity of some new pyridazine derivatives: bis-pyridazine. Part IV. Infect Dis-Drug Targets 2013;13:344–51
- Mantu D, Luca C, Moldoveanu C, et al. Synthesis and antituberculosis activity of some new pyridazine derivatives. Part II. Eur J Med Chem 2010;45:5164–8
- Luca MC, Tura V, Mangalagiu II. Considerations concerning design and mechanism of action of a new class of anticancer dual DNA intercalators. Med Hyp 2010;75:627–9
- Antoci V, Mantu D, Cozma DG, et al. Hybrid anticancer 1,2-diazine derivatives with multiple mechanism of action. Part 3. Med Hyp 2010;82:11–15
- Balan AM, Miron A, Tuchilus C, et al. Synthesis and in vitro analysis of novel dihydroxyacetophenone derivatives with antimicrobial and antitumor activities. Med Chem 2014;10:476–83
- Zbancioc AM, Zbancioc Gh, Tanase C, et al. Design, synthesis and in vitro anticancer activity of a new class of bifunctional DNA intercalators. Lett Drug Des Discov 2010;7:644–9
- CrysAlis RED, Oxford Diffraction Ltd.,Version 1.171.36.32, 2003
- Dolomanov OV, Bourhis LJ, Gildea RJ, et al. OLEX2: a complete structure solution, refinement and analysis program. J Appl Cryst 2009;42:339–41
- Sheldrick GM. A short history of SHELX. Acta Cryst 2008;A64:112−22
- Ollinger J, Bailey MA, Moraski GC, et al. A dual read-out assay to evaluate the potency of compounds active against Mycobacterium tuberculosis. PLoS One 2013;8:e60531
- Zelmer A, Carroll P, Andreu N, et al. A new in vivo model to test anti-tuberculosis drugs using fluorescence imaging. J Antimicrob Chemother 2012;67:1948–60
- Lambert RJ, Pearson J. Susceptibility testing: accurate and reproducible minimum inhibitory concentration (MIC) and non-inhibitory concentration (NIC) values. J Appl Microbiol 2000;88:784–90
- Al Matarneh CM, Ciobanu C-I, Mangalagiu II, Danac R. Design, synthesis and antimycobacterial activity of some new azaheterocycles: 4,7-phenanthroline with p-halogeno-benzoyl skeleton. Part VI. submitted
- Rescifina A, Zagni C, Varrica MG, et al. Recent advances in small organic molecules as DNA intercalating agents: synthesis, activity, and modeling. Eur J Med Chem 2014;74:95–115
- Mantu D, Maftei D, Iurea D, et al. Synthesis, structure, and in vitro anticancer activity of new polycyclic 1,2-diazines. Med Chem Res 2014;23:2909–15
- Cappoen D, Jacobs J, Van TN, et al. Straightforward palladium-mediated synthesis and biological evaluation of benzo[j]phenanthridine-7,12-diones as anti-tuberculosis agents. Eur J Med Chem 2012;48:57–68
- Nagesh HN, Suresh N, Naidu KM, et al. Synthesis and evaluation of anti-tubercular activity of 6-(4-substitutedpiperazin-1-yl) phenanthridine analogues. Eur J Med Chem 2014;74:333–9
- Douros J, Suffness M. New natural products under development at the National Cancer Institute. Recent Results Cancer Res 1981;76:153–75
- Kragh Larsen A, Grondard L, Couprie J, et al. The antileukemic alkaloid fagaronine is an inhibitor of DNA topoisomerases I and II. Biochem Pharmacol 1993;46:1403–12
- Wang L-K, Johnson RK, Hecht SM. Inhibition of topoisomerase I function by nitidine and fagaronine. Chem Res Toxicol 1993;6:813–18
- Holden JA, Wall ME, Wani MC, Manikumar G. Human DNA topoisomerase I: quantitative analysis of the effects of camptothecin analogs and the benzophenanthridine alkaloids nitidine and 6-ethoxydihydronitidine on DNA topoisomerase I-induced DNA strand breakage. Arch Biochem Biophys 1999;370:66–76
- Bevan CD, Lloyd RS. A high-throughput screening method for the determination of aqueous drug solubility using laser nephelometry in microtiter plates. Anal Chem 2000;72:1781–7
- Carroll P, Schreuder LJ, Muwanguzi-Karugaba J, et al. Sensitive detection of gene expression in mycobacteria under replicating and non-replicating conditions using optimized Far-Red reporters. PLoS One 2010;5:e9823
- Shoemaker RH. The NCI60 human tumour cell line anticancer drug screen. Nat Rev 2006;6:813–23
- Monks A, Scudiero D, Skehan P, et al. Feasibility of a high-flux anticancer drug screen utilizing a diverse panel of human tumour cell lines in culture. J Natl Cancer Inst 1991;83:757–66
- Boyd RM, Paull KD, Some practical considerations and applications of the national cancer institute in vitro anticancer drug discovery screen. Drug Dev Res 1995;34:91–109