
Abstract
Azoles are a promising class of the new generation of HIV-1 non-nucleoside reverse transcriptase inhibitors (NNRTIs). From thousands of reported compounds, many possess the same basic structure of an aryl substituted azole ring linked by a thioglycolamide chain with another aromatic ring. In order to find novel extensions for this basic scaffold, we explored the 5-position substitution pattern of triazole NNRTIs using molecular docking followed by the synthesis of selected compounds. We found that heterocyclic substituents in the 5-position of the triazole ring are detrimental to the inhibitory activity of compounds with four-membered thioglycolamide linker and this substitution seems to be viable only for compounds with shorter two-membered linker. Promising compound, N-(4-carboxy-2-chlorophenyl)-2-((4-benzyl-5-methyl-4H-1,2,4-triazol-3-yl)sulfanyl)acetamide, with potent inhibitory activity and acceptable aqueous solubility has been identified in this study that could serve as lead scaffold for the development of novel water-soluble salts of triazole NNRTIs.
Introduction
Acquired immunodeficiency syndrome (AIDS) caused by the HIV virus is a major health problem of our civilization. Current treatment standard for HIV infection is to combine several drugs with a different mode of action, known as the highly active antiretroviral therapy (HAART)Citation1–3. One of the drug groups used in such combinations are the non-nucleoside reverse transcriptase inhibitors (NNRTIs)Citation4–7. These compounds upon binding to the reverse transcriptase (RT) stop the replication of the virus. A very high mutation rate of the HIV-1 virus causes development of mutants resistant to currently used NNRTIs such as nevirapine and efavirenzCitation8–10. Thus, there is a continuous need for new inhibitors that are effective on resistant mutants. A large number of NNRTIs belonging to chemically different groups has been reported in literature. An excellent review of various NNRTI groups was written by Zhan et al.Citation7 An interesting group of compounds are azole-based inhibitors with thioglycolamide linker, many of which possess a high activity, low toxicity and good resistance profileCitation11–20. In many such compounds, the azole ring is a 1,2,4-triazole.
Azole NNRTIs can be divided into three main groups: disubstituted azoles, where the substituents are aromatic rings linked with the azole by one-atom chain ()Citation21,Citation22; azoles with the thioglycolanilide moiety, commonly referred to as “azole thioacetanilides” ()Citation12–18; and other with miscellaneous structure, such as capravirine, lersivirine and 4-benzyl-3-(benzylsulfanyl)-5-(thiophen-2-yl)-4H-1,2,4-triazoles ()Citation11.
Figure 1. Three main groups of azole NNRTIs: aryl–CH2–(–NH–, –O–) disubstituted azoles (a), azole-thioglycolanilide derivatives (b) and other (c).
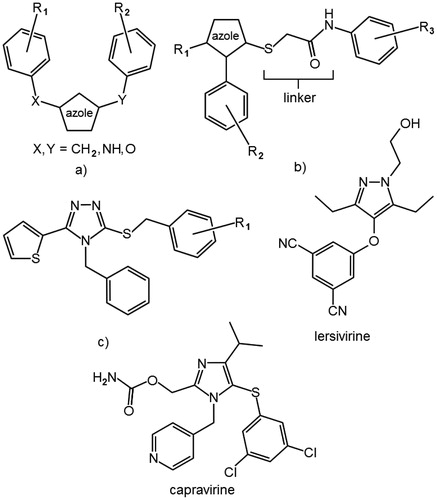
We are mainly interested in triazoles belonging to the second group. Triazoles in contrast to some other azoles such as tetrazoles or thiadiazoles can be triple substituted and therefore provide a more versatile core for NNRTIs. The RDEA806, a compound belonging to this group that completed phase 2a clinical trials, shows good activity against broad spectrum of resistant mutants, has good pharmacokinetics, and is well tolerated by patientsCitation19. A vast majority of reported azole NNRTIs with the thioglycolamide moiety differs only in the substitution of aromatic rings. Therefore, there is a lot of structure–activity relationship data available for the substituents in 3- and 4-position of the azole core, but very little is known about an effect on the activity of a substitution in 5-positionCitation12–18. Predominately, no or a small substituent like methyl is encountered in this position, and any larger group seems to reduce the inhibitory activityCitation12,Citation17. A crystallographic structure of 4-benzyl-3-(((2-chlorophenyl)methyl)sulfanyl)-5-(thiophen-2-yl)-4H-1,2,4-triazole ligated to HIV-1 RT was published (PDB code 2RKI). By examining this structure, it can be seen that the thiophene ring occupies a hydrophobic pocked formed by Val179, Ile180, Val189 (chain A) and Glu138 (chain B). This pocket in fact is an entrance to a partially closed tunnel that links NNRTIs binding site with the surface of the enzyme. By looking into other structures of NNRTI-reverse transcriptase complexes, such as PDB code 4O44, it can be seen that with a long substituent the tunnel can be opened, giving the possibility of additional van der Waals interactions, which can improve the ligand bindingCitation23. Molecular docking experiments suggested a similar binding mode of azoles with thioglycolamide linker and the ligand from the 2RKI crystallographic structure. We hypothesized that it would be possible to combine structural features of the two groups of compounds to obtain novel scaffolds. Therefore, the main goal of our study was to determine if extending the triazole NNRTIs by a larger substituent in the 5-position can open new possibilities for designing RT inhibitors. In this article, we report the results of these investigations that allow us to conclude that large five-membered heterocyclic substituents at the 5-position of the triazole core are incompatible with the thioglycolamide linker and such combinations should be avoided when designing novel inhibitors. A promising compound, N-(4-carboxy-2-chlorophenyl)-2-((4-benzyl-5-methyl-4H-1,2,4-triazol-3-yl)sulfanyl)acetamide, with potent inhibitory activity and acceptable aqueous solubility has been identified in this study that could serve as a scaffold for the development of novel water-soluble salts of triazole NNRTIs.
Methods
Chemistry
General comments
All reagents and solvents of analytical grade or higher were purchased from commercial sources and were used without purification unless otherwise stated. NMR spectra were recorded using Bruker Avance spectrometers (250, 300 and 700 MHz). Analytical thin layer chromatography was performed with Merck 60F254 silica gel plates (Darmstadt, Germany) and visualized by UV irradiation (254 nm). Melting points were determined on a Fischer–Johns block and are uncorrected. Elemental analyses were determined by an AMZ-CHX elemental analyzer (are within ± 0.5% of the theoretical values). Mass spectra (ESI TOF) were recorded using Waters LCT Premier XE mass spectrometer (Milford, MA).
General procedure for synthesis of 3,4-disubstituted-4,5-dihydro-1H-1,2,4-triazole-5-thiones 1a-3a, 5a, 6a, 8a, 9a, 11a, 12a, 29a and 31a
A solution of the corresponding thiosemicarbazide derivative (0.01 mol) in 2% sodium hydroxide (10 mL) was refluxed and progress of the reaction was monitored by thin layer chromatography. After 2 h, the reaction was completed and the reaction mixture was cooled and then acidified with 3 M hydrochloric acid. The precipitate was filtered, washed with water and crystallized from ethanol.
4-(2,4-dimethylphenyl)-3-(thiophen-2-yl)-4,5-dihydro-1H-1,2,4-triazole-5-thione 1a
(CAS 364596-54-3). Yield 89%, m.p. 254–256 °C, 1H NMR (300 MHz) (DMSO-d6) δ (ppm): 2.03 (s, 3H, CH3), 2.44 (s, 3H, CH3), 6.80–6.84 (m, 1H, Ar-H), 7.04–7.09 (m, 1H, Ar-H), 7.28 (s, 2H, Ar-H), 7.35 (s, 1H, Ar-H), 7.72–7.74 (d, 1H, Ar-H), 14.21 (s, 1H, NH).
4-(3-(trifluoromethyl)phenyl)-3-(thiophen-2-yl)-4,5-dihydro-1H-1,2,4-triazole-5-thione 2a
(CAS 690246-87-8) Yield 94%, m.p. 274–276 °C, 1H NMR (300 MHz) (DMSO-d6) δ (ppm): 6.83–6.85 (m, 1H, Ar-H), 7.06–7.10 (m, 1H, Ar-H), 7.74–7.77 (m, 1H, Ar-H), 7.88–7.90 (m, 2H, Ar-H), 8.03–8.08 (m, 2H, Ar-H), 14.28 (s, 1H, NH).
4-(4-methylphenyl)-3-(4-methyl-1,2,3-thiadiazol-5-yl)-4,5-dihydro-1H-1,2,4-triazole-5-thione 3a
Yield 88%, m.p. 111–113 °C, 2.34 (s, 3H, CH3), 3.71 (s, 3H, CH3), 7.09–7.20 (m, 2H, Ar-H), 7.23–7.45 (m, 2H, Ar-H), 14.51 (s, 1H, NH).
4-(4-methoxyphenyl)-3-(thiophen-2-yl)-4,5-dihydro-1H-1,2,4-triazole-5-thione 4a
(CAS 209670-96-2). Yield 91%, m.p. 263–265 °C, 3.82 (s, 3H, OCH3), 6.78–6.80 (m, 1H, Ar-H), 6.98–7.02 (m, 1H, Ar-H), 7.08–7.12 (m, 2H, Ar-H), 7.32–7.36 (m, 2H, Ar-H), 7.65–7.67 (m, 1H, Ar-H), 14.05 (s, 1H, NH).
4-(2,4-dichlorophenyl)-3-(4-methylimidazol-5-yl)-4,5-dihydro-1H-1,2,4-triazole-5-thione 5a
Yield 85%, m.p. 303–305 °C, 1H NMR (300 MHz) (DMSO-d6) δ (ppm): 2.36 (s, 3H, CH3), 7.45–7.55 (m, 3H, Ar-H), 7.76–7.77 (m, 1H, Ar-H), 12.11 (s, 1H, NH), 13.96 (s, 1H, NH).
4-(3-(trifluoromethyl)phenyl)-3-(4-methylimidazol-5-yl)-4,5-dihydro-1H-1,2,4-triazole- 5-thione 6a
Yield 80%, m.p. 308–310 °C, 1H NMR (300 MHz) (DMSO-d6) δ (ppm): 2.06 (s, 3H, CH3), 7.68–7.71 (m, 2H, Ar-H), 7.82–7.86 (m, 1H, Ar-H), 7.91 (s, 1H, Ar-H), 8.89 (s, 1H, Ar-H), 12.31 (s, 1H, NH), 14.56 (s, 1H, NH).
4-(4-methylphenyl)-3-(thiophen-2-yl)-4,5-dihydro-1H-1,2,4-triazole-5-thione 8a
(CAS 496787-40-7) Yield 91%, m.p. 286–288 °C, 1H NMR (300 MHz) (DMSO-d6) δ (ppm): 2.40 (s, 3H, CH3), 6.79–6.81 (m, 1H, Ar-H), 6.95–7.03 (m, 1H, Ar-H), 7.05–7.11 (m, 2H, Ar-H), 7.36–7.40 (m, 2H, Ar-H), 7.61–7.63 (m, 1H, Ar-H), 13.98 (s, 1H, NH).
3-(4-methyl-1,2,3-thiadiazol-5-yl)-4-(2,4-dimethylphenyl)-4,5-dihydro-1H-1,2,4-triazole-5-thione 11a
Yield 88%, m.p. 281–283 °C, 1H NMR (300 MHz) (DMSO-d6) δ (ppm): 1.95 (s, 3H, CH3), 2.41 (s, 3H, CH3), 2.91 (s, 3H, CH3), 7.24–7.27 (m, 2H, Ar-H), 7.32 (s, 1H, Ar-H), 14.64 (s, 1H, NH).
3-(4-methylimidazol-5-yl)-4-(2,4-dimethylphenyl)-4,5-dihydro-1H-1,2,4-triazole-5-thione 12a
Yield 77%, m.p. 200–202 °C, 1H NMR (300 MHz) (DMSO-d6) δ (ppm): 1.97 (s, 3H, CH3), 2.29 (s, 3H, CH3), 2.31 (s, 3H, CH3), 6.89–6.93 (m, 1H, Ar-H), 6.98–7.01 (m, 1H, Ar-H), 7.08 (s, 1H, Ar-H), 7.37 (s, 1H, Ar-H), 12.30 (s, 1H, NH), 13.89 (s, 1H, NH).
Physicochemical characterization of remaining 3,4-disubstituted-4,5-dihydro-1H- 1,2,4-triazole-5-thiones that were used as building blocks for synthesis final triazoles, that are 4-(4-methylphenyl)-3-(2-methylfuran-3-yl)-4,5-dihydro-1H-1,2,4-triazole-5-thione (9a), 3-methyl-4-(2,4-dimethylphenyl)-4,5-dihydro-1H-1,2,4-triazole-5-thione (29a), 4-benzyl-3-(thiophen-2-yl)-4,5-dihydro-1H-1,2,4-triazole-5-thione (31a), were reported elsewhereCitation12,Citation24–26.
Synthesis of 2-bromo-N-(2-chloro-4-sulfamoylphenyl)acetamide
Fifteen milligrams (1 equivalent) of 4-amino-3-chlorobenzene-1-sulfonamide (Sigma-Aldrich, Poznan, Poland) was dissolved in 2 ml of acetonitrile and cooled down to 0 °C. Thirty milligrams of potassium carbonate (3 equivalents) was added and 9.5 μl of freshly distilled bromoacetyl bromide (1.5 equivalent, Sigma-Aldrich) was added dropwise for 1–3 min. After complete addition, the reaction mixture was stirred for around 10 min (controlled by TLC, chloroform–methanol 29:1). Reaction mixture was centrifuged and the precipitate was washed with acetonitrile and discarded. Combined solutions were concentrated under reduced pressure. Addition of methylene chloride caused most of the product to precipitate and n-pentane addition further increased the yield. The product was centrifuged, washed thrice with n-pentane and dried. Yield 98%; m.p. 150–152 °C; 1H NMR (250 MHz, acetone-d6) δ (ppm): 4.36 (s, 2H, CH2), 6.79 (br s, 2H, NH2), 7.93 (dd, J = 8.7, 2.1 Hz, 1H), 8.02 (d, J = 2.1 Hz, 1H), 8,46 (dd, J = 8.7, 2.6 Hz, 1H), 9.40 (br s, 1H, NH).
Synthesis of 2-bromo-N-(2-nitrophenyl)acetamide
The compound was synthesized from o-nitroaniline according to the same procedure as for 2-bromo-N-(2-chloro-4-sulfamoylphenyl)acetamide using methylene chloride as a solvent. After the reaction was completed, the precipitate was centrifuged, washed with methylene chloride and discarded. Combined extracts were concentrated under reduced pressure and the product was precipitated with n-heptane, centrifuged, washed thrice with n-pentane and dried. Physicochemical characterization of the compound was reported elsewhereCitation27,Citation28.
Synthesis of 2-bromo-N-(4-carboxy-2-chlorophenyl)acetamide
Thirty-nine milligrams of 4-amino-3-chlorobenzoic acid (Sigma-Aldrich) was suspended in 2 ml of methylene chloride, 40 μl of diisopropylethylamine was added and the mixture was cooled in an ice bath. Twenty microliters of distilled bromoacetyl bromide was added slowly. The reaction mixture was stirred for 30 min controlled by TLC. The reaction mixture was diluted with methylene chloride, extracted thrice with water and evaporated. The residue was washed once again with water, dried under vacuum and dissolved in methylene chloride. The product was precipitated with n-pentane, washed with n-pentane and dried. Yield 60%, m.p. 200–202 °C. 1H NMR (250 MHz, acetone-d6) δ (ppm): 4.21 (s, 2H), 7.87 (dd, J = 8.6, 1.8, 1H), 7.95 (d, J = 1.8, 1H), 7.99 (dd, J = 8.6, 4.0, 1H), 10.05 (br s, 1H, OH).
General procedure for synthesis of compounds 1–12, 28–31 and 34
One equivalent of the corresponding 3,4-disubstituted-4,5-dihydro-1H-1,2,4-triazole-5-thione and 1.7 mg of potassium iodide (or tetrabutylammonium iodide) (0.3 eq.) were dissolved/suspended in 2 ml of methanol. 5.2 mg of K2CO3 (1.1 eq.) was added and the mixture was heated to 60 °C under reflux for 20 min. After cooling to < 30 °C, 1.2 equivalent of the corresponding 2-bromo-N-arylacetamide or benzyl bromide (Merck) was added and the reaction mixture was stirred for 1–2 h, monitored by TLC. The reaction mixture was filtered, evaporated, dissolved in methylene chloride (in case of compounds with carboxyl group the dry residue was dissolved in water, acidified with diluted hydrochloric acid and extracted with methylene chloride), filtered, concentrated and layered with n-heptane. The product precipitated or crystallized within 24 h was washed with n-heptane and dried. If product separated as an oil, addition of 1–2 drops of methylene chloride often caused solidification.
N-(2-nitrophenyl)-2-((4-(2,4-dimethylphenyl)-5-(thiophen-2-yl)-4H-1,2,4-triazol-3-yl)sulfanyl)acetamide 1
m.p. – (amorphous); 1H NMR (250 MHz, acetone-d6) δ (ppm): 1.96 (s, 3H, CH3), 2.46 (s, 3H, CH3), 4.29 (ABq, ΔδAB = 0.04, J = 15.6 Hz, 2H, CH2), 6.79 (dd, J = 3.6, 1.1 Hz, 1H), 7.07 (m, 1H), 7.33 (m, 1H), 7.41–7.48 (m, 2H), 7.70 (dd, J = 5.2, 1.2 Hz, 1H), 7.78–7.88 (m, 2H), 8.06 (m, 1H), 10.81 (br s, 1H, NH).
N-(2-nitrophenyl)-2-((5-(thiophen-2-yl)-4-(3-(trifluoromethyl)phenyl)-4H-1,2,4-triazol-3-yl)sulfanyl)acetamide 2
m.p. 187–189 °C; 1H NMR (250 MHz, acetone-d6) δ (ppm): 4.14 (s, 2H, CH2), 6.71 (dd, J = 3.7, 1.2 Hz, 1H), 6.86 (m, 1H), 7.22 (m, 1H), 7.44 (dd, J = 5.1, 1.2 Hz, 1H), 7.64 (m, 1H), 7.83 (m, 2H), 7.97 (m, 2H), 8.05 (dd, J = 8.4, 1.5 Hz, 1H), 8.41 (dd, J = 8.4, 1.4 Hz, 1H), 10.78 (br s, 1H, NH).
N-(2-nitrophenyl)-2-((4-(4-methylphenyl)-5-(4-methyl-1,2,3-thiadiazol-5-yl)-4H-1,2,4-triazol-3-yl)sulfanyl)acetamide 3
m.p. 181–182 °C; 1H NMR (250 MHz, acetone-d6) δ (ppm): 2.41 (s, 3H, CH3), 2.72 (s, 3H, CH3), 4.22 (s, 2H, CH2), 7.26 (m, 1H), 7.39 (m, 4H), 7.68 (m, 1H), 8.08 (dd, J = 8.5, 1.7 Hz, 1H), 8.46 (dd, J = 8.5, 1.2 Hz, 1H), 10.76 (br s, 1H, NH).
N-(2-chloro-4-sulfamoylphenyl))-2-((5-(thiophen-2-yl)-4-(4-methoxyphenyl)-4H-1,2,4-triazol-3-yl)sulfanyl)acetamide 4
m.p. 193–194 °C; 1H NMR (250 MHz, acetone-d6) δ (ppm): 3.95 (s, 3H, CH3), 4.16 (s, 2H, CH2), 5.78 (br s, 2H, NH2), 6.93 (m, 1H), 7.05 (m, 1H), 7.19 (m, 2H), 7.45 (m, 2H), 7.55 (m, 1H), 7.86 (dd, J = 8.9, 2.2, 1H), 8.00 (d, J = 2.2 Hz, 1H), 8.56 (d, J = 8.9 Hz, 1H), 9.96 (s, 1H, NH).
N-(2-nitrophenyl)-2-((4-(2,4-dichlorophenyl)-5-(4-methylimidazol-5-yl)-4H-1,2,4-triazol-3-yl)sulfanyl)acetamide 5
m.p. 246–248 °C; 1H NMR (250 MHz, acetone-d6) δ (ppm): 2.50 (s, 3H, CH3), 4.06 (ABq, ΔδAB = 0.06, J = 15.9 Hz, 2H, CH2), 7.20 (s, 1H), 7.28 (m, 1H), 7.46 (m, 2H), 7.64 (m, 1H), 7.71 (m, 1H), 8.14 (dd, J = 8.5, 1.5 Hz, 1H), 8.47 (dd, J = 8.5, 1.2 Hz, 1H), 10.25 (br s, 1H, NH), 10.72 (br s, 1H, NH).
N-(2-nitrophenyl)-2-((4-(3-(trifluoromethyl)phenyl)-5-(4-methylimidazol-5-yl)-4H-1,2,4-triazol-3-yl)sulfanyl)acetamide 6
m.p. 190–192 °C; 1H NMR (250 MHz, acetone-d6) δ (ppm): 2.15 (s, 3H, CH3), 4.32 (s, 2H, CH2), 7.46 (m, 1H), 7.82 (m, 4H), 7.99–8.07 (m, 3H), 8.67 (br s, 1H), 10.83 (s, 1H, NH).
N-(2-nitrophenyl)-2-((4-(4-methoxyphenyl)-5-(thiophen-2-yl)-4H-1,2,4-triazol-3-yl)-sulfanyl)acetamide 7
m.p. 137–138 °C; 1H NMR (250 MHz, acetone-d6) δ (ppm): 4.01 (s, 3H, CH3), 4.30 (s, 2H, CH2), 6.92 (dd, J = 3.8, 1.3 Hz, 1H), 7.05 (m, 1H), 7.27 (m, 2H), 7.42 (m, 1H), 7.56–7.61 (m, 3H), 7.84 (m, 1H), 8.25 (dd, J = 8.4, 1.6 Hz, 1H), 8.63 (dd, J = 8.5, 1.3 Hz, 1H), 11.00 (br s, 1H, NH).
N-(2-nitrophenyl)-2-((4-(4-methylphenyl)-5-(thiophen-2-yl)-4H-1,2,4-triazol-3-yl)-sulfanyl)acetamide 8
m.p. 151–152 °C; 1H NMR (250 MHz, CDCl3) δ (ppm): 2.39 (s, 3H, CH3), 4.09 (s, 2H, CH2), 6.81 (s, 1H), 6.82 (d, J = 0.7 Hz, 1H), 7.10 (m, 1H), 7.19–7.31 (m, 5H), 7.55 (m, 1H), 8.08 (dd, J = 8.5, 1.5 Hz, 1H), 8.56 (dd, J = 8.5, 1.3 Hz, 1H), 11.11 (br s, 1H, NH).
N-(2-nitrophenyl)-2-((4-(4-methylphenyl)-5-(2-methylfuran-3-yl)-4H-1,2,4-triazol-3-yl)-sulfanyl)acetamide 9
m.p. 170–172 °C; 1H NMR (250 MHz, DMSO-d6) δ (ppm): 2.45 (s, 3H, CH3), 2.49 (s, 3H, CH3), 4.24 (s, 2H, CH2), 5.75 (d, J = 2.1 Hz, 1H), 7.48–7.35 (m, 5H), 7.51 (d, J = 2.1 Hz, 1H), 7.80 (m, 1H), 7.89 (m, 1H), 8.07 (dd, J = 8.3, 1.2 Hz, 1H), 10.80 (s, 1H, NH).
N-(2-chloro-4-sulfamoylphenyl)-2-((4-(2,4-dimethylphenyl)-5-(thiophen-2-yl)-4H-1,2,4-triazol-3-yl)sulfanyl)acetamide 10
m.p. 130–133 °C; 1H NMR (700 MHz, DMSO-d6) δ (ppm): 1.91 (s, 3H, CH3), 2.40 (s, 3H, CH3), 4.30 (ABq, ΔδAB = 0.05, J = 15.4 Hz, 2H, CH2), 6.75 (dd, J = 3.7, 1.2 Hz, 1H), 7.01 (dd, J = 5.1, 3.7 Hz, 1H), 7.26 (d, J = 7.9 Hz, 1H), 7.35 (m, 2H), 7.45 (s, 2H, NH2), 7.64 (dd, J = 5.1, 1.2 Hz, 1H), 7.76 (dd, J = 8.7, 2.2 Hz, 1H), 7.90 (d, J = 2.2 Hz, 1H), 8.07 (d, J = 8.7 Hz, 1H), 10.16 (s, 1H, NH).
N-(2-chloro-4-sulfamoylphenyl)-2-((4-(2,4-dimethylphenyl)-5-(4-methyl-1,2,3-thiadiazol-5-yl)-4H-1,2,4-triazol-3-yl)sulfanyl)acetamide 11
m.p. 199–201 °C; 1H NMR (700 MHz, DMSO-d6) δ (ppm): 1.88 (s, 3H, CH3), 2.43 (s, 3H, CH3), 3.01 (s, 3H, CH3), 4.40 (ABq, ΔδAB = 0.07, J = 15.4 Hz, 2H, CH2), 7.31 (d, J = 8.5 Hz, 1H), 7.39 (m, 2H), 7.46 (s, 2H, NH2), 7.76 (dd, J = 8.5, 2.2 Hz, 1H), 7.90 (d, 2.2 Hz, 1H), 8.06 (d, J = 8.5 Hz, 1H), 10.18 (s, 1H, NH).
N-(2-chloro-4-sulfamoylphenyl)-2-((4-(2,4-dimethylphenyl)-5-(4-methyl-imidazol-5-yl- 4H-1,2,4-triazol-3-yl)sulfanyl)acetamide 12
1H NMR (700 MHz, DMSO-d6) δ (ppm): 1.90 (s, 3H, CH3), 2.33 (s, 3H, CH3), 2.41 (s, 3H, CH3), 4.21 (ABq, ΔδAB = 0.04, J = 15.3 Hz, 2H, CH2), 7.07 (m, 2H), 7.16 (s, 1H), 7.37 (s, 1H), 7.42 (br s, 2H, NH2), 7.73 (d, J = 8.3 Hz, 1H), 7.87 (s, 1H), 8.09 (d, J = 8.3 Hz, 1H), 8.53 (s, 1H, NH), 12.03 (br s, 1H, NH).
N-(4-carboxy-2-chlorophenyl)-2-((4-(2,4-dimethylphenyl)-5-(4-methyl-1,2,3-thiadiazol-5-yl)-4H-1,2,4-triazol-3-yl)sulfanyl)acetamide 28
m.p. 189–190 °C (potassium salt); 1H NMR (250 MHz, CD3OD) δ (ppm): 1.91 (s, 3H, CH3), 2.46 (s, 3H, CH3), 3.03 (s, 3H, CH3), 7.22–7.37 (m, 3H), 7.84 (dd, J = 8.5, 1.9 Hz, 1H), 7.97–8.04 (m, 2H).
N-(2-chloro-4-sulfamoylphenyl)-2-((4-(2,4-dimethylphenyl)-5-methyl-)-4H-1,2,4-triazol-3-yl)sulfanyl)acetamide 29
1H NMR (250 MHz, acetone-d6) δ (ppm): 10.28 (s, 1H, NH), 8.39 (d, J = 8.7 Hz, 1H), 7.79 (d, J = 2.1 Hz, 1H), 7.69 (dd, J = 8.7, 2.1 Hz, 1H), 7.19 (s, 1H), 7.12 (m, 2H), 6.57 (br s, 2H, NH2), 4.01 (s, 1H, CH2), 2.27 (s, 3H, CH3), 2.04 (s, 3H, CH3), 1.88 (s, 3H, CH3).
N-(4-carboxy-2-chlorophenyl)-2-((4-(2,4-dimethylphenyl)-5-methyl-)-4H-1,2,4-triazol-3-yl)sulfanyl)acetamide 30
m.p.173–175 °C (potassium salt);1H NMR (250 MHz, CD3OD) δ (ppm): 8.02 (d, J = 8.5 Hz, 1H), 7.98 (d, J = 1.8 Hz, 1H), 7.84 (dd, J = 8.5, 1.8 Hz, 1H), 7.27 (s, 1H), 7.21–7.11 (m, 2H), 2.38 (s, 3H, CH3), 2.19 (s, 3H, CH3), 2.0 (s, 3H, CH3).
N-(4-carboxy-2-chlorophenyl)-2-((4-benzyl-5-(thiophen-2-yl)-4H-1,2,4-triazol-3-yl)-sulfanyl)acetamide 31
m.p. 157–160 °C (potassium salt);1H NMR (250 MHz, CD3OD) δ (ppm): 4.17 (s, 2H, CH2), 5.5 (s, 2H, CH2), 7.0–7.38 (m, 7H), 7.65 (d, J = 5.2 Hz, 1H), 7.8–8.04 (m, 3H).
3-(benzylsulfanyl)-4-(2,4-dimethylphenyl)-5-methyl-4H-1,2,4-triazole 34
m.p. – (oil). 1H NMR (250 MHz, acetone-d6) δ (ppm): 1.84 (s, 3H, CH3), 2.09 (s, 3H, CH3), 2.36 (s, 3H, CH3), 4.37 (ABq, ΔδAB = 0.05, J = 13 Hz, 2H, CH2), 6.99 (d, J = 8.0 Hz, 1H), 7.13–7.38 (m, 7H).
Synthesis of N-(4-carboxy-2-chlorophenyl)-2-((4-benzyl-5-methyl-4H-1,2,4-triazol-3-yl)sulfanyl)acetamide 32
Seven milligrams of 4-benzyl-3-methyl-1,2,4-triazole-5-thione and 1.7 mg of potassium iodide were dissolved in 2 ml of methanol. 5.2 mg of potassium carbonate was added and the mixture was heated to 60 °C under reflux for 20 min. After cooling to ∼25 °C, 10 mg of 4-(2-bromoacetamido)-3-chlorobenzoic acid was added and the reaction mixture was stirred for 60 min, monitored by TLC (chloroform–methanol 29:1). The reaction mixture was filtered, evaporated under reduced pressure, dissolved in water and acidified with 0.5 M HCl. The solution was extracted several times with methylene chloride. The extract was concentrated to 1–2 ml and layered with several milliliters of n-heptane. Product separated as a white precipitate or small crystals. Yield 60%. A potassium salt of 32 was prepared by dissolving the product in methanol, adding 0.5 equivalent of potassium carbonate and stirring for 30 min; m.p. 165–167 °C (potassium salt); 1H NMR (700 MHz, CD3OD) δ (ppm): 2.38 (s, 3H, CH3), 4.11 (s, 2H, CH2), 5.27 (s, 2H, CH2), 7.14 (d, J = 7.4 Hz, 2H), 7.29–7.35 (m, 3H), 7.87 (dd, J = 8.5, 1.9 Hz, 1H), 7.99 (s, 1H), 8.00 (d, J = 1.8 Hz, 1H); 13C NMR (175 MHz, CD3OD) δ (ppm): 9.6, 37.1, 122.8, 124.6, 126.5, 128, 128.8, 130.1, 134.6, 134.7, 136.2, 150.3, 154, 167.3, 170.8; HRMS (ESI): m/z 417.0882 [M+H]+ (Calculated for C19H18N4O3SCl : 417.0788).
Enzymatic assay
Nevirapine (>98%) was purchased from Tokyo Chemical Industry, Japan. Reverse transcriptase assay kits were purchased from Roche (Warsaw, Poland) and Innovagen (Lund, Sweden).
The enzymatic assay using Roche kit was performed according to the manufacturer's instruction with the only modification that 2 ng (instead of suggested 4–6 ng which caused the reaction to run too fast) of supplied enzyme was used for each reaction. The compounds were prepared in concentrations ranging from 0.1 to 100 μM using a stock solution containing 5% DMSO. IC50 values was obtained from an interpolation of the inhibition curve and averaged from three separate measurements.
Surface plasmon resonance
Surface plasmon resonance (SPR) measurements were performed using Biacore3000 according to the published methodCitation29. Wild-type HIV-RT (Worthington) was immobilized on CM5 sensor chip via amine group coupling. Examined compounds were used in concentrations from 0.315 to 10 µM or 1.875 to 60 µM (for nevirapine). Sample injection time was 60 s. After 180 s of dissociation, the sensor surface was washed with HBS-EP buffer with 20% of DMSO. Obtained data was analyzed with BIAevaluation program assuming three possible binding models: single-step binding [Langmuir binding, Equation (1)], heterogenous binding [Equation (2)] and induced-fit [Equation (3)]Citation29,Citation30.
(1)
(2)
(3)
Molecular modeling
The docking study was performed using Glide SP and XP protocols using the same methodology as described earlierCitation31–33. Receptor grids were prepared from 2RKI and 3DLG crystallographic structures. Grid size was assigned automatically, van der Waals radii of atoms with the absolute value of partial charge less than 0.2 were scaled by 0.9. The docking was performed using default settings, three best poses were saved. All the returned ligand poses were inspected manually and those with unlikely binding mode were removed. Only for 2RKI-based receptor, Glide SP and XP returned at least one plausible pose for all (or almost all) of the compounds.
Results and discussion
Initially, the molecular docking study was performed to identify which of the available building blocks should be used for the synthesis of triazole NNRTIs. Our previous docking study of triazole/azole NNRTIs and HIV reverse transcriptase allowed us to choose the most reliable docking protocols for both pose prediction and scoringCitation31. From many available crystallographic structures of RT, the best results were achieved for 3DLG and 2RKI. The latter structure is the only 1,2,4-triazole-RT complex deposited in PDB to date, and receptors based on this structure give the most consistent ligand poses for majority of different azole NNRTIs. Another conclusion from our previous study was that combination of molecular docking with appropriate scoring functions can be used to select a smaller group of compounds from a larger ensemble of potential inhibitorsCitation31. In this study, Glide SP gave more reproducible and consistent ligand poses while Glide XP failed to return a valid pose in some cases. The first batch of nine compounds was selected from triazoles available in our laboratory based on their docking scores obtained for Glide SP protocol in 2RKI receptor. Their structure and docking scores are shown in .
Table 1. Considered R1, R2 and R3 substituents for triazole NNRTIs and selected results for Glide SP protocol in 2RKI receptor.
Subsequently, triazoles 1–9 were tested for their inhibitory activity against HIV-1 reverse transcriptase using RetroSys™ RT Activity Kit (Innovagen). This screening gave the results shown in . As can be seen, the highest inhibitory activity was shown by 1, 2, 3 and 4 with IC50 in the range of 6–15 µM. Another compound 5 was active at much higher concentration (IC50 at 43.5 µM) while remaining compounds did not show any significant inhibitory activity (IC50 > 100 µM).
Table 2. Inhibitory activity (in µM) of triazoles 1–9 against HIV-1 RT (wild type, Innovagen kit).
Based on these results, it was possible to draw the following conclusions, which were utilized in the design of the next series of inhibitors: (i) addition of the 2-methyl group into the 4-methylphenyl substituent at the 4-position of triazole resulted in an improved inhibitory activity. This effect may result from increased van der Waals interactions, as predicted by the docking protocol where the 2-methyl substituted analogs scored on average 1.45 lower evdw value (van der Waals interaction component of the scoring function); (ii) the 2-chloro-4-sulfamoylphenyl group at the thioglycolamide linker is better than the 2-nitrophenyl. This observation is also in agreement with docking results; compounds with the 2-chloro-4-sulfamoylphenyl substituent had the gscore and emodel on average 1.35 and 13.11 lower, respectively, than 2-nitrophenyl substituted analogs. Another important observation during the enzymatic study was that the compounds with the 2-nitrophenyl substitution exhibited poor solubility under experimental protocol. Also, there are indications that o-nitroaniline derivatives are metabolically unstable and show extremely rapid serum clearanceCitation13. Changing the 2-nitrophenyl group into the 2-chloro-4-sulfamoylphenyl resulted in the more active compounds. In addition, introducing the 4-carboxy-2-chlorophenyl allowed to form water-soluble salts of the inhibitors, hence compounds with the 2-nitrophenyl substituent were excluded from our further studies. Thus, the remaining substituent to be optimized is the one in the position 5 of the triazole core. We, therefore, repeated the docking protocol and the results for 5-position substituted variants are shown in (compounds 10–28). For comparison 5-methyl substituted triazoles 29 and 30 were also included, however these compounds do not present any novelty as they were already included in a patentCitation34 as potential antiviral agents.
Table 3. Structures of the 5-position substitution variants used for docking experiments and selected results for Glide SP protocol in 2RKI receptor.
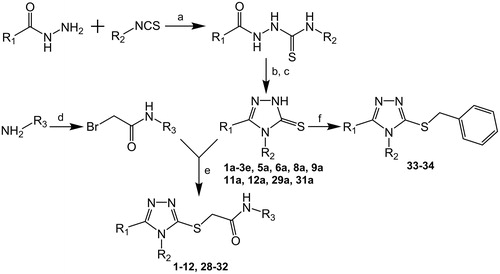
According to the scoring function values, piperidine, thiophene, imidazole, triazole and thiadiazole groups at the 5-position of triazole core were selected as the most promising substituents. It should be noted that thiophen-2-yl, 4-methyl-1,2,3-thiadiazol-5-yl and 4-methylimidazol-5-yl groups obtained better scores than a methyl substituent (compounds 10–12 versus 29), which is most likely a result of larger hydrophobic surface area. The 1,2,4-triazol-3-yl, pyrrol-2-yl and pyrazin-2-yl substituents received similar docking scores, however the latter two groups showed much worse van der Waals interactions (evdw −43.45 and −45.76 for pyrrol-2-yl and pyrazin-2-yl, respectively, versus −50.15 for the 1,2,4-triazol-3-yl), thus the 1,2,4-triazol-3-yl substituent was suspected to be a better option. Based on these docking results, the compounds designed in as 10, 11, 12, 14 and 28 were selected for synthesis. General synthetic route is showed in Scheme 1. As the graph indicates, an appropriate five-membered heteroaryl carboxylic acid hydrazide was reacted with aryl isothiocyanate to form a thiosemicarbazide intermediate, which was cyclized by an action of a base. After an acidification, 3,4-disubstituted-4,5-dihydro-1H-1,2,4-triazole-5-thione was obtained. In turn, an appropriate aniline derivative was acylated by an action of 2-bromoacetyl bromide in the presence of a base. The obtained 2-bromo-N-aryl acetamide was reacted with the 3,4-disubstituted-4,5-dihydro-1H-1,2,4-triazole-5-thione to form final compounds 10, 11, 12 and 28. Unfortunately, compound 14 could not be obtained under the synthetic protocol due to the formation of an unidentified derivative with dimethylphenyl attached to the second triazole ring in position 5.
Scheme 1. General synthetic route and structures of examined compoundsa. R1, R2 and R3 substituents are presented in and . aReagents and conditions: (a) diethyl ether, 48 h, rt. (b) 2% NaOH, reflux, 2h. (c) 3M HCl, rt. (d) BrCH2COBr, K2CO3, acetonitrile, 0 °C. (e) K2CO3, KI, methanol, 1–2 h, rt. (f) PhCH2Br, K2CO3, KI, methanol, 1–2 h, rt.
Subsequently, compounds 10, 11, 12 and 28 were submitted for an enzymatic assay. The HIV-1 RT inhibitory activity was measured using the more convenient colorimetric Reverse Transcriptase Assay Kit from Roche. The compounds were used in concentrations ranging from 0.1 to 100 μM. As mentioned before, the modeling study indicated that heterocyclic substituents at the 5-position of triazole should increase favorable van der Waals interactions and result in compounds that bind stronger to the enzyme than 5-methyl analogues 29 and 30. Results from the enzymatic experiments, however, show that large substituents at this position are detrimental to the activity. As indicates that the IC50 values of 10, 11 and 28 were estimated at 76, 53 and 45.8 μM, respectively, while 12 did not show any significant inhibitory activity against HIV-1 RT (IC50 > 100 μM).
Table 4. Inhibitory activity (in µM) of triazoles 10–12, 28–34 against HIV-1 RT (wild type, Roche kit).
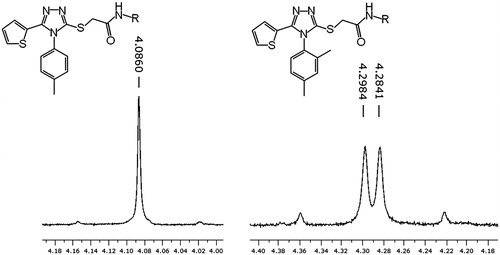
To explain the discrepancy between experimental and computational results several possibilities were considered. The simplest explanation would be an inaccuracy of the employed computational methodology, for example inadequate modeling of protein flexibility. HIV reverse transcriptase is known to be a flexible protein and movements of its domains are crucial for its enzymatic activity and NNRTIs act by locking the so-called thumb domain in an inactive positionCitation35. Modeling protein flexibility is a complex problem and can be done with molecular docking only to a limited extent. Another explanation is the fact that NNRTIs act as allosteric inhibitors of the reverse transcriptase, therefore binding with the enzyme is not necessarily correlated with its inhibition. We might be able to observe correlation between inhibitory activity and binding free energy for a series of compounds provided that they bind to the same conformation of the enzyme. In our case, the analyzed compounds had significant differences in substituents size therefore it is unlikely that all of the compounds bound to the same conformation of the enzyme. Unfortunately, to the best of our knowledge, there is no model that can quantitatively correlate enzymatic activity of HIV reverse transcriptase with its conformation. We also suspected that the negative effect on the inhibitory activity of a large, five-membered heterocyclic substituent in the 5-position of a triazole results from an increased rigidity of the core aromatic ring system. The phenyl ring in the 4-position and a heteroaryl ring in the 5-position of the triazole affect each other's rotations. The rigidity of the compounds is especially visible for compounds with the 2,4-dimethylphenyl substitution. This group prevents a full rotation of the phenyl ring which possibly results in formation of two atropisomers. The existence of atropisomers is suggested by 1H-NMR spectra, where the signal for methylene group of thioglycolamide linker changes from a singlet to an AB pattern when the 2,4-dimethylphenyl is present ().
Figure 2. 1H-NMR spectra (250 MHz) showing the splitting of the signal of methylene protons resulting from constraining the phenyl group rotation by the 2-methyl substituent.
To test whether the core rigidity was responsible for the decreased activity, two compounds with the benzyl group at the 4-position and the thiophene ring (31) or the methyl group (32) at the 5-position were prepared and tested. Compound 31 with IC50 of 18.5 µM was found to be more active than any other compound with a heteroaryl ring at the 5-position and the thioglycolamide linker (10, 11, 12, 28, IC50 at 45.8 µM or higher), but less active than the known inhibitor 33Citation11 (Scheme 1) which has the thiophene ring at the 5-position but also has a shorter sulfanylmethyl linker (IC50 = 3.3 µM). In turn, substituting the thiophene ring in 31 with the methyl group resulted in somewhat more active compound 32 (IC50 = 7.1 µM). This compound, however, was still less active than its analogue 30 with the 2,4-dimethylphenyl group at the 4-position (IC50 = 0.3 µM). Finally, both compound 34 (IC50 ∼100 µM) and 4-benzyl-3-benzylsulfanyl-5-methyl-4H-1,2,4-triazole described earlier in literatureCitation11 (EC50 23.5 µM) which are 5-methyl substituted and have the sulfanylmethyl linker, exhibited weaker activity. All these observations are consistent and lead to the following conclusions. Increasing the size of a substituent at the 5-position is beneficial for the activity only if a short linker such as the sulfanylmethyl is present, and there are no steric clashes with the substituent at the 4-position. For triazoles with longer linker such as the thioglycolamide linker, small substituents at the 5-position, like a methyl, seem to be optimal. Larger substituents are incompatible with the thioglycolamide linker and such combinations should be avoided when designing novel inhibitors.
Finally, to shed some light on the interaction between the most potent inhibitor found in our studies 32 and HIV-1 reverse transcriptase, we set up an SPR biosensor assay. For comparison, two other inhibitors 10 (highest docking score) and 28 (highest inhibitory activity from the compounds with a heterocycle in 5-position and the thioglycolamide linker) were also included in the assay. It was reported that SPR biosensor screening methodology provides an efficient way to examine binding kinetics for small molecules binding to HIV-1 RTCitation29. Examined compounds were used in concentrations from 0.315 to 10 µM and 1.875 to 60 µM for nevirapine. As can be seen from results collected in , the binding of examined compounds is best described by the simplest one-step model. KD values for all compounds including nevirapine are of the similar order, but there is no correlation with RT inhibition activity. However, 10 which showed the lowest KD had also the best docking score. As both the molecular modeling and SPR measurements indicate that 10 binds well to the enzyme, it suggests that the reverse transcriptase is able to retain most of its activity even after binding with the inhibitor. This is consistent with the anticipated allosteric inhibition mechanism of the examined triazoles, where binding of a ligand to the NNRTI site does not necessarily lead to a complete inactivation of the enzyme. It was shown by other authors that the resistance mechanism of some of the RT mutants is not a result of decreased binding of inhibitors but of the restored enzyme's natural flexibilityCitation36,Citation37. It is therefore theoretically possible that an appropriate structural modification of described compounds can increase their inhibitory potency by altering their allosteric effect on the enzyme without changing their binding strength.
Table 5. Kinetic constants for the interaction between HIV-1 RT and triazoles 10, 28 and 32.
Conclusion
In this study, molecular docking approach combined with enzymatic studies was used to explore the 5-position substitution pattern of triazole NNRTIs. We found that heterocyclic substituents in the 5-position of the triazole ring are detrimental to the inhibitory activity of compounds with four-membered thioglycolamide linker and this substitution seems to be viable only for compounds with shorter two-membered linker. Our results also suggest that some of the analyzed compounds can bind to the reverse transcriptase without inactivating it completely due to the allosteric character of RT–NNRTIs interactions. A promising compound, N-(4-carboxy-2-chlorophenyl)-2-((4-benzyl-5-methyl-4H-1,2,4-triazol-3-yl)sulfanyl)acetamide potassium salt, with good inhibitory activity and aqueous solubility has been identified, and can serve as a lead scaffold for the development of novel water-soluble salts of triazole NNRTIs. Our next step will be the synthesis and modeling of various substitution variants of the identified scaffold.
Acknowledgements
The authors acknowledge Mr. Ł. Piotrowski of the Institute of Applied Radiation Chemistry for mass spectra.
Declaration of interest
The authors declare no conflicts of interest. The reported studies are supported by the grant 2011/02/A/ST4/00246 (2012–2017) from the Polish National Science Centre (NCN).
References
- Barbaro G, Scozzafava A, Mastrolorenzo A, Supuran CT. Highly active antiretroviral therapy: current state of the art, new agents and their pharmacological interactions useful for improving therapeutic outcome. Curr Pharm Des 2005;11:1805–43
- Mocroft A, Ledergerber B, Katlama C, et al. Decline in the AIDS and death rates in the EuroSIDA study: an observational study. Lancet 2003;362:22–9
- Hammer SM, Eron JJ Jr, Reiss P, et al. Antiretroviral treatment of adult HIV infection: 2008 recommendations of the International AIDS Society-USA panel. J Am Med Assoc 2008;300:555–70
- Tronchet JM, Seman M. Nonnucleoside inhibitors of HIV-1 reverse transcriptase: from the biology of reverse transcription to molecular design. Curr Top Med Chem 2003;3:1496–511
- Martins S, Ramos MJ, Fernandes PA. The current status of the NNRTI family of antiretrovirals used in the HAART regime against HIV infection. Curr Med Chem 2008;15:1083–95
- De Clercq E. The role of non-nucleoside reverse transcriptase inhibitors (NNRTIs) in the therapy of HIV-1 infection. Antiviral Res 1998;38:153–79
- Zhan P, Chen X, Li D, et al. HIV-1 NNRTIs: structural diversity, pharmacophore similarity, and implications for drug design. Med Res Rev 2013;33:E1–72
- Bennett DE, Camacho RJ, Otelea D, et al. Drug resistance mutations for surveillance of transmitted HIV-1 drug-resistance: 2009 update. PLoS One 2009;4:e4724
- Bacheler LT, Anton ED, Kudish P, et al. Human immunodeficiency virus type 1 mutations selected in patients failing efavirenz combination therapy. Antimicrob Agents Chemother 2000;44:2475–84
- Mansky LM. The mutation rate of human immunodeficiency virus type 1 is influenced by the vpr gene. Virology 1996;222:391–400
- Kirschberg TA, Balakrishnan M, Huang W, et al. Triazole derivatives as non-nucleoside inhibitors of HIV-1 reverse transcriptase – structure-activity relationships and crystallographic analysis. Bioorg Med Chem Lett 2008;18:1131–4
- De La Rosa M, Kim HW, Gunic E, et al. Tri-substituted triazoles as potent non-nucleoside inhibitors of the HIV-1 reverse transcriptase. Bioorg Med Chem Lett 2006;16:4444–9
- Muraglia E, Kinzel OD, Laufer R, et al. Tetrazolethioacetanilides: potent non-nucleoside inhibitors of WT HIV reverse transcriptase and its K103N mutant. Bioorg Med Chem Lett 2006;16:2748–52
- Zhan P, Liu X, Li Z, et al. Novel 1,2,3-thiadiazole derivatives as HIV-1 NNRTIs with improved potency: synthesis and preliminary SAR studies. Bioorg Med Chem 2009;17:5920–7
- Zhan P, Liu X, Li Z, et al. Synthesis and biological evaluation of imidazole thioacetanilides as novel non-nucleoside HIV-1 reverse transcriptase inhibitors. Bioorg Med Chem 2009;17:5775–81
- Zhan P, Wang L, Liu H, et al. Arylazolyl(azinyl)thioacetanilide. Part 9: Synthesis and biological investigation of thiazolylthioacetamides derivatives as a novel class of potential antiviral agents. Arch Pharm Res 2012;35:975–86
- Wang Z, Wu B, Kuhen KL, et al. Synthesis and biological evaluations of sulfanyltriazoles as novel HIV-1 non-nucleoside reverse transcriptase inhibitors. Bioorg Med Chem Lett 2006;16:4174–7
- Zhan P, Liu X, Cao Y, et al. 1,2,3-Thiadiazole thioacetanilides as a novel class of potent HIV-1 non-nucleoside reverse transcriptase inhibitors. Bioorg Med Chem Lett 2008;18:5368–71
- Moyle G, Boffito M, Stoehr A, et al. Phase 2a randomized controlled trial of short-term activity, safety, and pharmacokinetics of a novel nonnucleoside reverse transcriptase inhibitor, RDEA806, in HIV-1-positive, antiretroviral-naive subjects. Antimicrob Agents Chemother 2010;54:3170–8
- Zhan P, Li D, Chen X, et al. Functional roles of azoles motif in anti-HIV agents. Curr Med Chem 2011;18:29–46
- Leung CS, Zeevaart JG, Domaoal RA, et al. Eastern extension of azoles as non-nucleoside inhibitors of HIV-1 reverse transcriptase; cyano group alternatives. Bioorg Med Chem Lett 2010;20:2485–8
- Zeevaart JG, Wang L, Thakur VV, et al. Optimization of azoles as anti-human immunodeficiency virus agents guided by free-energy calculations. J Am Chem Soc 2008;130:9492–9
- Mislak AC, Frey KM, Bollini M, et al. A mechanistic and structural investigation of modified derivatives of the diaryltriazine class of NNRTIs targeting HIV-1 reverse transcriptase. Biochim Biophys Acta 2014;1840:2203–11
- Siwek A, Wujec M, Stefanska J, Paneth P. Antimicrobial properties of 4-aryl-3-(2-methyl-furan-3-yl)-Δ2-1,2,4-triazoline-5-thiones. Phosphorus Sulfur Silicon Relat Elem 2009;184:3149–59
- El-Ashmawy MB. The reaction of 3-(2-thienyl)-5-mercapto-4H-1,2,4-triazole with aryl halides. Mansoura J Pharm Sci 1991;7:168–77
- Malbec F, Milcent R, Barbier G. Derivatives of 2,4-dihydro-1,2,4-triazole-3-thione and 2-amino-1,3,4-thiadiazole from thiosemicarbazones of esters. J Heterocycl Chem 1984;21:1689–98
- Benno R, Hildegard B. Splitting of hexamethylenetetramine addition compounds of N-haloacetylated substituted anilines be means of sulfur dioxide. Pharmazie 1949;4:149–50
- Xie H, Ng D, Savionv SN, et al. Structure-activity relationships in the binding of chemically derivatized CD4 to gp120 from human immunodeficiency virus. J Med Chem 2007;50:4898–908
- Geitmann M, Unge T, Danielson H. Biosensor-based kinetic characterization of the interaction between HIV-1 reverse transcriptase and non-nucleoside inhibitors. J Med Chem 2006;49:2367–74
- Elinder M, Selhorst P, Vanham G, et al. Inhibition of HIV-1 by non-nucleoside reverse transcriptase inhibitors via an induced fit mechanism – importance of slow dissociation and relaxation rates for antiviral efficacy. Biochem Pharmacol 2010;80:1133–40
- Frączek T, Siwek A, Paneth P. Assessing molecular docking tools for relative biological activity prediction: a case study of triazole HIV-1 NNRTIs. J Chem Inf Model 2013;53:3326–42
- Friesner RA, Banks JL, Murphy RB, et al. Glide: a new approach for rapid, accurate docking and scoring. 1. Method and assessment of docking accuracy. J Med Chem 2004;47:1739–49
- Glide, version 5.7. New York, NY: Schrödinger, LLC; 2011
- Girardet JL, Koh YH, de la Rosa M, et al. S-triazole alpha-mercaptoacetanilides as inhibitors of HIV reverse transcriptase. US patent 7947721 B2, May 24, 2011
- Esnouf R, Ren J, Ross C, et al. Mechanism of inhibition of HIV-1 reverse transcriptase by non-nucleoside inhibitors. Nat Struct Biol 1995;2:303–8
- Schauer GD, Huber KD, Leuba SH, Sluis-Cremer N. Mechanism of allosteric inhibition of HIV-1 reverse transcriptase revealed by single-molecule and ensemble fluorescence. Nucleic Acid Res 2014;42:11687–96
- Seckler JM, Leioatts N, Miao H, Grossfield A. The interplay of structure and dynamics: insights from a survey of HIV-1 reverse transcriptase crystal structures. Proteins 2013;81:1792–801