
Abstract
Peptidylglycine α-amidating monooxygenase (PAM) is a bifunctional enzyme that catalyzes the final reaction in the maturation of α-amidated peptide hormones. Peptidylglycine α-hydroxylating monooxygenase (PHM) is the PAM domain responsible for the copper-, ascorbate- and O2-dependent hydroxylation of a glycine-extended peptide. Peptidylamidoglycolate lyase is the PAM domain responsible for the Zn(II)-dependent dealkylation of the α-hydroxyglycine-containing precursor to the final α-amidated peptide. We report herein that cinnamic acid and cinnamic acid analogs are inhibitors or inactivators of PHM. The inactivation chemistry exhibited by the cinnamates exhibits all the attributes of a suicide-substrate. However, we find no evidence for the formation of an irreversible linkage between cinnamate and PHM in the inactivated enzyme. Our data support the reversible formation of a Michael adduct between an active site nucleophile and cinnamate that leads to inactive enzyme. Our data are of significance given that cinnamates are found in foods, perfumes, cosmetics and pharmaceuticals.
Introduction
Peptidylglycine α-hydroxylating monooxygenase (PHM) is a copper-, O2- and ascorbate-dependent enzyme responsible for the oxidative cleavage of C-terminal glycine-extended precursor peptides to the active α-amidated peptide and glyoxylateCitation1,Citation2. This reaction proceeds by the abstraction of hydrogen from the glycyl Cα followed by the O2-dependent hydroxylation to generate a C-terminal α-hydroxyglycine-extended peptideCitation3,Citation4. Dealkylation of the α-hydroxyglycine-extended peptides is catalyzed by peptidylamidoglycolate lyase (PAL), a zinc- and calcium-dependent enzyme ()Citation4–6. The relationship between PHM and PAL is complex and species-dependent. Vertebrates produce a bifunctional protein, referred to as peptidylglycine α-amidating monooxygenase (PAM), which contains one PHM and one PALCitation7, while invertebrates generate the monofunctional PHM and PAL enzymes from separate genesCitation8–10. Bifunctional and pentafunctional (four PHM to one PAL) enzymes are reported from different mollusk speciesCitation11,Citation12. Furthermore, alternative splicing of the PAM mRNA and proteolytic processing of the multifunctional PAM proteins results in a diverse array of catalytically active forms of PAM, PHM, and PAL in vivoCitation13,Citation14. The significance of the complexities within the PAM/PHM/PAL system is currently unclear, but hints at a regulatory strategy for the production of α-amidated peptides or toward a biological function for C-terminal α-hydroxylated peptides.
Figure 1. The reactions catalyzed by PHM and PAL (A), the cinnamate-derived vinyl radical (B) and the 4-phenyl-3-buteneoate-derived allylic radical (C). Bifunctional PAM is compromised of separate monofunctional enzymes, PHM and PAL. In vivo, the substrate for PHM and PAL is a C-terminal glycine extended peptide (R = a peptide) to generate an α-amidated peptide.
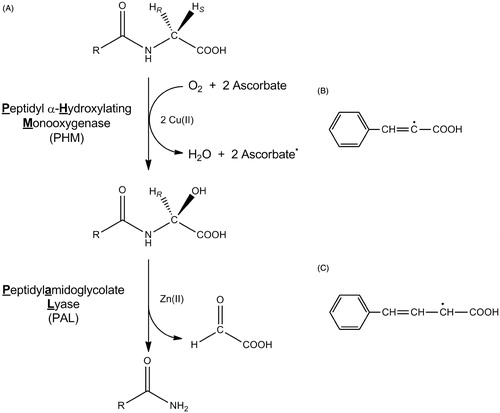
In mammals, PAM activity has been found in the blood and in many tissuesCitation15, with the highest levels being found in pituitary, central nervous system and the atrium of the heartCitation7,Citation14–16. A large percentage of mammalian bioactive peptides, ∼50%, possess an α-amidated C-terminus. As a consequence, a number of pathological conditions correlate to dysfunction in the expression of specific α-amidated peptides or one of the PAM/PHM/PAL proteins, including cancerCitation17,Citation18, arthritisCitation19, inflammationCitation20,Citation21 and Alzheimer's diseaseCitation22. Peptide amidation is important in insects as well because ≥80% of all insect bioactive peptides are α-amidatedCitation23. Considerable effort has been expended to develop PAM/PHM/PAL inhibitors as such compounds could prove valuable as either insecticides or drugs to treat diseases related to imbalances in α-amidated peptide productionCitation24–37. Included amongst these works are reports that cinnamates and ring-substituted cinnamates are irreversible inactivators of PHMCitation32,Citation33. Intriguingly, Bradbury et al.Citation33 suggest that cinnamate-mediated inactivation of PHM results from the formation of a vinyl radical (). This chemistry is related to the irreversible inactivation of PHM by trans-benzoylacrylateCitation25, trans-4-phenyl-3-butenoate (PBA, also known as trans-styrylacetate)Citation36,Citation37, and trans-styrylthioacetateCitation38; PBA inactivation is proposed to occur via the formation of an allylic radical ()Citation35,Citation37. The formation of vinyl radical during the cinnamate-mediated inactivation of PHM is mechanistically relevant because this would require the formation of a PAM-based oxidant that is sufficiently strong to abstract a hydrogen atom from the α-carbon of cinnamate. If true, this may force a revision of the current models for PHM catalysisCitation39–42.
In addition to the potential mechanistic significance of the cinnamate-mediated inactivation of PHM, man has a long history of the exposure to and uses for cinnamate and cinnamate derivatives. Cinnamates are found in fruits, fruit juices, vegetables and flowers and are used in perfumes, cosmetics and pharmaceuticalsCitation43–46. Medically, cinnamates are known to lower blood glucose levelsCitation47 and have potential as treatments for cancer, tuberculosis and other human diseasesCitation48,Citation49. The inactivation of PHM by cinnamate merits further investigation given the important role of the PAM/PHM/PAL system in the biosynthesis of α-amidated peptide hormonesCitation1,Citation2. Herein, we report that cinnamate and a number of cinnamate analogs are PHM inhibitors, information that contributes toward the rational design of PHM-directed therapeutics. A subset of these compounds, cinnamate and three ring-substituted cinnamates, are also turnover-dependent inactivators of PHM. We find that inactivated PHM is indistinguishable from untreated enzyme and find no evidence for cinnamate oxidation during the inactivation reaction. The reversible Michael addition of an active site nucleophile to cinnamate could account for our data and may explain the inactivation of PHM by a variety of acrylatesCitation20,Citation30,Citation35, and the 2- and 3-alkenoatesCitation32 as well as provide an explanation for the lack of PHM-labeling by 3H-PBACitation37.
Materials and methods
Chemicals and instrumentation
Cinnamic acid, 2-trifluorocinnamic acid, 3-(3-pyridyl)acrylic acid, phenylpropiolic acid, 3,4-methylenedioxycinnamic acid, N,N-dimethylaminocinnamic acid, urocanic acid, 4-aminocinnamic acid and perdeuterated cinnamic acid (C6D5–CD=CD–COOH) were from Sigma (St. Louis, MO); bovine catalase was from Worthington (Lakewood, NJ); and [ring-14C6]-trans-cinnamic acid (specific activity of 50–60 mCi/mmol and 0.1 mCi/mL) was from American Radiolabeled Chemicals (St. Louis, MO). Recombinant rat bifunctional PAM was a gift from Unigene Laboratories, Inc. (Fairfield, NJ), was greater than 95% pure as judged by SDS polyacrylamide electrophoresis (Figure S1, Supplementary material) and had a specific activity of ≥4.0 μmol of O2 consumed/min/mg at 37 °C under standard assay conditionsCitation50. All other reagents were of the highest quality commercially available.
Oxygen consumption was monitored using a Yellow Springs Instrument Model 5300 (Yellow Springs, OH) oxygen monitor. Spectrophotometric studies were performed on a JASCO UV-VIS spectrophotometer (Easton, MD). HPLC separations were performed on an Agilent HP 1100 (Santa Clara, CA), equipped with a four-channel solvent mixing system, a quaternary pump and either a variable wavelength UV detector or a Gilson Model 121 fluorimeter.
Synthesis of N-dansyl-4-aminocinnamic acid
A solution of dansyl chloride (400 mg, 1.5 mmol) in a minimal volume of dry pyridine (∼2 mL) was added dropwise to a N2-purged solution of 4-aminocinnamic acid (500 mg, 3.1 mmol) in 50 mL of dry pyridine at 60 °C. After 24 h, the reaction was diluted and extracted with Et2O (100 mL × 3), yielding a yellow oil upon concentration. Crystallization of the final N-dansyl-4-aminocinnamic acid was performed with methanol/H2O (285 mg, 48%). 1H NMR (400 MHz, CDCl3): δ 8.68 (s, 2H), 8.50 (d, J = 8.5 Hz, 3H), 8.31 (d, J = 8.5 Hz, 3H), 8.23 (d, J = 7.4 Hz, 3H), 7.86–7.81 (m, 1H), 7.63–7.53 (m, 7H), 7.48–7.37 (m, 7H), 7.30 (d, J = 8.6 Hz, 7H), 7.17 (d, J = 7.6 Hz, 3H), 6.99 (d, J = 8.6 Hz, 6H), 6.25 (d, J = 16.0 Hz, 3H), 2.85 (s, 21H). m/z observed: 397.1. The 1H NMR spectrum of N-dansyl-4-aminocinnamic acid is included in the Supplementary materials (Figure S2, Supplementary materials).
Measurement of the PHM-dependent consumption of O2
Enzyme concentrations were determined by the Bradford method using bovine serum albumin as a standardCitation51. The enzymatic reactions were initiated by the addition of PAM (0.5 nmol, 35 µg) into 2.0 mL of 100 mM MES/NaOH pH 6.0, 30 mM NaCl, 1% (v/v) ethanol, 0.001% (v/v) Triton X-100, 10 μg/mL bovine catalase, 1.0 μM Cu(NO3)2, 5.0 mM sodium ascorbate at 37.0 ± 0.1 °C. N-Acetylglycine (8 mM) served as the oxidizable substrate for the inhibition studiesCitation45. Initial rates were measured by following the PAM-dependent consumption of O2 using an oxygen electrode. All kinetic data were determined in duplicate and are reported as mean ± standard deviation.
PHM inhibition
The initial rates of O2 consumption by PHM were measured at eight different concentrations of the oxidizable substrate in the absence and presence of three different inhibitor concentrations. N-Acetylglycine served as the oxidizable substrate at concentrations of 1.0–45.0 mM. The initial velocities from the inhibition experiments were fit to the equations for competitive, non-competitive and uncompetitive inhibition, respectively, using SigmaPlot 9.0 (Systat Software, San Jose, CA). All the inhibition data contained herein were the best fit to the one-site competitive inhibition model for the calculation of KI,obs.
PHM inactivation
Reactions consisting of 100 µL containing 100 mM MES/NaOH pH 6.0, 30 mM NaCl, 1% (v/v) ethanol, 0.001% (v/v) Triton X-100, 10 µg/ml bovine catalase, 1.0 µM Cu(NO3)2, 5.0 mM sodium ascorbate and the desired concentration of the inactivator were initiated by the addition of 1.0 nmol (70 μg) enzyme and incubated at 37 °C. Aliquots of 15 μL were withdrawn at various intervals and diluted into 2.0 mL reactions containing 100 mM MES/NaOH pH 6.0, 30 mM NaCl, 1% (v/v) ethanol, 0.001% (v/v) Triton X-100, 10 µg/mL bovine catalase, 1.0 μM Cu(NO3)2, 5.0 mM sodium ascorbate, and 20 mM N-acetylglycine and monitored for O2 consumption. The inactivator concentrations employed were based on their respective KI,obs values that had been determined in separate experiments.
PAL inactivation
Inactivation of PAL activity within bifunctional PAM was evaluated by measuring the initial rates of glyoxylate formation from α-hydroxyhippurate. Reactions of 0.3 nmol (20 µg) of PAM in a 100 μL solution containing 100 mM MES/NaOH pH 6.0, 30 mM NaCl, 1.0% (v/v) ethanol, 0.001% (v/v) Triton X-100, and a concentration of cinnamate or analog equal to 5 × IC50 were incubated for 2 h. A 20 µL aliquot was removed and used to initiate a 1.0 mL reaction containing 100 mM MES/NaOH pH 6.0, 30 mM NaCl, 1.0% (v/v) ethanol, 0.001% (v/v) Triton X-100, and 10 mM α-hydroxyhippurate. At 10 min intervals from 10 to 60 min, a 100 µL aliquot was removed, added to a vial containing 20 µL of 6% (v/v) trifluoroacetic acid (TFA) to terminate the reaction, and the concentration of glyoxylate measured in the acidified samples.
Glyoxylate was determined by the spectrophotometric method of Christman et al.Citation52 as modified by Katopodis and MayCitation25. Standard curves of [glyoxylate] versus A520 were constructed in the appropriate buffers using a glyoxylate solution that had been calibrated by measuring the glyoxylate-dependent oxidation of NADH (Δε340 = 6.22 × 103 M−1 cm−1) as catalyzed by lactate dehydrogenase.
Tiopronin protection against the cinnamate-mediated inactivation of PHM
Protection of PHM against the cinnamate-mediated inactivation by tiopronin was evaluated by comparing the rate of glyoxylate formation from N-acetylglycine in the absence of cinnamate (no inactivator control), in the presence of cinnamate, and in the presence of cinnamate and tiopronin. Reactions of 0.3 nmol (20 µg) of PAM in a 100 μL solution containing 100 mM MES/NaOH pH 6.0, 30 mM NaCl, 1.0% (v/v) ethanol, 0.001% (v/v) Triton X-100, 0 or 7.5 mM cinnamate, and 0 or 60 μM tiopronin were incubated for 2 h. A 20 µL aliquot was removed and used to initiate a 1.0 mL reaction containing 100 mM MES/NaOH pH 6.0, 30 mM NaCl, 1% (v/v) ethanol, 0.001% (v/v) Triton X-100, 10 µg/mL bovine catalase, 1.0 μM Cu(NO3)2, 5.0 mM sodium ascorbate and 8 mM N-acetylglycine and monitored for glyoxylate production. At 10 min intervals from 10 to 90 min, a 100 µL aliquot was removed, added to a vial containing 20 µL of 6% (v/v) TFA to terminate the reaction, and the concentration of glyoxylate measured in the acidified samples as described above.
Reversibility of inactivation
To investigate the reversibility of inactivation, a reaction mixture of 250 µL containing 100 mM MES/NaOH pH 6.0, 30 mM NaCl, 1% (v/v) ethanol, 0.001% (v/v) Triton X-100, 10 µg/mL bovine catalase, 1.0 µM Cu(NO3)2, 5.0 mM sodium ascorbate and 7 mM cinnamate (2 × KI,obs) was initiated by the addition of enzyme and incubated at 37 °C for 2 h. The reaction was then extensively dialyzed and concentrated to 50 μL. The concentrated, cinnamate-treated enzyme was then used to initiate a 1.0 mL reaction containing 100 mM MES/NaOH pH 6.0, 30 mM NaCl, 1.0% (v/v) ethanol, 0.001% (v/v) Triton X-100 and 10 mM hippurate. At various time intervals, the recovery of PHM activity was tested after quenching a 100 µL aliquot with 1% (v/v) TFA. The concentration of glyoxylate produced in the acidified samples was used to test for PHM reactivation.
Investigation of enzyme modification by the cinnamates
Covalent modification of PAM was investigated using N-dansyl-4-aminocinnamate, a fluorescent molecule, as an inactivator. A reaction (0.5 mL) containing 100 M MES/NaOH pH 6.0, 30 mM NaCl, 1% (v/v) ethanol, 0.001% (v/v) Triton X-100, 10 µg/mL bovine catalase, 1.0 µM Cu(NO3)2, 5.0 mM sodium ascorbate and 1.0 mM N-dansyl-4-aminocinnamate was initiated by the addition of 0.7 nmol (50 µg) of enzyme and incubated at 37 °C for 3 h. Under these conditions, the mole ratio of N-dansyl-4-aminocinnamate/PAM was 710. The reaction mixture was dialyzed against 100 mL of 100 mM MES/NaOH pH 6.0, 30 mM NaCl, 1% (v/v) ethanol and 0.001% (v/v) Triton X-100 for 4 h changing the dialysis buffer every hour. The reaction mixture was then concentrated by ultra-filtration to ∼100 µL. This sample was then trypsin digested and the peptide fragments were analyzed by reverse phase HPLC (Phenomenex Luna 5 μ C18 column, 250 mm × 4.6 mm) utilizing the in-line fluorescence detector and compared against a standard sample prepared in parallel in the absence of N-dansyl-4-aminocinnamate.
Enzyme modification was also examined using [ring-14C6]-trans-cinnamate. A 250 µL reaction containing 100 mM MES/NaOH pH 6.0, 30 mM NaCl, 1% (v/v) ethanol, 0.001% (v/v) Triton X-100, 10 µg/mL bovine catalase, 1.0 µM Cu(NO3)2, 5.0 mM sodium ascorbate and 3 µCi (50–60 nmol) of 14C-cinnamate was initiated by the addition of 0.24–0.70 nmol (18–52 µg) of PAM. Under these conditions, the mole ratio of [ring-14C6]-trans-cinnamate/PAM ranged from 70 to 210. The reaction was allowed to incubate at 37 °C for 3 h before ultra-filtration was performed. The reaction was then washed with 200 µL of 100 mM MES/NaOH pH 6.0, 30 mM NaCl, 1% (v/v) ethanol and 0.001% (v/v) Triton X-100 and ultra-filtration repeated. The underside of the filtration membrane was then washed with cold poly(ethylene glycol) to remove excess non-enzyme bound radio-labeled cinnamate. Counts per minute were then compared using a scintillation counter to a “no-PAM” control.
Modification of the PAM active site was also investigated as a possible means of cinnamate-mediated inactivation. A 100 µL reaction containing 100 mM MES/NaOH pH 6.0, 30 mM NaCl, 1% (v/v) ethanol, 0.001% (v/v) Triton X-100, 1.0 µM Cu(NO3)2, 5.0 mM sodium ascorbate and 3 mM cinnamate was initiated with 0.3 nmol (20 µg) of PAM and incubated at 37 °C for 12 h. The enzyme was then dialyzed against 100 mM MES/NaOH pH 6.0, 30 mM NaCl, 1% (v/v) ethanol, 0.001% (v/v) Triton X-100 for 4 h and the volume reduced to ∼40 µL by ultra-filtration. The enzyme sample was analyzed for covalent modification using LC/MS and MALDI-TOF and compared to an enzyme control that lacked cinnamate. The reactions were performed in duplicate and subjected to trypsin digestion. The sample and the no cinnamate control were digested with sequencing grade modified porcine trypsin in 50 mM ammonium bicarbonate buffer (pH 8.2) at a PAM–trypsin ratio of 50:1. Proteolytic digestion was carried out overnight at 37 °C. The resulting peptide mixture were mixed with α-cyano-4-hydroxycinnamic acid solution (4 mg/mL in 50% (v/v) acetonitrile and 5% (v/v) isopropanol) in 1:1 ratio and spotted onto a blank stainless steel MALDI plate. MALDI-TOF-MS was carried out on 4700 Proteomics Analyzer (Applied Biosystems, Carlsbad, CA) using reflective positive mode. The data were externally calibrated with 4700 Mass Standards (Applied Biosystems, Carlsbad, CA) from the manufacturer.
Molecular docking studies
Molecular docking studies were performed using the First Discovery 3.0 suite (www.schrodinger.com) on a dual 3.2 GHz Xeon workstation running Fedora Core 4 to investigate the possible binding modes of the inhibitors. The crystal structure for reduced PHMCitation53 was obtained from the Protein Data Bank (http://www.rcsb.org/pdb/, 1SDW). All co-crystallized species determined to be redundant for ligand binding were removed (nickel, water, glycerol and substrate). Formal charges for enzyme-bound copper ions and bond orders were corrected, and hydrogen atoms were added using Maestro. Further receptor refinements were carried out utilizing ProteinPrep from within Maestro. Investigation of inhibitor binding modes was performed using GlideCitation54 and QsiteCitation55 jointly for quantum-polarized ligand docking. This method generates highly accurate binding modes by quantum mechanically calculating the partial atomic charges of the docked ligand using B3LYP/6-31G* within the receptor and, subsequently, re-docking the ligandCitation56,Citation57. All default settings were used.
Density functional theory investigation of bond dissociation energies
The relative bond dissociation energies of Cα-H for select cinnamic acid analogs were calculated using JaguarCitation58 to reveal any correlation between these values and the inactivation kinetic parameters. Density functional theory calculations were performed with the B3LYP hybrid exchange-correlation functional and the 6-31G* basis set. The geometries were optimized for all molecules in the gas phase. The Cα-H bond was then increased from 1.08 to 3.0 Å with a step-size of 0.1 Å, while holding the molecule rigid, and single point energies were calculated.
Results
PAM inhibition by cinnamic acid analogs
Cinnamate and several cinnamate analogs were initially investigated as reversible PHM inhibitors. Cinnamate competitively inhibited the PHM-dependent consumption of O2, using N-acetylglycine as the oxidizable substrate, as shown in . Steady-state inhibition constants for cinnamate and the cinnamate analogs are included in . The observed inhibition constants, KI,obs, ranged by a factor of 1000 from 10 μM for N-dansyl-4-aminocinnamate to 10 mM for urocanic acid. The relatively tight-binding of N-dansyl-4-aminocinnamate to PHM is consistent with earlier structure–activity studies indicating that peptide substrates possessing a hydrophobic amino acid in the penultimate position have the highest (V/K)app valuesCitation59.
Figure 2. Inhibition of PHM by cinnamic acid. Assays were performed as described in “Methods” section for the initial rate determination. The points are initial rates and the lines were computer fit using SigmaPlot 9.0. Initial rates of O2 consumption were measured for ≤3 min, during which the inactivation mediated by cinnamate makes a neglible contribution to the observed inhibition.
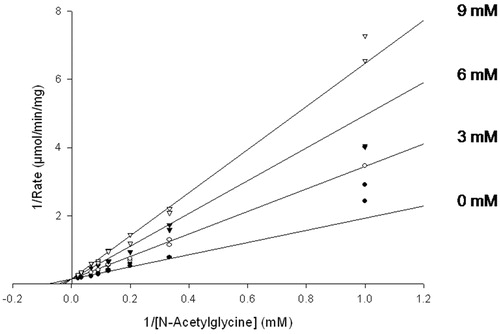
Table 1. KI,obs values for the inhibition of PHM by cinnamate and the cinnamate analogsa.
PHM-specific inactivation by cinnamate and cinnamate analogs
Cinnamate and the cinnamate analogs were first evaluated as PHM inactivators by the dilution method, a common method for determining the kinetic parameters for time-dependent inactivatorsCitation60. In agreement with the earlier report by Bradbury et al.Citation33, we found cinnamate to inactivate the PHM-catalyzed consumption of O2 in the presence of N-acetylglycine and ascorbate (). The 1/kinact,obs versus 1/[cinnamate] replot was linear ()Citation56.Footnote† In addition to cinnamate, only three other compounds, N-dansyl-4-aminocinnamate, 4-aminocinnamate and N,N-dimethyl-4-aminocinnamate were found to inactivate PHM (). No deuterium isotope effect was observed on the cinnamate-mediated inactivation as the (kinact/KI)obs value obtained using perdeuterated cinnamate was identical to that for cinnamate within experimental error (). The steady-state inhibition constant, KI,obs, for cinnamate, perdeuterated cinnamate, N-dansyl-4-aminocinnamate, 4-aminocinnamate, and N,N-dimethyl-4-aminocinnamate (), was measured under conditions (relatively short-time frames) such that the rate of the PHM inactivation contributed little to the observed inhibition.
Figure 3. Inactivation of PHM by cinnamic acid. (A) PHM was incubated at 37 °C in the presence of the indicated concentration of cinnamate as described in the “Methods” section. At the indicated times, an aliquot was removed and assayed for residual O2 consumption activity. Lines are linear regression fits to the equation: log[(vt/v0)] = (−kobs/2.303)t + C, where v0 is the average initial rate of O2 consumption in the absence of cinnamate, vt is the initial O2 consumption rate at time = t, and C is a constant which should be within experimental error of 2.0. The error bars represent the standard deviation of the duplicate measurements. (B) A reciprocal replot of the inactivation data from panel A.
![Figure 3. Inactivation of PHM by cinnamic acid. (A) PHM was incubated at 37 °C in the presence of the indicated concentration of cinnamate as described in the “Methods” section. At the indicated times, an aliquot was removed and assayed for residual O2 consumption activity. Lines are linear regression fits to the equation: log[(vt/v0)] = (−kobs/2.303)t + C, where v0 is the average initial rate of O2 consumption in the absence of cinnamate, vt is the initial O2 consumption rate at time = t, and C is a constant which should be within experimental error of 2.0. The error bars represent the standard deviation of the duplicate measurements. (B) A reciprocal replot of the inactivation data from panel A.](/cms/asset/bdc0b08a-df68-4d82-81b3-e4bbd1b729bb/ienz_a_1046064_f0003_c.jpg)
Table 2. Values for kinact,obs and (kinact/KI)obs for cinnamate and selected cinnamate analogs.
PHM inactivation by cinnamate () was pseudo first order and was dependent upon the initial concentration of the inactivator, O2, and ascorbate ( and Figure S3, Supplementary materials). In addition, tiopronin, a PHM substrateCitation61, did protect PHM from inactivation by the cinnamate-mediated inactivation (Figure S4, Supplementary materials).
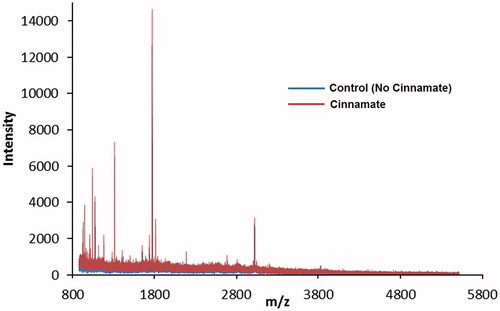
The enzyme used for our experiments was bifunctional rat PAM comprised the separate and catalytically active PHM and PAL domainsCitation50. Consumption of O2 in the presence of ascorbate and N-acetylglycine represents the reaction catalyzed by PHM; thus, inactivation of O2 consumption by the cinnamates only reports on the inactivation of the PHM domain of bifunctional PAM. The cinnamate and a number of cinnamate analogs were tested as PAL inactivators to investigate whether PAM inactivation was specific to the PHM domain. The data indicate that PAL is unaffected by the various cinnamates tested and retained 100% activity when compared to the control reaction (Figure S5, Supplementary materials). Extensive dialysis of bifunctional PAM incubated with cinnamate yielded inactive enzyme unable to catalyze the production of glyoxylate from hippuric acid. All attempts to generate cinnamate-labeled enzyme were unsuccessful. MALDI-TOF analysis of cinnamate-inactivated PAM showed no differences between the treated enzyme and the untreated control (). Incubation of PAM with 14C-cinnamate in the presence of O2 and ascorbate yielded unlabeled protein, again devoid of PHM activity. Similarly, the incubation of bifunctional PAM with N-dansyl-4-aminocinnamate, in the presence of O2 and ascorbate, yielded enzyme devoid of PHM activity and also lacking any fluorescent label.
Figure 4. MALDI-TOF overlay of trypsin-digested active and cinnamate-inactivated PHM. The catalytically active control is shown in blue, while the inactivated PAM is shown in red. Inactivation was achieved through incubation with cinnamate as described in the “Methods” section. This figure is best viewed in color, which is only available online.
Key to fully understanding the impact of our labeling experiments is (a) the limit of detection of fluorescently labeled enzyme potentially generated by inactivation with N-dansyl-4-aminocinnamate and (b) the limit of detection of 14C-labeled enzyme potentially generated by inactivation with 14C-cinnamate. Our level of detection of N-dansyl-4-aminocinnamate is 20 pmol. We used 700 pmol of enzyme in our experiments to identify fluorescently labeled peptides from PHM inactivated with N-dansyl-4-aminocinnamate. We applied the tryptic digest of the N-dansyl-4-aminocinnamate inactivated PHM directly to the RP-HPLC column to identify any labeled peptides using our in-line fluorescent detector and no fluorescently labeled peptides were found. Assuming that our peptide losses are low and that a 1:1 cinnamate–PHM adduct forms, we would expect ∼700 pmol of fluorescently labeled peptide, which is ∼35-fold above our level of detection. These experiments show that labeled PHM cannot be a major product of the N-dansyl-4-aminocinnamate inactivation chemistry. However, a very low extent of labeling, an <0.05:1 cinnamate–PHM adduct, would be below our level of detection. The background level for the 14C labeling experiment was 20–30 cpm. We incubated 0.24–0.70 nmol of PHM with 50–60 nmol of [ring-14C6]-cinnamate, removed the unreacted 14C-cinnamate after the inactivation reaction by ultrafiltration, and found no 14C-label in PHM above background. Again, assuming a low loss of PHM and formation of a 1:1 14C-cinnamate–PHM adduct, we expected to generate 0.24–0.70 nmol of 14C-labeled PHM possessing 10–40 nCi of radioactivity based on specific radioactivity of the 14C-cinnamate (50–60 mCi/mmol). With a quenching efficiency of 60–80% for 14C, we would have found 14C-labeled PHM in the range of 13 000–70 000 cpm – far above background. Only a very low extent of 14C-labeling, an < 0.001:1 cinnamate–PHM adduct, would be below our level of detection. The most reasonable conclusion based on our inability to find labeled PHM upon the inactivation with N-dansyl-4-aminocinnamate or [ring-14C6]-cinnamate is that no labeled enzyme is produced. A result consistent with our analysis of inactivated PHM by mass spectrometry and that obtained for the incubation of PHM with 3H-PBACitation37.
Computational chemistry and modeling
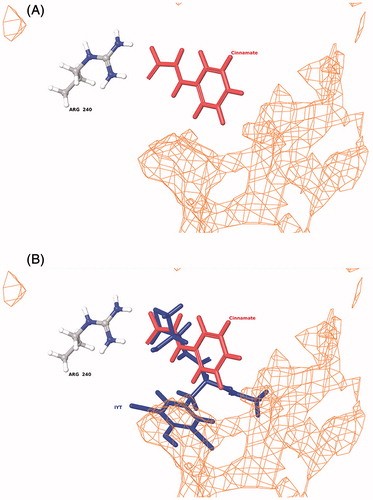
In silico docking of cinnamate and the cinnamate analogs indicated that all bind in the PHM active site consistent with the competitive inhibition observed for cinnamate (). The docking pose for cinnamate is shown in and the position of cinnamate in the PHM active site relative to bound substrate is shown in . All of the compounds form a salt bridge between the carboxy terminus and the guanidino group of R240 similar to that observed for the glycine-extended substratesCitation53,Citation62. This interaction aligns the Cα-H in close proximity to CuM, one of the two copper atoms bound to PHMCitation53,Citation62. However, the lack of glycyl amide hydrogen bonding with the N316 coupled with the relatively small size of the compounds prevents interaction with the nearby hydrophobic pocket, suggesting greater mobility when compared to glycine-extended peptide substrates.
Figure 5. Binding of cinnamate into the PHM active site (A) and the relative orientation of cinnamate to bound substrate (N-α-acetyl-3,5-diiodotyrosylglycine, IYT) in PHM (B). The orientation of both the IYT (blue) and cinnamate (red) ligands is superimposed to compare recognized electrostatic (Arg-240) and hydrophobic (orange mesh) interactions near the CuM domain associated with productive ligand bindingCitation53,Citation62. PHM contains two bound copper atoms, CuM and CuHCitation53.
Discussion
Structure–activity data
Cinnamate is structurally related to two PHM substrates, N-cinnamoylglycine and N-hydrocinnamoylglycineCitation24, and two PHM inactivators, PBA and trans-benzoylacrylateCitation25,Citation34,Citation36,Citation37. These compounds bind to PHM with higher affinity than cinnamate (KI = 4 mM): KM,hydrocinnamoylglycine = 0.9 mM, KM,cinnamoylglycine = 0.2 mM, KI,PBA = 0.05 mM and KI,benzoylacrylate = 0.2 mM, at least to the extent that KM and KI values reflect relative binding affinities to PHM. Our modeling results indicate that the carboxylate moiety of cinnamate interacts with R240 in the PHM active site (), as was demonstrated for the C-terminus of the peptide substratesCitation62. The PHM active site possesses a hydrophobic pocketCitation53,Citation63 and our modeling results indicate that the phenyl moiety of cinnamate cannot reach from R240 to this hydrophobic pocket (). The presence of a longer spacer between the carboxylate moiety and the phenyl ring, like that found in N-cinnamoylglycine, N-hydrocinnamoylglycine, PBA, and trans-benzoylacrylate, enables the phenyl moiety to interact with the hydrophobic pocket within the PHM active site and, probably, is the main reason these four compounds are bound to PHM with higher affinity than cinnamate (). The relatively tight binding of N-dansyl-4-aminocinnamate to PHM may also result from the interaction of N,N-dimethyl-1-naphthylamine moiety of the dansyl group with the hydrophobic pocket of PHM. The small difference in the KI values of cinnamate and phenylpropiolic acid is likely a result of the differential positioning of the phenyl ring within the PHM active site. Cinnamate analogs with a replacement of the phenyl ring with either a pyridine ring (3-(3-pyridyl)acrylate) or an imidazole ring (urocanate) bind to PHM with lower affinity. The pyridine and imidazole rings must interact less favorably with amino acids within the PHM active site, leading to higher KI values for 3-(3-pyridyl)acrylate and urocanate relative to cinnamate.
Decoration of the phenyl ring of cinnamate with substituent groups results in changes in the affinity of PHM for the cinnamate derivative. 2-Trifluoromethyl-, 3,4-methylenedioxy-, 4-amino-, and 4-nitro-cinnamate all bind to PHM with greater affinity than cinnamate and N,N-dimethyl-4-aminocinnamate binds to PHM with approximately the same affinity as cinnamate (). The differences in PHM affinity observed for this set of compounds must be related to differences in interactions between the substituent group decorating the phenyl ring and active site amino acid(s). More definitive SAR results are not possible given the limited set of readily available ring-substituted cinnamates.
Cinnamate-mediated inactivation
Cinnamate, trans-benzoylacrylate, and PBA are not the only olefinic compounds known to inactivate PHM. Other olefinic inactivators of PHM include monoethyl fumarateCitation25, styrylthioacetateCitation38, 2- and 3-alkenoatesCitation32, vinylglycineCitation32, 3-substituted acrylatesCitation32, substituted 4-oxo-2-hexenoatesCitation30, and d-Phe-l-Phe-l-vinylglycineCitation64. Other monooxygenases, P450 and dopamine β-monooxygenase (DβM, structurally and mechanistically related to PHM), are inactivated by olefinsCitation65–68. Most of these olefinic inactivators exhibit characteristics of suicide substrates: the inactivation only occurs during enzyme turnover and substrates or competitive inhibitors protect the monooxygenases against inactivation. In addition, olefinic inactivation of PHM, DβM, and P450 is not mediated by the initial formation of the corresponding epoxideCitation37,Citation65,Citation67. The inactivation of DβM and P450 leads to both the generation of olefin-derived oxidation products and to the labeling of the inactivated enzyme. Padgette et al.Citation67 reported a partition ratio, kcat/kinact, of 900 for inactivation of DβM with phenyl(aminomethyl)ethane and found that inactivation with [3H]-phenyl(aminomethyl)ethane yields 3H-labeled DβM. The inactivation of P450 with 2-isopropyl-4-pentenamide exhibited a partition ratio of 200–300 and led to the alkylation of the enzyme-bound hemeCitation66. The olefinic inactivation of DβM and P450 was proposed to result from the formation of an olefin-derived radical intermediateCitation66,Citation67.
The chemistry for the inactivation of PHM by the cinnamates must be different. Incubation of PHM with PBA results in both O2 consumption and the generation of oxidized products, 2- and 4-hydroxy-4-phenyl-3-butenoate, with a partition ratio of 85–90Citation37. None of the compounds tested within this study showed any O2 consumption (above background) when screened for activity even though inactivation was O2- and ascorbate-dependent (Figure S3, Supplementary materials). A low background rate of O2 consumption is observed for PHM in the absence of an oxidizable substrate resulting from the presence of Cu(II) and ascorbate in an aerobic solutionCitation69. Incubation of N-dansyl-4-aminocinnamate with PHM in the presence of ascorbate and O2 does not result in any change in C18-reverse phase HPLC retention time for N-dansyl-4-aminocinnamate; additional evidence that the cinnamates are not PHM substrates. Finally, the lack of a deuterium kinetic isotope effect when using perdeuterated cinnamate as an inactivator on either kinact,obs or (kinact/KI)obs indicates that cinnamate-mediated inactivation of PHM does not involve rate-determining C–H bond cleavage.
Incubation of PHM with either [14C]-cinnamate or N-dansyl-4-aminocinnamate yielded inactive enzyme that was not labeled. Our comparison of trypsin-digested cinnamate-inactivated enzyme to control (untreated) enzyme by MALDI-TOF showed no differences between the samples. This suggests that inactivated PHM was not covalently decorated by an unlabeled fragment from either of these olefins. In other words, upon inactivation, the phenyl ring of [14C]-cinnamate or N-dansyl-4-aminocinnamate was not lost in some manner yielding modified PHM without incorporation of the 14C or the dansyl group. Despite the lack of labeling upon inactivation, our modeling and tiopronin protection studies indicate that the cinnamates are binding at the PHM active site.
The relatively low (kinact/KI)obs, values for cinnamate and the cinnamate analogs indicate that these compounds are more likely to dissociate from the PHM–inactivtor complex than inactivate PHM (). A comparison of the inactivation kinetic parameters () to the relative Cα–H bond dissociation energies () illustrates that there is no clear correlation between these values. For example, the Cα–H bond dissociation energy of N-dansyl-4-aminocinnamate is approximately 2 kcal/mol higher than that of cinnamate, yet (kinact/KI)obs value for N-dansyl-4-aminocinnamate is ∼500-fold higher than the (kinact/KI)obs value for cinnamate. The major difference in the (kinact/KI)obs ratios between N-dansyl-4-aminocinnamate and cinnamate stems from the contribution of binding affinity of each to PHM: KI,obs = 4 mM for cinnamate and 10 μM for N-dansyl-4-aminocinnamate. In sum, our data suggest that PHM-mediated inactivation by the cinnamate and the cinnamate analogs does not result from Cα–H bond cleavage: (a) no O2 consumption was observed when PHM was incubated with cinnamate in the presence of ascorbate and O2, (b) no evidence that an oxidized cinnamate is generated by incubation with PHM under turnover conditions, (c) no clear correlation between the inactivation kinetics and the Cα–H bond dissociation energies, (d) no kinetic isotope effect for the inactivation of PHM with perdeuterated cinnamate and (e) no labeled protein found when PHM was inactivated with [14C]-cinnamate or N-dansyl-4-aminocinnamate. Any mechanism that accounts for the cinnamate-mediated inactivation must be consistent with the data showing that inactive PHM has not been modified in an irreversible fashion, that cinnamate is not a PHM substrate, and that inactivation requires that PHM exist in a catalytically active state.
Table 3. Relative bond dissociation energies calculated for selected PHM inhibitors or inactivators.
Like the chemistry proposed for the olefinic inactivation of DβM and P450, it has been suggested that olefinic inactivation of PHM results from the formation of a non-epoxide oxidized species, likely an olefin-derived radicalCitation25,Citation33,Citation35–38,Citation64. This hypothesis is reasonable given that inactivation is turnover-dependent and that the PHM-catalyzed hydroxylation reaction likely proceeds through a substrate-based radicalCitation39–42. However, this proposed chemistry is inconsistent with our data and is thermodynamically challenging. The formation of a vinyl radical from cinnamate is ∼80 kJ/mol higher than formation of an allylic radical from PBACitation70. It is not clear that any of the reduced oxygen species proposed to form during PHM catalysis are of sufficient strength to generate a vinyl radical from cinnamate.
We propose that inactivation of PHM occurs via the Michael addition of an active site nucleophile to cinnamate (). α,β-Unsaturated carbonyls are known Michael acceptorsCitation71 and this chemistry is reversible, meaning that the covalently bound cinnamate could have been lost during the procedures used to evaluate inactive PHM. This would account for PHM being unlabeled after inactivation by either [14C]-cinnamate or N-dansyl-4-aminocinnamate. The tiopronin protection studies and our modeling results indicate that the cinnamates bind in the active site. Potential active site nucleophiles that could serve as Michael donors include Lys-134, Glu-313, and Tyr-318Citation36,Citation53,Citation62,Citation72. Although methionine is a weak Michael donor, the potential reaction between either of the two important active site methionine residues, Met-109 or Met-314Citation53,Citation62,Citation73, and the olefin might explain olefinic inactivation of PHM. A Met-olefin Michael adduct would be readily reversible; thus, accounting for the lack of PHM labeling by [14C]-cinnamate or N-dansyl-4-aminocinnamate. Formation of a reversible Michael adduct between an active site nucleophile and cinnamate is consistent with the lack of a kinetic isotope effect on kinact,obs because α-secondary deuterium effects are small (∼20%, see referenceCitation74) and the lack of significant substituent effects on kinact,obs because electron-donating or electron-withdrawing groups at the para-position of cinnamate would have only a small effect on the electropositive nature of the cinnamoyl β-carbon. The β-carbon of cinnamate is the most electropositive and is the carbon atom most susceptible to nucleophilic attack.
Figure 6. Proposed mechanism for the cinnamate-mediated inactivation of PHM. E-Nu represents an active site nucleophile, possibilities include Lys-134, Glu-313, Tyr-318 Met-109 or Met-314.
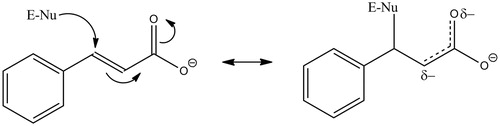
While the reversible Michael addition of an active site nucleophile to cinnamate is one plausible mechanism to explain PHM inactivation, two questions remain. Why does cinnamate inactivation require turnover conditions and why does cinnamate-inactivated PHM remain inactive? Studies of the kinetic mechanism show that the O2 and the oxidizable substrate only bind after the reduction of enzyme-bound 2Cu(II) to 2Cu(I) by ascorbateCitation75. We found that O2 binds after hippurate or benzyaldehyde imino-oxy acetateCitation76 rendering the O2-requirement for cinnamate inactivation perplexing. Either the order of binding for cinnamate is different, requiring O2 to bind to reduced PHM before cinnamate can bind, or O2 binding to the reduced PHM–cinnamate complex results in a subtle rearrangement in the active site to facilitate cinnamate attack by the active site nucleophile. The lack of reactivation of cinnamate-inactivated PHM would result from a relatively slow off-rate from native PHM. Thus, dialysis or dilution of the cinnamate-inactivated PHM would not show a significant regain in activity, consistent with our data. However, loss of the PHM native structure by denaturation or proteolysis would significantly increase the off-rate for cinnamate accounting for the lack of label in denaturated PHM after inactivation with either 14C-cinnamate or N-dansyl-4-aminocinnamate. Further complicating these experiments is the loss of PHM activity under turnover conditionsCitation77 meaning that reactivation of cinnamate-inactivated PHM under non-denaturing conditions cannot attain 100% of the original activity.
Another potential explanation for PHM inactivation is the cinnamate-dependent removal of copper from the enzyme. Copper-free PHM is catalytically inactiveCitation53,Citation62,Citation78. Cinnamates are known to form complexes with both Cu(I)Citation79,Citation80 and Cu(II)Citation81 and copper removal/chelation has been attributed to specific PHMCitation61 and DβM inhibitorsCitation68. PHM inactivation requires turnover; thus, inactivation of PHM by cinnamate could result from the formation of a relatively high affinity cinnamate–Cu(I) complex. Our data are inconsistent with the simple removal of the copper from PHM to yield inactive apo-enzyme because this mode of inactivation would be reversibleCitation78. The time-dependence in the cinnamate inactivation could result in the slow accumulation of a tight-binding E–2Cu(I)–cinnamate complex. The cinnamate would have to bind to PHM–2Cu(I) with sufficient affinity to explain the irreversibility of the inactivation. PHM denaturation would result in release of the cinnamate accounting for the lack of enzyme labeling with either [14C]-cinnamate or N-dansyl-4-aminocinnamate. Again, the O2-dependence of the cinnamate-mediated activation is problematic. Either cinnamate only binds to the E–2Cu(I)–O2 complex to form an E–2Cu(I)–O2–cinnamate complex or O2 binding results in a subtle conformational change that facilitates the formation of the a E–2Cu(I)–O2–cinnamate complex. It is entirely possible that the cinnamate-mediated inactivation of PHM results from a combination of both the formation of a Michael adduct between an active site nucleophile and cinnamate and the formation of tight enzyme-bound Cu(I)–cinnamate complex. Alternatively, Michael addition of an active site nucleophile could also subtly alter the PHM structure to enable the formation of a tight enzyme-bound Cu(I)–cinnamate complex that decays slowly relative to the retro-Michael reaction.
The Michael addition of an active site nucleophile to an unsaturated carbonyl compound might also account for the inactivation of PHM by the olefinic inactivators. The inactivation by PBA is more complex because, in contrast to the other olefinic inactivators, PBA can serve as a PHM substrate. Yet, PHM is not labeled by PBACitation37. We propose that the PBA-mediated inactivation of PHM occurs by more than one process. Inactivation by PBA could occur via Michael addition/Cu(I) chelation, like the chemistry we have outlined above, and via a reaction between an active site amino acid and PBA-derived radicalCitation35–37. PHM inactivation by a PBA-derived radical would lead, most likely, to a covalent bond between PBA and the enzyme. Hence, inactivation by [3H]-PBA should yield [3H]-enzyme, which was not found. Based on this result, we suggest that Michael addition/Cu(I) chelation is the predominant inactivation chemistry and that inactivation via a PBA-derived radical only accounts for a small percentage of the inactivation yielding levels of labeled enzyme that were below the level of detection.
In conclusion, we have demonstrated that cinnamate and other cinnamate analogs are capable of PHM inactivation. The most likely explanation for the inactivation of PHM mediated by the cinnamates is probably the result of the reversible Michael addition of an active site nucleophile at the α,β-unsaturated carbonyl of the cinnamates. The slow accumulation of a tight and inactive PHM–2Cu(I)–cinnamate–O2 complex could also contribute to the net inactivation of PHM. Similar chemistry may account for the inactivation of PHM by other olefins. The development of a drug possessing an α,β-unsaturated carbonyl should be evaluated as a PHM inactivator, an issue of particular concern given the importance of PHM in α-amidated peptide hormone biosynthesis. Also, these findings could prove therapeutically relevant as PHM dysfunction is correlated to a number of human diseasesCitation17–22, meaning that PHM could be a target for the development of novel olefinic-based drugs.
Supplementary material available online
Supplementary Figures S1-S5.
Acknowledgements
The authors acknowledge fruitful discussions of this work with Dr. Randy W. Larsen and technical support from Jason Cuce.
Declaration of interest
This work was supported, in part, by grants from the National Institutes of Health (R15-GM073659 and R03-DA03432), the Gustavus and Louise Pfieffer Research Foundation, the University of South Florida – Established Researcher Grant Program, the Wendy Will Case Cancer Fund, the Eppley Foundation for Research, the Milheim Foundation for Cancer Research, the Shin Foundation for Medical Research, and the Shirley W. & William L. Griffin Foundation to D.J.M.; a predoctoral fellowship from the American Heart Association (0415259B) and a postdoctoral fellowship from the National Science Foundation CI-TraCS Program (OCI-1122919) to E.W.L.; and from the National Institutes of Health (NIH-RCMI, 2G12MD007595-04 and 1SC3GM112558-01) and Louisiana Cancer Research Consortium to N.R.M. The contents are solely the responsibility of the authors and do not necessarily represent the official views of the Louisiana Cancer Research Consortium or the National Institutes of Health.
Notes
*A reviewer noted that the no cinnamate control in exhibited loss of activity. A repeat of the experiment to eliminate the activity loss in the no cinnamate control is difficult to accomplish because PHM exhibits a loss of activity under turnover conditions, probably the result of reactive, reduced O2-species that from during catalysis (see referenceCitation77). The inclusion of catalase in the inactivation experiments serves to decrease this problem as much as possible.
†The inactivation and inhibition parameters were measured at one fixed concentration of O2 and ascorbate and are, thus, reported as the kinact,obs, KI,obs, and (kinact/KI)obs.
References
- Bradbury AF, Finnie MDA, Smyth DG. Mechanism of C-terminal amide formation by pituitary enzymes. Nature 1982;298:686–8
- Eipper BA, Mains RE, Glembotski CC. Identification in pituitary tissue of a peptide α-amidation activity that acts on glycine-extended peptides and requires molecular oxygen, copper, and ascorbic acid. Proc Natl Acad Sci USA 1983;80:5144–8
- Young SD, Tamburini PP. Enzymatic peptidyl α-amidation proceeds through formation of an α-hydroxyglycine intermediate. J Am Chem Soc 1989;111:1933–4
- Katopodis AG, Ping D, May SW. A novel enzyme from bovine neurointermediate pituitary catalyzes dealkylation of α-hydroxyglycine derivatives, thereby functioning sequentially with peptidylglycine α-amidating monooxygenase in peptide amidation. Biochemistry 1990;29:6115–20
- Takahashi K, Harada S, Higashimoto Y, et al. Involvement of metals in enzymatic and nonenzymatic decomposition of C-terminal α-hydroglycine to amide: an implication for the catalytic role of enzyme-bound zinc in the peptidylamidoglycolate lyase reaction. Biochemistry 2009;48:1654–62
- Chufán EE, De M, Eipper BA, et al. Amidation of bioactive peptides: the structure of the lyase domain of the amidating enzyme. Structure 2009;17:965–73
- Eipper BA, Milgram SL, Husten EJ, et al. Peptidylglycine α-amidating monooxygenase: a multifunctional protein with catalytic, processing, and routing domains. Protein Sci 1993;2:489–97
- Zabriskie TM, Klinge M, Szymanski CM, et al. Peptide amidation in an invertebrate – purification, characterization and inhibition of peptidylglycine α-hydroxylating monooxygenase from the heads of honeybees (Apis mellifera). Arch Insect Biochem Physiol 1994;26:27–48
- Kolhekar AS, Roberts MS, Jiang N, et al. Neuropeptide amidation in Drosophila: separate genes encode the two enzymes catalyzing amidation. J Neurosci 1997;17:1363–76
- Hauser F, Williamson M, Grimmelikhuijzen CJP. Molecular cloning of a peptidylglycine α-hydroxylating monooxygenase from sea anemones. Biochem Biophys Res Commun 1997;241:509–12
- Fan X, Spijker S, Akalal D-BG, Nagle GT. Neuropeptide amidation: cloning of a bifunctional α-amidating enzyme from Aplysia. Brain Res Mol Brain Res 2000;82:25–34
- Spijker S, Smit AB, Eipper BA, et al. A molluscan peptide α-amidating enzyme precursor that generates five distinct enzymes. FASEB J 1999;13:735–48
- Eipper BA, Green CB-R, Campbell TA, et al. Alternative splicing and endoproteolytic processing generate tissue-specific forms of pituitary peptidylglycine α-amidating monooxygenase. J Biol Chem 1992;267:4008–15
- Stoffers DA, Green CB-R, Eipper BA. Alternative mRNA splicing generates multiple forms of peptidyl-glycine α-amidating monooxygenase in rat atrium. Proc Natl Acad Sci USA 1989;86:735–9
- Wand GS, Ney RL, Baylin S, et al. Characterization of peptide alpha-amidation activity in human tissue and plasma. Metabolism 1985;34:1044–52
- Eipper B, Mains RE. Peptide α-amidation. Annu Rev Physiol 1988;50:333–4
- Treston AM, Scott FM, Vos M, et al. Biochemical characterization of peptide α-amidation enzyme activities of human neuroendocrine lung cancer cell lines. Cell Growth Diff 1993;4:911–20
- Rocchi P, Boudouresque F, Zamora AJ, et al. Expression of adrenomedullin and peptide amidation activity in human prostate cancer and in human prostate cancer cell lines. Cancer Res 2001;61:1196–206
- Bauer JD, Sunman JA, Foster MS, et al. Anti-inflammatory effects of 4-phenyl-3-butenoic acid and 5-(acetylamino)-4-oxo-6-phenyl-2-hexenoic acid methyl ester, potential inhibitors of neuropeptide bioactivation. J Pharmacol Exp Ther 2007;320:1171–7
- Jeng AY, Fujimoto RA, Chou M, et al. Suppression of substance P biosynthesis in sensory neurons of dorsal root ganglion by prodrug esters of potent peptidylglycine a-amidating monooxygenase. J Biol Chem 1997;272:14666–71
- Ogonowski AA, May SW, Moore AB, et al. Antiinflammatory and analgesic activity of an inhibitor of neuropeptide amidation. J Pharmacol Exp Ther 1997;280:846–53
- Wand GS, May C, May V, et al. Alzheimer's disease: low levels of peptide α-amidation activity in brain and CSF. Neurology 1987;37:1057–61
- Scherkenbeck J, Zdobinsky T. Insect neuropeptides: structures, chemical modifications and potential for insect control. Bioorg Med Chem 2009;17:4071–84
- Merkler DJ, Asser AS, Baumgart LE, et al. Substituted hippurates and hippurate analogs as substrates and inhibitors of peptidylglycine α-hydroxylating monooxygenase (PHM). Bioorg Med Chem 2008;16:10061–74
- Katopodis AG, May SW. Novel substrates and inhibitors of peptidylglycine α-amidating monooxygenase. Biochemistry 1990;29:4541–8
- Erion MD, Tan J, Wong M, Jeng AY. Inhibition of peptidylglycine α-amidating monooxygenase by N-substituted homocysteine analogs. J Med Chem 1994;37:4430–7
- Mounier CE, Shi J, Sirimanne SR, et al. Pyruvate-extended amino acid derivatives as highly potent inhibitors of carboxy-terminal peptide amidation. J Biol Chem 1997;272:5016–23
- Andrews MD, O'Callaghan KA, Vederas JC. Synthesis of tripeptide inhibitors of peptidylglycine α-amidating monooxygenase (PAM) containing d- and l-styrylglycine. Tetrahedron 1997;53:8295–306
- Cao F, Gamble AB, Kim H-K, et al. Potent and selective inhibitors of human peptidylglycine α-amidating monooxygenase. MedChemComm 2011;2:760–3
- Foster MS, Oldham CD, May SW. Looking glass mechanism-based inhibition of peptidyglycine α-amidating monooxygenase. Tetrahedon Asymm 2011;22:283–93
- Morris KM, Cao F, Onagi H, et al. Prohormone-substrate peptide sequence recognition by peptidylglycine α-amidating monooxygenase and its reflection in increase glycolate inhibitor potency. Bioorg Med Chem Lett 2012;22:7015–18
- Rhodes CH, Honisnger C. Structure–activity relationships among inhibitors of peptidyglycine amidating monooxygenase. Ann NY Acad Sci 1993;689:663–6
- Bradbury AF, Mistry J, Smyth DG. Fluorescent inhibitors of peptidylglycine hydroxylase. In: Giralt E, Andreu D, eds. Peptides 1990, Proceedings of the Twenty-First European Peptide Symposium. Leiden: ESCOM Science Publishers B.V.; 1991:763–5
- Bradbury AF, Mistry J, Roos BA, Smyth DG. 4-Phenyl-3-butenoic acid, an in vivo inhibitor of peptidylglycine hydrolase (peptide amidating enzyme). Eur J Biochem 1990;189:363–8
- Cao, F, Easton CJ. Production and regulation of levels of amidated peptide hormones. Aust J Chem 2013;66:297–307
- Langella E, Pierre S, Ghattas W, et al. Probing the peptidylglycine α-hydroxylating monooxygenase active site with novel 4-phenyl-3-butenoic acid based inhibitors. ChemMedChem 2010;5:1568–76
- Driscoll WJ, König S, Fales HM, et al. Peptidylglycine α-hydroxylating monooxygenase generates two hydroxylated products from its mechanism-based suicide substrate, 4-phenyl-3-butenoic acid. Biochemistry 2000;39:8007–16
- Casara P, Ganzhorn A, Philippo C, et al. Unsaturated thioacetic acids as novel mechanism-based inhibitors of peptidylglcyine α-hydroxylating monooxygenase. Bioorg Med Chem Lett 1996;6:393–6
- Barrett BJW, Easton CJ, Henry DJ, et al. Inhibition of peptidylglycine α-amidating monooxygenase by exploitation of factors affecting the stability and ease of formation of glycyl radicals. J Am Chem Soc 2004;126:13306–11
- Chen P, Solomon EI. Oxygen activation by the noncoupled binuclear copper site in peptidylglycine α-hydroxylating monooxygenase. Reaction mechanism and role of the noncoupled nature of the active site. J Am Chem Soc 2004;126:4991–5000
- Crespo A, Martí MA, Roitberg AE, et al. The catalytic mechanism of peptidylglycine α-hydroxylating monooxygenase investigated by computer simulation. J Am Chem Soc 2006;128:12817–28
- Abad E, Rommel JB, Kästner J. Reaction mechanism of the bicopper enzyme peptidylglycine α-hydroxylating monooxygenase. J Biol Chem 2014;289:13726–38
- Hoskins JA. The occurrence, metabolism and toxicity of cinnamic acid and related compounds. J Appl Toxicol 1984;4:283–92
- Spanos GA, Wrolstand RE. Phenolics of apple, pear, and white grape juices and their changes with processing and storage – a review. J Agric Food Chem 1992;40:1478–87
- Letizia CS, Cocchiara J, Lapczynski A, et al. Fragrance material review on cinnamic acid. Food Chem Toxicol 2005;43:925–43
- Sharma P. Cinnamic acid derivatives: a new chapter of various pharmacological activities. J Chem Pharm Res 2011;3:403–23
- Babu R, Nabi SA, Swapna S, Apparao C. Cinnamic acid as one of the antidiabetic active principle(s) from the seeds of Syzygium alternifolium. Food Chem Toxicol 2012;50:1425–31
- De P, Baltas M, Bedos-Belval F. Cinnamic acid derivatives as anticancer agents – a review. Curr Med Chem 2011;18:1672–703
- De P, Bedos-Belval F, Vanucci-Bacque C, Baltas M. Cinnamic acid derivatives in tuberculosis, malaria, and cardiovascular diseases – a review. Curr Org Chem 2012;16:747–68
- Wilcox BJ, Ritenour-Rodgers KJ, Asser, AS, et al. N-Acylglycine amidation: implications for the biosynthesis of fatty acid primary amides. Biochemistry 1999;38:3235–45
- Bradford MM. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal Biochem 1976;72:248–54
- Christman AA, Foster PW, Esterer MB. The allantoin content of blood. J Biol Chem 1944;155:161–71
- Prigge ST, Eipper BA, Mains RE, Amzel LM. Dioxygen binds end-on to mononuclear copper in a precatalytic complex. Science 2004;304:864–7
- GLIDE. Portland, OR, NY: Schrodinger LLC; 2000
- Qsite. Portland, OR, NY: Schrodinger LLC; 2000
- Friesner RA, Banks JL, Murphy RB, et al. Glide: a new approach for rapid, accurate docking and scoring. 1. Method and assessment of docking accuracy. J Med Chem 2004;47:1739–49
- Cho AE, Guallar V, Berne BJ, Friesner R. Importance of accurate charges in molecular docking: quantum mechanical/molecular mechanical (QM/MM) approach. J Comput Chem 2005;26:915–31
- Jaguar, Version 6.5. New York: Schrodinger LLC; 2006
- Tamburini PP, Young SD, Jones BN, et al. Peptide substrate specificity of the α-amidating enzyme isolated from rat medullary thyroid CA-77 cells. Int J Pept Protein Res 1990;35:153–6
- Morrison JF, Stone SR. Approaches to the study and analysis of the inhibition of enzymes by slow- and tight-binding inhibitors. Comments Mol Cell Biophys 1985;2:347–369
- McIntyre NR, Lowe EW Jr, Chew GH, et al. Thiorphan, tiopronin, and related analogs as substrates and inhibitors of peptidylglycine α-amidating monooxygenase (PHM). FEBS Lett 2006;580:521–32
- Prigge ST, Kolhekar AS, Eipper BA, Amzel LM. Substrate-mediated electron transfer in peptidyl α-hydroxylating monooxygenase. Nat Struct Biol 1999;6:976–83
- McIntyre NR, Lowe EW Jr, Belof JL, et al. Evidence for substrate preorganization in the peptidylglycine α-amidating monooxygenase reaction describing the contribution of ground state tunneling. J Am Chem Soc 2010;132:16393–402
- Zabriskie TM, Cheng H, Vederas JC. Mechanism-based inactivation of peptidylglycine α-hydroxylating monooxygenase (PHM) by a substrate analogue, d-phenylalanyl-l-phenylalanyl-d-vinyglycine. J Am Chem Soc 1992;114:2270–2
- Ortiz de Montellano PR, Mico BA. Destruction of cytochrome P-450 by ethylene and other olefins. Mol Pharmacol 1980;18:128–35
- Ortiz de Montellano PR, Kunze KL, Beilan HS, Wheeler C. Destruction of cytochrome P-450 by vinyl fluoride, fluroxene, and acetylene. Evidence for a radical intermediate in olefin oxidation. Biochemistry 1982;21:1331–9
- Padgette SR, Wimalasena K, Herman HH, et al. Olefin oxygenation and N-dealkylation by dopamine β-monooxygenase: catalysis and mechanism-based inhibition. Biochemistry 1985;24:5826–39
- Sirimanne SR, May SW. Interaction of non-conjugated olefinic substrate analogues with dopamine β-monooxygenase: catalysis and mechanism-based inhibition. Biochem J 1995;306:77–85
- Mi L, Zuberbühler AD. Cuprous complexes and dioxygen. Part 12. Rate law and mechanism of the copper-catalyzed oxidation of ascorbic acid in aqueous acetonitrile. Helv Chim Acta 1992;75:1547–56
- Berkowitz J, Mayhew CA, Ruščić B. A photoionization study of the vinyl radical. J Chem Phys 1988;88:7396–404
- Amslinger S. The tunable functionality of α,β-unsaturated carbonyl compounds enables their differential application in biological systems. ChemMedChem 2010;5:351–6
- Kolhekar AS, Keutmann HT, Mains RE, et al. Peptidylglycine α-hydroxylating monooxygenase: active site residues, disulfide linkages, and a two-domain model of the catalytic core. Biochemistry 1997;36:10901–9
- Bauman AT, Broers BA, Kline CD, Blackburn NJ. A copper–methionine interaction controls the pH-dependent activation of peptidylglycine monooxygenase. Biochemistry 2011;50:10819–28
- Cleland WW. The use of isotope effects to determine enzyme mechanisms. Arch Biochem Biophys 2005;433:2–12
- Gilligan JP, Lovato SJ, Mehta NM, et al. Multiple forms of peptidyl α-amidating enzyme: purification from rat medullary thyroid carcinoma CA-cell-conditioned media. Endocrinology 1989;124:2729–36
- McIntyre NR, Lowe EW Jr, Merkler DJ. Imino-oxy acetic acid dealkylation as evidence for an inner-sphere alcohol intermediate in the reaction catalyzed by peptidylglycine α-hydroxylating monooxygenase. J Am Chem Soc 2009;131:10308–19
- Merkler DJ, Kulathila R, Tamburini PP, Young SD. Selective inactivation of the hydroxylase activity of bifunctional rat peptidylglycine α-amidating enzyme. Arch Biochem Biophys 1992;294:594–602
- Kulathila R, Consalvo AP, Fitzpatrick PF, et al. Bifunctional peptidylglycine α-amidating enzyme requires two copper atoms for maximum activity. Arch Biochem Biophys 1994;311:191–5
- Lorain C, Bolte M, Lemaire, J. Etude de la photoreactive des carboxylates de cuivre (I). I. Etude du cinnamate du cuivre (I). Nouv J Chim 1981;5:643–9
- Hallnemo G, Olsson T, Ullenius C. Initial formation of an olefin-copper(I) π-complex in conjugate addition of lithium dimethylcuprate to t-butyl cinnamate. J Organomet Chem 1984;265:C22–4
- Busnot A, Busnot F, Hemidy JF, Le Querler JF. New complexes of copper cinnamates and imidazole or substituted imidazole. Thermochim Acta 1993;228:219–30