
Abstract
An efficient synthesis of several N-[(1-benzyl-1,2,3-triazol-4-yl)methyl]carboxamides in the 13β- and 13α-d-secoestrone series is reported. Novel triazoles were synthesized via the Cu(I)-catalyzed azide–alkyne cycloaddition of steroidal alkynyl carboxamides and p-substituted benzyl azides. Each of the products was evaluated in vitro by means of MTT assays for antiproliferative activity against a panel of human adherent cancer cell lines (HeLa, MCF-7, A431 and A2780). Some of them exhibited activities similar to those of the reference agent cisplatin. On change of the substitution pattern of the benzyl group of the azide, great differences in the cell growth-inhibitory properties were observed. The p-alkylbenzyl-substituted triazoles selectively exerted high cytostatic action against A2780 cells, with IC50 values of 1 µM. We investigated the potential inhibitory action exerted on the human 17β-HSD1 activity of the new secosteroids. Three triazoles effectively suppressed the estrone to 17β-estradiol conversion with IC50 values in low micromolar range.
Introduction
The development of anticancer drugs based on naturally occurring biologically active substances is one of the most important objectives of modern medicinal chemistryCitation1–3. Structural modifications of steroidal hormones may lead to compounds with diverse biological activitiesCitation4–6. The challenge of such drug design is the discovery of lead compounds with selective biological potency: when the new action is not followed by the original hormonal activity. The literature reveals that the homologization or opening of ring D or inversion of the configuration at C-13 of the estrane skeleton may lead to the complete loss of estrogenic activityCitation7–9. We recently reported that 3-benzyloxy-d-secoestrone alcohols and oximes display high in vitro antiproliferative potential against a number of cancer cell lines, with IC50 values in the low micromolar or submicromolar rangeCitation10,Citation11. The most potent compounds were found to induce apoptosis via the enhancement of tubulin polymerization or to cause disturbance in a cell cycle. Furthermore, 16-oximes of 13α-estrone proved to inhibit the proliferation of particular cancer cell lines effectively, with high selectivity for cancer cell linesCitation12. As d-seco- and 13α-estrones have been reported to possess no estrogenic activity, but to exert growth-inhibitory action, we chose these compounds as candidates for the design of potentially cytostatic derivatives with selective biological activity. To diversify the structure, starting compounds bearing different protecting groups at C-3 were used. d-Secoestrone carboxylic acids are readily available in both the 13α- and the 13β-estrone seriesCitation10,Citation13–15. We therefore set out to build additional structural elements, such as a triazole moiety, onto the carboxylic acid scaffolds, in order to obtain compounds with enhanced antiproliferative propertiesCitation10. The triazole structure is a good mimic of peptide bonding with improved metabolic and proteolytic stabilityCitation16; moreover, there is literature evidence of steroidal triazoles with moderate or enhanced antiproliferative behaviourCitation17–21. Introduction of a terminal alkyne function onto the carboxyl group was planned by using propargylamine and peptide coupling reagents, followed by the formation of triazoles from the alkynes by using differently substituted benzyl azides as dipoles. The resulting N-[(1-benzyl-1,2,3-triazol-4-yl)methyl]carboxamide structural moiety is completely or partially present in a number of literature drug candidate examples: in small molecules and in triterpenoids with diverse biological activities, such as antitumour or antiviralCitation22–24. Certain carboxamido- or triazolyl-derivatives of estrone (C15 or C16 substituted) are able to inhibit the 17β-hydroxysteroid dehydrogenase type 1 (17β-HSD1) isozymeCitation5,Citation25,Citation26 and to block the estrone to 17β-estradiol conversion. This inhibition suppresses both the gonadal and the peripheral 17β-estradiol synthesis and the hormone ablation might be applied for the treatment of estrogen-dependent diseases such as endometriosis, as well as breast and ovarian cancers.
In view of these promising literature evidences, we planned to determine the in vitro antiproliferative activities of the newly synthesized secoestrones by means of MTT assays against a panel of human adherent cancer cell lines (HeLa, MCF-7, A431 and A2780). We also planned to study a potential inhibition of the human 17β-HSD1 enzyme.
Experimental
Chemistry
Melting points (mp) were determined with a Kofler hot-stage apparatus and are uncorrected. Elemental analyses were performed with a Perkin-Elmer CHN analyzer model 2400. Thin-layer chromatography: silica gel 60 F254; layer thickness 0.2 mm (Merck, Darmstadt, Germany); detection with iodine or UV (365 nm) after spraying with 5% phosphomolybdic acid in 50% aqueous phosphoric acid and heating at 100–120 °C for 10 min. Flash chromatography: silica gel 60, 40–63 μm (Merck, Darmstadt, Germany). 1H NMR spectra were recorded in CDCl3 solution (if not otherwise stated) with a Bruker DRX-500 instrument at 500 MHz, with Me4Si as internal standard. 13C NMR spectra were recorded with the same instrument at 125 MHz under the same conditions. Full scan mass spectra of the compounds were acquired in the range 50 to 800 m/z with an Agilent 500MS Ion trap mass spectrometer equipped with an electrospray ionization source.
General procedure for the preparation of N-(prop-2-yn-1-yl)carboxamides 5–8
Compounds 1 or 3 (314 mg, 1.00 mmol) or 2 or 4Citation15 (390 mg, 1.00 mmol) were dissolved in N,N-dimethylformamide (DMF, 5 ml), and 1-hydroxybenzotriazole (HOBt, 148.6 mg, 1.1 mmol), N,N,N′,N′-tetramethyl-O-(1H-benzotriazol-1-yl)uronium hexafluorophosphate (HBTU, 417.2 mg, 1.1 mmol) and N,N-diisopropylethylamine (DIPEA, 0.17 ml, 1.00 mmol) were added. After 15 minutes of stirring, propargylamine (0.06 ml, 1.00 mmol) in DMF (1 ml) was added. The mixture was stirred at room temperature for 2 h, and then diluted with water and extracted with CH2Cl2. The combined organic phases were washed with water until neutral and dried over Na2SO4. The crude products 5–8 were subjected to flash chromatography with CH2Cl2 as eluent.
3-Methyloxy-14β-(prop-2-en-yl)-N-(prop-2-yn-1-yl)-des-D-estra-1,3,5(10)-trien-13α-carboxamide 5
Compound 1 (314 mg, 1.00 mmol). Yield: 274 mg (78%), oil, Rf = 0.40 (dichloromethane). 1H NMR (500 MHz, CDCl3): δ [ppm] = 1.18(s, 3H, 18-H3); 2.24(s, 1H, C≡CH); 2.83(m, 2H, 6-H2); 3.78(s, 3H, OCH3); 4.06(m, 2H, HN–CH2); 4.97(m, 2H, 16 a-H2); 5.85(m, 1H, 16-H); 5.91(m, 1H, NH); 6.63(s, 1H, J = 2.2 Hz, 4-H); 6.71(dd, 1H, J = 8.6 Hz, J = 2.2 Hz, 2-H); 7.18(d, 1H, J = 8.6 Hz, 1-H). 13C NMR δ [ppm] = 15.2(C-18); 25.9; 27.2; 29.5; 30.2; 35.3; 37.9; 40.9; 42.9; 45.7; 47.3, 55.2(OCH3); 71.6(C≡CH); 79.8(C≡CH); 111.7(C-2); 113.5(C-4); 115.0(C-16a); 126.3(C-1); 132.2(C-10); 137.8(C-5); 138.9(C-16); 157.5(C-3); 177.9(C-17). MS m/z (%): 352 (MH+, 100), 236 (38). Anal. Calcd. for C23H29NO2: C, 78.59; H, 8.32. Found: C, 78.67;H, 8.45%.
Analytical data on the described propargyl derivatives 5–8 are presented in supporting information (SI).
General procedure for the synthesis of triazoles 10–13
To a stirred solution of the appropriate terminal alkyne (5–8, 1.00 mmol) in toluene (5 ml), benzyl azide (9, 1.00 mmol), triphenylphosphine (Ph3P) (52 mg, 0.2 mmol), copper(I) iodide (CuI) (19 mg, 0.1 mmol) and DIPEA (0.52 ml, 3.00 mmol) were added. The reaction mixture was refluxed for 2 h, the solvent was evaporated off, and the residue was subjected to flash chromatography over silica gel with 50% ethyl acetate/50% CH2Cl2 as eluent.
3-Methyloxy-N-[(1-benzyl-1H-1,2,3-triazol-4-yl)methyl]-14β-(prop-2-en-yl)-des-D-estra-1,3,5(10)-trien-13α-carboxamide 10a
Compound 5 (351 mg, 1.00 mmol). Yield: 464 mg (96%), mp 61–63 °C, Rf = 0.44 (50% ethyl acetate/50% dichloromethane). 1H NMR (500 MHz, DMSO-d6): δ [ppm] = 1.05(s, 3H, 18-H3); 2.74(m, 2H, 6-H2); 3.68(s, 3H, OCH3); 4.29(m, 2H, HN–CH2); 4.81(m, 2H, 16a-H2); 5.55(s, 2H, N-CH2); 5.71(m, 1H, 16-H); 6.59(d, 1H, J = 2.2 Hz, 4-H); 6.67(dd, 1H, J = 8.6 Hz, J = 2.2 Hz, 2-H); 7.16(d, 1H, J = 8.6 Hz, 1-H); 7.29–7.36 overlapping multiplets: 2″-H, 3″-H, 4″-H, 5″-H, 6″-H; 7.84(m, 1H, C = CH); 8.12(m, 1H, NH). 13C NMR δ [ppm] = 15.2(C-18); 25.8; 26.9; 29.6; 34.6; 34.7; 36.9; 40.8; 42.5; 45.0; 46.7; 52.6; 54.8(OCH3); 111.6(C-2); 113.0(C-4); 114.3(C-16a); 122.8(C = CH); 126.3(C-1); 127.8(2C: C-2″, C-6″); 128.0(C-4″); 128.7(2C, C-3″, C-5″); 131.8(C-10); 136.1(C-1″); 137.3(C-5); 139.5(C-16); 145.8(C = CH); 157.0(C-3); 177.4(C-17). MS m/z (%): 485 (MH+, 100), 486 (36). Anal. Calcd. for C30H36N4O2: C, 74.35; H, 7.49. Found: C, 74.51; H, 7.63%.
Analytical data on the described triazolyl compounds 10–13 are presented in supporting information (SI).
MTT assay for determination of antiproliferative activity
The growth-inhibitory effects of the test compounds were determined on HeLa, MCF-7, A2780 and A431 cells isolated from cervical, breast, ovarian and skin cancer, respectivelyCitation27. These cell lines were obtained from the European Collection of Cell Cultures, Salisbury, UK. The cells were cultivated in minimal essential medium (Sigma-Aldrich, Budapest, Hungary) supplemented with 10% fetal bovine serum, 1% non-essential amino acids and an antibiotic-antimycotic mixture. All media and supplements were obtained from Life Technologies (Paisley, Scotland, UK). All cell types were seeded into 96-well plates at a density of 5000 cells/well and incubated with two concentrations (10 and 30 µM) of the compounds at 37 °C under cell culturing conditions. After a 72-h incubation, cells were treated with 5.0 mg/ml MTT [3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide] solution for 4 h. The precipitated formazan crystals were dissolved in dimethyl sulfoxide and the absorbance was read at 545 nm with a microplate reader. Wells with untreated cells were utilized as controls. For the selected molecules the assay was repeated with a set of dilutions and sigmoidal concentration–response curves were fitted to the measured data, and IC50 values were calculated by means of GraphPad Prism 4 (GraphPad Software, San Diego, CA). Cisplatin was used as a positive control in same concentrations as the test compounds.
Determination of 17β-HSD1 activity and its inhibition in the human placenta cytosol
Radioactive [6,7-3H(N)]estrone, S. A. = 50 Ci/mmol was purchased from the American Radiolabeled Chemicals (St. Louis, MO). Non-radioactive estrone and 17β-estradiol standards, NADH coenzyme, other chemicals and solvents with purity of analytical grade were purchased from Sigma (St. Louis, MO) and Fluka (Buchs, Switzerland). Kieselgel-G TLC layers (Si 254 F, 0.25 mm thick) were obtained from Merck (Darmstadt, Germany). Human term placenta specimens were collected and used under ethical approval of the institutional Human Investigation Review Board. Inhibitory effects exerted on the 17β-HSD1 activity by the newly synthesized compounds 5–8, and 10–13 were investigated via conversion of estrone to 17β-estradiol in vitro. Our previously published radioincubation methods for 3β-hydroxysteroid dehydrogenase/Δ5-Δ4-isomerase (Δ5-3β-HSD)Citation28,Citation29 and the 17β-hydroxysteroid dehydrogenase type 2 (17β-HSD2)Citation30 were modified and adapted to the 17β-HSD1 measurements. Human placental cytosol served as feasible source of the isozymeCitation31–33. Human term placenta specimens were combined and homogenized with an Ultra-Turrax in 0.1 M HEPES buffer (pH = 7.3) containing 1 mM EDTA and 1 mM dithiotreitol and the cytosol were obtained with fractionated centrifugation. The substrate was added to the incubate in 10 µl of a 25 v/v% propylene glycol in HEPES buffer solution, whereas test compounds were applied in 10 µl of dimethyl sulfoxide solution. (These organic solvent contents of 200 µl final volume of the HEPES buffer incubation medium did not reduce the enzyme activity substantially.) The incubates contained estrone substrate in 1µM final concentration with 250 000 dpm activity of tritium labelled tracer, and the coenzyme NADH was used in an excess 100 µM concentration. Enzymatic reaction was started by the addition of the cytosol aliquots. Incubation was carried out at 37 °C for 2.5 min and then was stopped by the addition of ethyl acetate and freezing. After extraction, unlabelled carriers of estrone and the product 17β-estradiol were added to the samples. The two steroids were separated by TLC with the solvent system dichloromethane/diisopropyl ether/ethyl acetate (70:15:15 v/v) and UV spots were used to trace the separated steroids. Spots were cut out and the radioactivity of the 17β-estradiol formed and of the estrone remaining was measured by means of liquid scintillation counting. Control incubates without test substances were prepared in every series. In these non-inhibited incubations 7–9% of estrone was transformed, therefore the substrate concentration could be accepted constant during the enzyme reaction. Test compounds were applied at 10 µM and their inhibitory effects were expressed by relative conversions, in comparison to the transformation measured in non-inhibited control incubation. IC50 values were determined for the most effective test compounds. In these cases, conversions were measured at six-eight different concentrations between 0.1–10 µM. IC50 results were calculated using an unweighted iterative leastsquares method for four-parameter logistic curve fitting. IC50 of unlabelled estrone was measured as reference.
Results and discussion
Chemistry
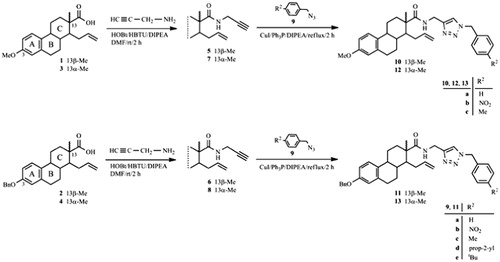
Two epimeric pairs of steroidal carboxylic acids 1–4 were chosen as starting materials for the introduction of a terminal alkyne function. The starting δ-alkenyl carboxylic acids 1–4 were obtained from the appropriate δ-alkenyl aldehydes via Jones oxidation, as described earlierCitation15. The coupling reactions were carried out by using propargylamine and peptide coupling reagents: HOBt and HBTU (Scheme 1) and proceeded in a chemoselective manner, yielding the desired carboxamides 5–8 in high yields, without the formation of side-products. The resulting N-(prop-2-yn-1-yl)carboxamides 5–8 were transformed into triazoles 10–13 with benzyl azides (9) under “click” reaction conditions (using CuI as catalyst, triphenylphosphane as stabilizing ligand, DIPEA as base and toluene as solvent). Benzyl azides 9a–e were prepared from benzyl bromides as described in the literatureCitation34–37. The structures of the triazoles were diversified by using benzyl azides containing different p-substituents, with the aim of determining the dependence of the biological activity on the structure of the triazoles. All the cycloadditions proceeded with full conversion of the starting compounds, no side-products were formed and the nature of the p-substituent of the benzyl ring did not influence the efficiency or the rate of the reactions. The structures of the newly synthesized compounds 5–8 and 10–13 were established from 1H and 13C NMR measurements.
Scheme 1. Synthesis of N-(prop-2-yn-1-yl)carboxamides 5–8 and the related triazoles 10–13.
Determination of antiproliferative properties of the newly synthesized compounds
Compounds 5–8 and 10–13 were evaluated in vitro by means of MTT assays for antiproliferative activity against HeLa (cervical carcinoma), A2780 (ovarian carcinoma), A431 (skin epidermoid carcinoma) and MCF-7 (breast adenocarcinoma) cells. Compounds 5–8 did not display substantial antiproliferative effects against the tested tumour cell lines (). The inhibitions of proliferation were generally under 50% at both applied concentrations, the 3-benzyl ether derivatives 6 and 8 proving to be slightly more potent than their 3-methyl ether counterparts 5, 7. Compound 6 at 30 μM exerted a close to 70% growth-inhibitory effect against A431 cells, but was inactive on HeLa, the proliferation of which was effectively inhibited by the epimeric counterpart (8). The inhibitory potencies of the triazoles 10–13, however, were greatly affected by the structural differences between the heterocyclic products. As concerns the 3-methyl ether derivatives 10 and 12, the determining moiety is the p-substituent on the N-benzyl ring. The p-methyl compound in both the 13α- and 13β-series 12c and 10c displayed the highest activity, 90% inhibition of the MCF-7 and A2780 cell lines being attained at 30 μM. The difference in growth-inhibitory behaviour depended on the orientation of the angular methyl group and was most marked in the case of the 3-benzyloxytriazoles 11 and 13. The results of the MTT assays of the 13α derivatives 13 revealed their substantially lower activities than those of the 13β compounds 11, and only 13c at 30 μM exhibited >50% inhibition on HeLa cells. In contrast, the three 13β-triazoles 11a–c selectively displayed high activity against A2780 cells. In view of the latter promising results, two additional p-alkyl-N-benzyl-substituted derivatives 11d,e were synthesized in order to examine whether the alkyl group facilitates the biological action. The MTT results revealed improved growth-inhibitory effects for 11d and 11e (95% inhibition at 30 μM on A2780 cells).
Table 1. Antiproliferative properties of the synthetized compounds 5–8 and 10–13.
Inhibition of 17β-HSD1
Argenta et al. assayed high activity of 17β-HSD1 in a series of ovarian epithelial carcinomasCitation38 and they concluded that 17β-HSD1-specific inhibitors may have role in the therapy of ovarian cancer. Reed et al. reported that the 17β-HSD1 mRNA is expressed in MCF-7 and T47D breast cancer cell lines, and there is high association between mRNA expression and enzyme activityCitation39. The literature reveals that selective inhibition of 17β-HSD1 reduces estrogen responsive cell growth of the mentioned cell linesCitation40. Since our present results of the MTT assays demonstrated remarkable activities for the potent compounds on ovarian cancer (A2780) and breast cancer (MCF-7) cell lines, herein we investigated potential in vitro inhibitory effect of the new secoestrone derivatives 5–8 and 10–13 towards the human placental 17β-HSD1. Compounds 10b,c and 13a exerted considerable inhibitions, IC50 values were found 1.3, 4.5 and 3.6 µM, respectively (). Inhibition potential of the p-nitrophenyl-derivative 10b is similar to the unlabelled estrone, for which 1.8 µM reference IC50 was determined. Other compounds displayed weak inhibitions, relative conversions were decreased to 53–89% at the 10 µM test concentration.
Table 2. In vitro inhibition of human placenta cytosol 17β-HSD1 activity by test compounds 5–8 and 10–13.
Conclusion
Novel antiproliferative triazolyl D-secoestrone derivatives were synthesized. Introduction of the N-(prop-2-yn-1-yl)carboxamide structural element led to compounds 5–8 with moderate cytostatic behaviour, but through the “clicking” of benzyl azides to the terminal alkyne function, the antiproliferative action was intensified. Among the 3-methoxytriazoles 10 and 12, irrespectively of the orientation of the angular methyl group, the substitution pattern on the N-benzyl ring substantially influenced the antiproliferative behaviour: the presence of the p-alkyl group appeared to be advantageous. In contrast, in the 3-benzyl ether series 11 and 13, the configuration of C-13 was the determining moiety: the 13β derivatives 11 exhibited much higher potency than that of their epimer counterparts 13, and displayed selective growth-inhibitory action against A2780 cells. It can be concluded that the combination of the latter structural components in the d-secoestrone series, i.e. the 3-benzyl protecting group, the triazolyl function and the 13β-methyl group, enhances the cytostatic potential. 17β-HSD1 inhibition tests performed in vitro with human placenta cytosol revealed that 10b,c and 13a effectively suppress estrone–estradiol conversion catalyzed by this isozyme. 17β-HSD1 inhibitory potential of d-secoestrone derivatives is a novel finding.
Although there is no correlation between the effects of the compounds on enzyme activity and on cell proliferation, secoestrones with high 17β-HSD1 inhibitory potential are promising scaffolds for the design of estrone-based antitumor agents with no estrogenic activity. To the best of our knowledge, no 17β-HSD1 inhibitor has entered the clinical trial step, because of the retained estrogenicity of the candidate compounds. Further investigations with secoestrones can be interesting and may provide valuable inhibitor structure-activity data.
Supplementary material available online
IENZ_1050008_Supp.pdf
Download PDF (314 KB)Declaration of interest
The authors report no declarations of interest. The authors are grateful for financial support from the Hungarian Scientific Research Fund (OTKA K101659 and K109293).
References
- Newman DJ, Cragg GM. Natural products, derivatives and mimics as antitumor agents. R Soc Chem 2011;320:3–36
- Saha SK, Khuda-Bukhsh AR. Molecular approaches towards development of purified natural products and their structurally known derivatives as efficient anti-cancer drugs: current trends. Eur J Pharm 2013;714:239–48
- Gupta A, Kumar SB, Negi AS. Current status on development of steroids as anticancer agents. J Steroid Biochem Mol Biol 2013;137:242–70
- Numazawa M, Ando M, Watari Y, et al. Structure–activity relationships of 2-, 4-, or 6-substituted estrogens as aromatase inhibitors. J Steroid Biochem Mol Biol 2005;96:51–8
- Poirier D. Contribution to the development of inhibitors of 17β-hydroxysteroid dehydrogenase types 1 and 7: key tools for studying and treating estrogen-dependent diseases. J Steroid Biochem Mol Biol 2011;125:83–94
- Lordan S, Mackrill JJ, O’Brien NM. Oxysterols and mechanisms of apoptopic signalling: implications in the pathology of degenerative diseases. J Nutr Biochem 2009;20:321–36
- Minorics R, Bozsity N, Wölfling J, et al. Antiproliferative effect of normal and 13-epi-D-homoestrone and their 3-methyl ethers on human reproductive cancer cell lines. J Steroid Biochem Mol Biol 2012;132:168–75
- Jovanovic-Santa S, Petrovic J, Andric S, et al. Synthesis, structure, and screening of estrogenic and antiestrogenic activity of new 3,17-substituted-16,17-seco-estratriene derivatives. Bioorg Chem 2003;31:475–84
- Ayan D, Roy J, Maltais R, Poirier D. Impact of estradiol structural modifications (18-methyl and/or 17-hydroxy inversion of configuration) on the in vitro and in vivo estrogenic activity. J Steroid Biochem 2011;127:324–30
- Mernyak E, Szabo J, Huber J, et al. Synthesis and antiproliferative effects of D-homo- and D-secoestrones. Steroids 2014;87:128–36
- Mernyak E, Fiser G, Szabo J, et al. Synthesis and in vitro antiproliferative evaluation of D-secooxime derivatives of 13β- and 13α-estrone. Steroids 2014;89:47–55
- Berenyi A, Minorics R, Ivanyi Z, et al. Synthesis and investigation of the anticancer effects of estrone-16-oxime ethers in vitro. Steroids 2013;78:69–78
- Schneider G, Hackler L, Sohar P. Steroids, XXXVIII. Neighboring group participation. IX. Preparation of 16a-(hydroxymethyl)-3-methoxyestra-1,3,5(10)-trien-17a-ol and solvolysis investigations. Liebigs Ann Chem 1988:679–83
- Wölfling J, Mernyák E, Forgó P, Schneider G. Stereoselective halogenation of the 16-hydroxymethyl-3-methoxy-13α-estra-1,3,5(10)-trien-17-ols and their solvolytic investigation. Steroids 2003;68:451–8
- Frank É, Mernyák E, Wölfling J, Schneider Gy. Stereoselective approach to some novel 16-methylated and 16-halomethylated tetrahydropyran and δ-lactone derivatives in both the normal and the 13α-estrone series. Syn Lett 2002;11:1803–6
- Pedersen DS, Abell A. 1,2,3-Triazoles in peptidomimetic chemistry. Eur J Org Chem 2011;2011:2399–411
- Kadar Z, Molnar J, Schneider G, et al. A facile “click” approach to novel 15β-triazolyl-5α-androstane derivatives, and an evaluation of their antiproliferative activities in vitro. Bioorg Med Chem 2012;20:1396–402
- Frank E, Molnar J, Zupko I, Wölfling J. Synthesis of novel steroidal 17α-triazolyl derivatives via Cu(I)-catalyzed azide-alkyne cycloaddition, and an evaluation of their cytotoxic activity in vitro. Steroids 2011;76:1141–8
- Kadar Z, Baji A, Zupko I, et al. Efficient approach to novel 1α-triazolyl-5α-androstane derivatives as potent antiproliferative agents. Org Biomol Chem 2011;9:8051–7
- Kadar Z, Frank E, Schneider G, et al. Efficient synthesis of novel A-ring-substituted 1,2,3-triazolylcholestane derivatives via catalytic azide-alkyne cycloaddition. Arkivoc 2012;3:279–96
- Jurasek M, Dzubak P, Sedlak D, et al. Preparation, preliminary screening of new types of steroid conjugates and their activities on steroid receptors. Steroids 2013;78:356–61
- Yu F, Peng Y, Wang Q, et al. Development of bivalent oleanane-type triterpenes as potent HCV entry inhibitors. Eur J Med Chem 2014;77:258–68
- Miller MJ, Moraski GC, Stefely J. Triazole derivatives as anticancer agents and preparation thereof. 2009; WO 2009111502
- Haiying S, Zaneta N-C, Jianfeng L, et al. Design, synthesis and characterization of a potent, non-peptide, cell-permeable, bivalent smac mimetic that concurrently targets both the BIR2 and BIR3 domains in XIAP. J Am Chem Soc 2007;129:15279–94
- Allan GM, Lawrence HR, Cornet J, et al. Modification of estrone at the 6, 16, and 17 positions: novel potent inhibitors of 17β-hydroxysteroid dehydrogenase type 1. J Med Chem 2006;49:1325–45
- Messinger J, Husen B, Koskimies P, et al. Estrone C15 derivatives–A new class of 17b-hydroxysteroid dehydrogenase type 1 inhibitors. Mol Cell Endocrinol 2009;301:216–24
- Mosmann T. Rapid colorimetric assay for cellular growth and survival: application to proliferation and cytotoxicity assays. J Immunol Methods 1983;65:55–63
- Tóth I, Szécsi M, Julesz J, Faredin I. Activity and inhibition of 3-beta-hydroxysteroid dehydrogenase/delta-5-4-isomerase in human skin. Skin Pharmacol 1997;10:160–8
- Darvas B, Székács A, Fónagy A, et al. Progesterone in Periplaneta americana and Neobellieria bullata adults from the procuticle phase until first progeny production. Gen Comp Endocrinol 1997;107:450–60
- Djurendic EA, Klisurić O, Szécsi M, et al. X-ray structural analysis and antitumor activity of new salicylic acid derivatives. Struct Chem 2014;25:1747–58
- Tremblay MR, Auger S, Poirier D. Synthesis of 16-(bromoalkyl)-estradiols having inhibitory effect on human placental estradiol 17 beta-hydroxysteroid dehydrogenase (17 beta-HSD type 1). Bioorg Med Chem 1995;3:505–23
- Poirier D, Dionne P, Auger S. A 6beta-(thiaheptanamide) derivative of estradiol as inhibitor of 17beta-hydroxysteroid dehydrogenase type 1. J Steroid Biochem Mol Biol 1998;64:83–90
- Wetzel M, Gargano EM, Hinsberger S, et al. Discovery of a new class of bicyclic substituted hydroxyphenylmethanones as 17β-hydroxysteroid dehydrogenase type 2 (17β-HSD2) inhibitors for the treatment of osteoporosis. Eur J Med Chem 2012;47:1–17
- Pardin C, Roy I, Lubell WD, Keillor JW. Reversible and competitive cinnamoyl triazole inhibitors of tissue transglutaminase. Chem Biol Drug Des 2008;72:189–96 (9a, 9b)
- Maycock CD, Santos JP, Duarte MF, et al. Study of selected benzyl azides by UV photoelectron spectroscopy and mass spectrometry. J Mol Struct 2010;980:163–71 (9a, 9c)
- Pötzsch R, Voit B. Thermal and photochemical crosslinking of hyperbranched polyphenylene with organic azides. Macromol Rapid Commun 2012;33:635–9 (9d)
- Barr L, Lincoln SF, Easton CJ. A cyclodextrin molecular reactor for the regioselective synthesis of 1,5-disubstituted-1,2,3-triazoles. Supramol Chem 2005;17:547–55 (9e)
- Chura JC, Ryu HS, Simard M, et al. Steroid-converting enzymes in human ovarian carcinomas. Mol Cell Endocrinol 2009;301:51–8
- Day JM, Tutill HJ, Newman SP, et al. 17β-hydroxysteroid dehydrogenase type 1 and type 2: association between mRNA expression and activity in cell lines. Mol Cell Endocrinol 2006;248:246–9
- Kruchten P, Werth R, Bey E, et al. Selective inhibition of 17β-hydroxysteroid dehydrogenase type 1 (17βHSD1) reduces estrogen responsive cell growth of T47-D breast cancer cells. J Steroid Biochem Mol Biol 2009;114:200–6